
The Roles of Fatty Acids and Apolipoproteins in the Kidneys


Abstract
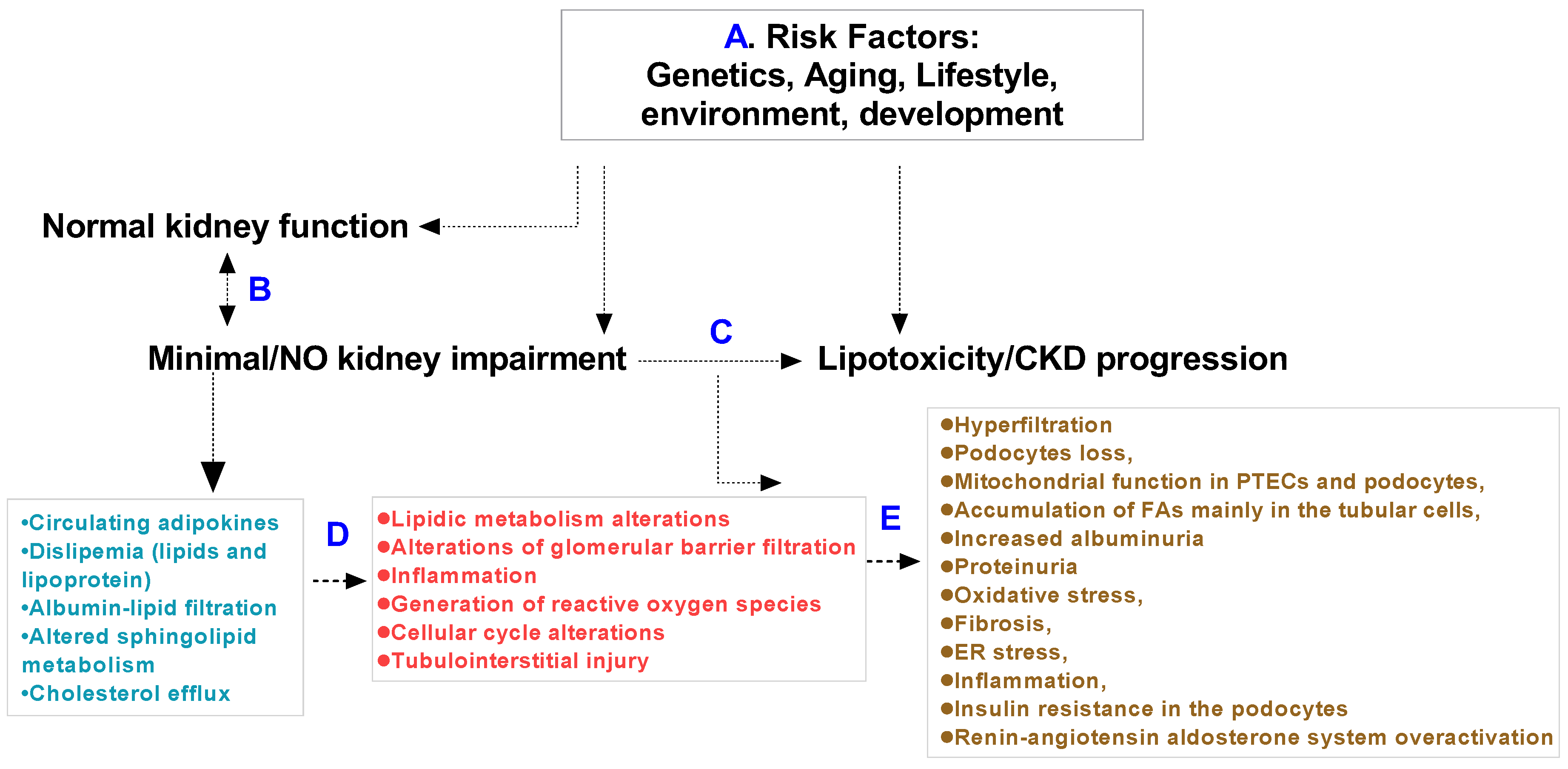
:1. Introduction
2. Physiological Roles of Fatty Acids in the Kidneys
3. Molecular and Pathological Roles of Fatty Acids in Renal Lipotoxicity and CKD
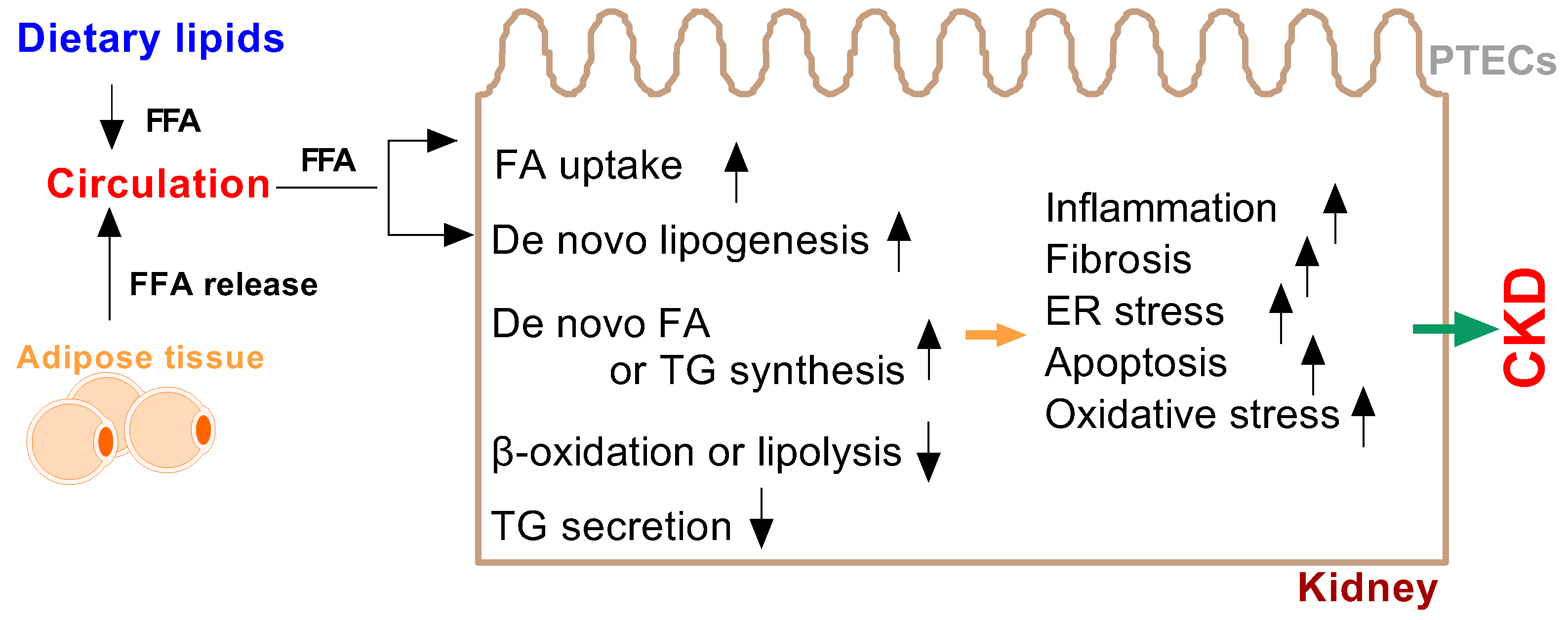
3.1. Molecular Mechanism of Fatty Acids in Renal Lipotoxicity
3.2. Glycotoxicity and Lipotoxicity
3.3. DKD and CKD
3.4. Different Single Cell Function and CKD
4. Roles and Biological and Pathological Functions of Apolipoproteins in the Kidneys and CKD
Pathological Role of Apolipoproteins in Kidney Diseases
5. Future Perspectives and Conclusions
Funding
Conflicts of Interest
References
- Grundy, S.M.; Benjamin, I.J.; Burke, G.L.; Chait, A.; Eckel, R.H.; Howard, B.V.; Mitch, W.; Smith, S.C., Jr.; Sowers, J.R. Diabetes and cardiovascular disease: A statement for healthcare professionals from the American Heart Association. Circulation 1999, 100, 1134–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meguid El Nahas, A.; Bello, A.K. Chronic kidney disease: The global challenge. Lancet 2005, 365, 331–340. [Google Scholar] [CrossRef]
- Wahba, I.M.; Mak, R.H. Obesity and obesity-initiated metabolic syndrome: Mechanistic links to chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 550–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maric-Bilkan, C. Obesity and diabetic kidney disease. Med. Clin. N. Am. 2013, 97, 59–74. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Komers, R. Pathophysiology of the diabetic kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the United States. JAMA 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Luyckx, V.A.; Tonelli, M.; Stanifer, J.W. The global burden of kidney disease and the sustainable development goals. Bull. World Health Organ. 2018, 96, 414D–422D. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Rodriguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermudez-Lopez, M.; Sanchez-Nino, M.D.; Perez-Fernandez, M.; Ortiz, A. Atherosclerosis in Chronic Kidney Disease: More, Less, or Just Different? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kon, V. Mechanisms for increased cardiovascular disease in chronic kidney dysfunction. Curr. Opin. Nephrol. Hypertens. 2009, 18, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Visconti, L.; Benvenga, S.; Lacquaniti, A.; Cernaro, V.; Bruzzese, A.; Conti, G.; Buemi, M.; Santoro, D. Lipid disorders in patients with renal failure: Role in cardiovascular events and progression of chronic kidney disease. J. Clin. Transl. Endocrinol. 2016, 6, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, A.B.; Voloshyna, I.; De Leon, J.; Miyawaki, N.; Mattana, J. Cholesterol Metabolism in CKD. Am. J. Kidney Dis. 2015, 66, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, B.C.; Kronenberg, F.; Beddhu, S.; Cheung, A.K. Lipoprotein metabolism and lipid management in chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Bulbul, M.C.; Dagel, T.; Afsar, B.; Ulusu, N.N.; Kuwabara, M.; Covic, A.; Kanbay, M. Disorders of Lipid Metabolism in Chronic Kidney Disease. Blood Purif. 2018, 46, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Pan, X. Cholesterol Metabolism in Chronic Kidney Disease: Physiology, Pathologic Mechanisms, and Treatment. Adv. Exp. Med. Biol. 2022, 1372, 119–143. [Google Scholar] [PubMed]
- Bajaj, A.; Damrauer, S.M.; Anderson, A.H.; Xie, D.; Budoff, M.J.; Go, A.S.; He, J.; Lash, J.P.; Ojo, A.; Post, W.S.; et al. Chronic Renal Insufficiency Cohort Study, I. Lipoprotein(a) and Risk of Myocardial Infarction and Death in Chronic Kidney Disease: Findings From the CRIC Study (Chronic Renal Insufficiency Cohort). Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1971–1978. [Google Scholar] [CrossRef] [Green Version]
- Purnell, J.Q.; Marcovina, S.M.; Hokanson, J.E.; Kennedy, H.; Cleary, P.A.; Steffes, M.W.; Brunzell, J.D. Levels of lipoprotein(a), apolipoprotein B, and lipoprotein cholesterol distribution in IDDM. Results from follow-up in the Diabetes Control and Complications Trial. Diabetes 1995, 44, 1218–1226. [Google Scholar] [CrossRef]
- Schiffrin, E.L.; Lipman, M.L.; Mann, J.F. Chronic kidney disease: Effects on the cardiovascular system. Circulation 2007, 116, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D. Dyslipidemia of chronic renal failure: The nature, mechanisms, and potential consequences. Am. J. Physiol. Renal. Physiol. 2006, 290, F262–F272. [Google Scholar] [CrossRef]
- Vaziri, N.D. Disorders of lipid metabolism in nephrotic syndrome: Mechanisms and consequences. Kidney Int 2016, 90, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Noels, H.; Lehrke, M.; Vanholder, R.; Jankowski, J. Lipoproteins and fatty acids in chronic kidney disease: Molecular and metabolic alterations. Nat. Rev. Nephrol. 2021, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Lipotoxicity and impaired high density lipoprotein-mediated reverse cholesterol transport in chronic kidney disease. J. Ren. Nutr. 2010, 20 (Suppl. S5), S35–S43. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diab. Rep. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferro, C.J.; Mark, P.B.; Kanbay, M.; Sarafidis, P.; Heine, G.H.; Rossignol, P.; Massy, Z.A.; Mallamaci, F.; Valdivielso, J.M.; Malyszko, J.; et al. Lipid management in patients with chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 727–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Xie, Y.; Shao, X.; Ni, Z.; Mou, S. L-FABP: A novel biomarker of kidney disease. Clin. Chim. Acta 2015, 445, 85–90. [Google Scholar] [CrossRef]
- Minami, S.; Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Namba, T.; Matsuda, J.; Kimura, T.; Kaimori, J.Y.; Matsui, I.; Hamano, T.; et al. Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy 2017, 13, 1629–1647. [Google Scholar] [CrossRef]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARalpha and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef] [Green Version]
- Baek, J.; He, C.; Afshinnia, F.; Michailidis, G.; Pennathur, S. Lipidomic approaches to dissect dysregulated lipid metabolism in kidney disease. Nat Rev Nephrol 2021, 18, 38–55. [Google Scholar] [CrossRef]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Cohney, S.; De Michieli, F.; Pinach, S.; Saba, F.; Gambino, R. Fatty Liver and Chronic Kidney Disease: Novel Mechanistic Insights and Therapeutic Opportunities. Diabetes Care 2016, 39, 1830–1845. [Google Scholar] [CrossRef] [Green Version]
- Scerbo, D.; Son, N.H.; Sirwi, A.; Zeng, L.; Sas, K.M.; Cifarelli, V.; Schoiswohl, G.; Huggins, L.A.; Gumaste, N.; Hu, Y.; et al. Kidney triglyceride accumulation in the fasted mouse is dependent upon serum free fatty acids. J. Lipid. Res. 2017, 58, 1132–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, A.M.; Schwartz, J.H.; Kroshian, V.M.; Tercyak, A.M.; Laraia, J.; Masino, S.; Lieberthal, W. Renal mouse proximal tubular cells are more susceptible than MDCK cells to chemical anoxia. Am. J. Physiol. 1993, 265 Pt 2, F342–F3450. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Oikawa, S.; Sato, H.; Sato, T.; Ito, S.; Sasaki, J. Lipoprotein glomerulopathy: Significance of lipoprotein and ultrastructural features. Kidney Int. Suppl. 1999, 71, S37–S41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrass, C.K. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am. J. Nephrol. 2004, 24, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.B.A.; Foresto-Neto, O.; Saraiva-Camara, N.O.; Sanders-Pinheiro, H. Renal lipotoxicity: Insights from experimental models. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Libby, A.E.; Jones, B.; Lopez-Santiago, I.; Rowland, E.; Levi, M. Nuclear receptors in the kidney during health and disease. Mol. Aspects Med. 2021, 78, 100935. [Google Scholar] [CrossRef]
- Vlad, C.E.; Foia, L.; Popescu, R.; Ivanov, I.; Luca, M.C.; Delianu, C.; Toma, V.; Statescu, C.; Rezus, C.; Florea, L. Apolipoproteins A and B and PCSK9: Nontraditional Cardiovascular Risk Factors in Chronic Kidney Disease and in End-Stage Renal Disease. J Diabetes Res 2019, 2019, 6906278. [Google Scholar] [CrossRef]
- Kwiterovich, P.O., Jr.; Sloan, H.R.; Fredrickson, D.S. Glycolipids and other lipid constituents of normal human liver. J. Lipid Res. 1970, 11, 322–330. [Google Scholar] [CrossRef]
- Bobulescu, I.A. Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, M.J.; Krebs, H.A. The fuel of respiration of rat kidney cortex. Biochem. J. 1969, 112, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, A.; Kimura, K.; Sugaya, T.; Yamanouchi, M.; Hase, H.; Kaneko, T.; Hirata, Y.; Goto, A.; Fujita, T.; Omata, M. Urinary free fatty acids bound to albumin aggravate tubulointerstitial damage. Kidney. Int. 2002, 62, 1628–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, D.; Wenzel, D.G. Injury produced by free fatty acids to lysosomes and mitochondria in cultured heart muscle and endothelial cells. Atherosclerosis 1974, 20, 417–426. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Werneburg, N.W.; Li, Z.; Bronk, S.F.; Gores, G.J. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am. J. Physiol. Gastrointest Liver. Physiol. 2006, 290, G1339–G1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.J.; Huber, T.B.; Godel, M.; Jarad, G.; Hartleben, B.; Kwoh, C.; Keil, A.; Karpitskiy, A.; Hu, J.; Huh, C.J.; et al. Albumin-associated free fatty acids induce macropinocytosis in podocytes. J. Clin. Investig. 2015, 125, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Liebman, S.E.; Lucia, M.S.; Li, J.; Levi, M. Role of altered renal lipid metabolism and the sterol regulatory element binding proteins in the pathogenesis of age-related renal disease. Kidney Int. 2005, 68, 2608–2620. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, D.; Levi, M. Kidney aging—Inevitable or preventable? Nat. Rev. Nephrol. 2011, 7, 706–717. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Liebman, S.E.; Lucia, M.S.; Phillips, C.L.; Levi, M. Calorie restriction modulates renal expression of sterol regulatory element binding proteins, lipid accumulation, and age-related renal disease. J. Am. Soc. Nephrol. 2005, 16, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.H.; Duann, P. Dyslipidemia in Kidney Disorders: Perspectives on Mitochondria Homeostasis and Therapeutic Opportunities. Front. Physiol. 2020, 11, 1050. [Google Scholar] [CrossRef] [PubMed]
- Moorhead, J.F.; Chan, M.K.; El-Nahas, M.; Varghese, Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 1982, 2, 1309–1311. [Google Scholar] [CrossRef]
- Guder, W.G.; Schmolke, M.; Wirthensohn, G. Carbohydrate and lipid metabolism of the renal tubule in diabetes mellitus. Eur. J. Clin. Chem. Clin. Biochem. 1992, 30, 669–674. [Google Scholar] [PubMed]
- Joles, J.A.; Kunter, U.; Janssen, U.; Kriz, W.; Rabelink, T.J.; Koomans, H.A.; Floege, J. Early mechanisms of renal injury in hypercholesterolemic or hypertriglyceridemic rats. J. Am. Soc. Nephrol. 2000, 11, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M. Lipotoxicity. Kidney Int. 2006, 70, 1560–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Moradi, H. Mechanisms of dyslipidemia of chronic renal failure. Hemodial. Int. 2006, 10, 1–7. [Google Scholar] [CrossRef]
- Ruan, X.Z.; Varghese, Z.; Moorhead, J.F. An update on the lipid nephrotoxicity hypothesis. Nat. Rev. Nephrol. 2009, 5, 713–721. [Google Scholar] [CrossRef]
- Rutledge, J.C.; Ng, K.F.; Aung, H.H.; Wilson, D.W. Role of triglyceride-rich lipoproteins in diabetic nephropathy. Nat. Rev. Nephrol. 2010, 6, 361–370. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; de Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic kidney disease: A report from an ADA Consensus Conference. Diabetes Care 2014, 37, 2864–2883. [Google Scholar] [CrossRef] [Green Version]
- Wahl, P.; Ducasa, G.M.; Fornoni, A. Systemic and renal lipids in kidney disease development and progression. Am. J. Physiol. Renal. Physiol. 2016, 310, F433–F445. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P.; Furth, S.L.; Zoccali, C.; World Kidney Day Steering, C. Obesity and kidney disease: Hidden consequences of the epidemic. Clin. Kidney J. 2017, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef] [Green Version]
- Opazo-Rios, L.; Mas, S.; Marin-Royo, G.; Mezzano, S.; Gomez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Juszczak, F.; Caron, N.; Mathew, A.V.; Decleves, A.E. Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 7994. [Google Scholar] [CrossRef]
- Ruster, C.; Wolf, G. Adipokines promote chronic kidney disease. Nephrol. Dial. Transplant. 2013, 28 (Suppl. S4), iv8–iv14. [Google Scholar] [CrossRef]
- Mount, P.; Davies, M.; Choy, S.W.; Cook, N.; Power, D. Obesity-Related Chronic Kidney Disease—The Role of Lipid Metabolism. Metabolites 2015, 5, 720–732. [Google Scholar] [CrossRef]
- Yang, M.; Geng, C.A.; Liu, X.; Guan, M. Lipid Disorders in NAFLD and Chronic Kidney Disease. Biomedicines 2021, 9, 1405. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, M.K.; De Nardo, W.; Watt, M.J. Impact of Lipotoxicity on Tissue “Cross Talk” and Metabolic Regulation. Physiology 2019, 34, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Ajay, A.K.; Chang, J.H.; Mou, S.; Zhao, H.; Kishi, S.; Li, J.; Brooks, C.R.; Xiao, S.; Woo, H.M.; et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 2021, 33, 1042–1061.e7. [Google Scholar] [CrossRef] [PubMed]
- Novikov, A.; Vallon, V. Sodium glucose cotransporter 2 inhibition in the diabetic kidney: An update. Curr. Opin. Nephrol. Hypertens. 2016, 25, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Kim, J.A.; Montagnani, M.; Chandrasekran, S.; Quon, M.J. Role of lipotoxicity in endothelial dysfunction. Heart Fail. Clin. 2012, 8, 589–607. [Google Scholar] [CrossRef] [Green Version]
- Dumas, S.J.; Meta, E.; Borri, M.; Luo, Y.; Li, X.; Rabelink, T.J.; Carmeliet, P. Phenotypic diversity and metabolic specialization of renal endothelial cells. Nat. Rev. Nephrol. 2021, 17, 441–464. [Google Scholar] [CrossRef]
- Bartlett, C.S.; Jeansson, M.; Quaggin, S.E. Vascular Growth Factors and Glomerular Disease. Annu. Rev. Physiol. 2016, 78, 437–461. [Google Scholar] [CrossRef]
- Eom, M.; Hudkins, K.L.; Alpers, C.E. Foam cells and the pathogenesis of kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ku, H.; Zhao, L.; Wheeler, D.C.; Li, L.C.; Li, Q.; Varghese, Z.; Moorhead, J.F.; Powis, S.H.; Huang, A.; et al. Inflammatory stress induces statin resistance by disrupting 3-hydroxy-3-methylglutaryl-CoA reductase feedback regulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, X.Z.; Varghese, Z.; Powis, S.H.; Moorhead, J.F. Dysregulation of LDL receptor under the influence of inflammatory cytokines: A new pathway for foam cell formation. Kidney Int. 2001, 60, 1716–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Ruan, X.Z.; Li, Q.; Huang, A.; Moorhead, J.F.; Powis, S.H.; Varghese, Z. Inflammatory cytokines disrupt LDL-receptor feedback regulation and cause statin resistance: A comparative study in human hepatic cells and mesangial cells. Am. J. Physiol. Renal. Physiol. 2007, 293, F680–F687. [Google Scholar] [CrossRef] [Green Version]
- Alelign, T.; Petros, B. Kidney Stone Disease: An Update on Current Concepts. Adv. Urol. 2018, 2018, 3068365. [Google Scholar] [CrossRef]
- Grundy, S.M.; Denke, M.A. Dietary influences on serum lipids and lipoproteins. J. Lipid. Res. 1990, 31, 1149–1172. [Google Scholar] [CrossRef]
- Linsel-Nitschke, P.; Tall, A.R. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat. Rev. Drug. Discov. 2005, 4, 193–205. [Google Scholar] [CrossRef]
- Navab, M.; Anantharamaiah, G.M.; Reddy, S.T.; Fogelman, A.M. Apolipoprotein A-I mimetic peptides and their role in atherosclerosis prevention. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 540–547. [Google Scholar] [CrossRef]
- Degoma, E.M.; Rader, D.J. Novel HDL-directed pharmacotherapeutic strategies. Nat. Rev. Cardiol. 2011, 8, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Shapiro, M.D. Apolipoproteins in vascular biology and atherosclerotic disease. Nat. Rev. Cardiol. 2022, 19, 168–179. [Google Scholar] [CrossRef]
- Sparks, D.L.; Lund-Katz, S.; Phillips, M.C. The charge and structural stability of apolipoprotein A-I in discoidal and spherical recombinant high density lipoprotein particles. J. Biol. Chem. 1992, 267, 25839–25847. [Google Scholar] [CrossRef]
- Yang, H.; Fogo, A.B.; Kon, V. Kidneys: Key modulators of high-density lipoprotein levels and function. Curr. Opin. Nephrol. Hypertens. 2016, 25, 174–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, V.; Yang, H.C.; Smith, L.E.; Vickers, K.C.; Linton, M.F. High-Density Lipoproteins in Kidney Disease. Int. J. Mol. Sci. 2021, 22, 8201. [Google Scholar] [CrossRef] [PubMed]
- Barrios, C.; Zierer, J.; Wurtz, P.; Haller, T.; Metspalu, A.; Gieger, C.; Thorand, B.; Meisinger, C.; Waldenberger, M.; Raitakari, O.; et al. Circulating metabolic biomarkers of renal function in diabetic and non-diabetic populations. Sci. Rep. 2018, 8, 15249. [Google Scholar] [CrossRef]
- Jacobs-Cacha, C.; Puig-Gay, N.; Helm, D.; Rettel, M.; Sellares, J.; Meseguer, A.; Savitski, M.M.; Moreso, F.J.; Soler, M.J.; Seron, D.; et al. A misprocessed form of Apolipoprotein A-I is specifically associated with recurrent Focal Segmental Glomerulosclerosis. Sci. Rep. 2020, 10, 1159. [Google Scholar] [CrossRef] [Green Version]
- Saraf, S.L.; Zhang, X.; Shah, B.N.; Raslan, R.; Tayo, B.O.; Lash, J.P.; Franceschini, N.; Gordeuk, V.R. Engulfment and cell motility 1 (ELMO1) and apolipoprotein A1 (APOA1) as candidate genes for sickle cell nephropathy. Br. J. Haematol. 2021, 193, 628–632. [Google Scholar] [CrossRef]
- Zhao, W.B.; Zhu, L.; Rahman, T. Increased serum concentration of apolipoprotein B is associated with an increased risk of reaching renal replacement therapy in patients with diabetic kidney disease. Ren. Fail. 2020, 42, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Zewinger, S.; Speer, T.; Kleber, M.E.; Scharnagl, H.; Woitas, R.; Lepper, P.M.; Pfahler, K.; Seiler, S.; Heine, G.H.; Marz, W.; et al. HDL cholesterol is not associated with lower mortality in patients with kidney dysfunction. J. Am. Soc. Nephrol. 2014, 25, 1073–1082. [Google Scholar] [CrossRef] [Green Version]
- Wolska, A.; Reimund, M.; Sviridov, D.O.; Amar, M.J.; Remaley, A.T. Apolipoprotein Mimetic Peptides: Potential New Therapies for Cardiovascular Diseases. Cells 2021, 10, 597. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Kim, H.J.; Moradi, H.; Farmand, F.; Navab, K.; Navab, M.; Hama, S.; Fogelman, A.M.; Quiroz, Y.; Rodriguez-Iturbe, B. Amelioration of nephropathy with apoA-1 mimetic peptide in apoE-deficient mice. Nephrol. Dial. Transplant. 2010, 25, 3525–3534. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Bai, Y.; Yuan, J.; Said, H.L.; Sigala, W.; Ni, Z. ApoA-1 mimetic peptide reverses uremia-induced upregulation of pro-atherogenic pathways in the aorta. Am. J. Nephrol. 2010, 32, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Bandarian, F.; Daneshpour, M.S.; Hedayati, M.; Naseri, M.; Azizi, F. Identification of Sequence Variation in the Apolipoprotein A2 Gene and Their Relationship with Serum High-Density Lipoprotein Cholesterol Levels. Iran. Biomed. J. 2016, 20, 84–90. [Google Scholar] [PubMed]
- Weisgraber, K.H.; Mahley, R.W. Apoprotein (E—A-II) complex of human plasma lipoproteins. I. Characterization of this mixed disulfide and its identification in a high density lipoprotein subfraction. J. Biol. Chem. 1978, 253, 6281–6288. [Google Scholar] [CrossRef]
- Borghini, I.; James, R.W.; Blatter, M.C.; Pometta, D. Distribution of apolipoprotein E between free and A-II complexed forms in very-low- and high-density lipoproteins: Functional implications. Biochim. Biophys. Acta 1991, 1083, 139–146. [Google Scholar] [CrossRef]
- Torricelli, F.C.; De, S.K.; Gebreselassie, S.; Li, I.; Sarkissian, C.; Monga, M. Dyslipidemia and kidney stone risk. J. Urol. 2014, 191, 667–672. [Google Scholar] [CrossRef]
- Dugue-Pujol, S.; Rousset, X.; Pastier, D.; Quang, N.T.; Pautre, V.; Chambaz, J.; Chabert, M.; Kalopissis, A.D. Human apolipoprotein A-II associates with triglyceride-rich lipoproteins in plasma and impairs their catabolism. J. Lipid Res. 2006, 47, 2631–2639. [Google Scholar] [CrossRef] [Green Version]
- Dugue-Pujol, S.; Rousset, X.; Chateau, D.; Pastier, D.; Klein, C.; Demeurie, J.; Cywiner-Golenzer, C.; Chabert, M.; Verroust, P.; Chambaz, J.; et al. Apolipoprotein A-II is catabolized in the kidney as a function of its plasma concentration. J. Lipid Res. 2007, 48, 2151–2161. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, C.; Wallenius, K.; Walentinsson, A.; Greasley, P.J.; Miliotis, T.; Hammar, M.; Iaconelli, A.; Tapani, S.; Raffaelli, M.; Mingrone, G.; et al. Identification of Proteins Associated with the Early Restoration of Insulin Sensitivity After Biliopancreatic Diversion. J. Clin. Endocrinol. Metab. 2020, 105, e4157–e4168. [Google Scholar] [CrossRef]
- Prokaeva, T.; Akar, H.; Spencer, B.; Havasi, A.; Cui, H.; O’Hara, C.J.; Gursky, O.; Leszyk, J.; Steffen, M.; Browning, S.; et al. Hereditary Renal Amyloidosis Associated With a Novel Apolipoprotein A-II Variant. Kidney Int. Rep. 2017, 2, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- Rysz, J.; Gluba-Brzozka, A.; Rysz-Gorzynska, M.; Franczyk, B. The Role and Function of HDL in Patients with Chronic Kidney Disease and the Risk of Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 601. [Google Scholar] [CrossRef] [Green Version]
- Silbernagel, G.; Genser, B.; Drechsler, C.; Scharnagl, H.; Grammer, T.B.; Stojakovic, T.; Krane, V.; Ritz, E.; Wanner, C.; Marz, W. HDL cholesterol, apolipoproteins, and cardiovascular risk in hemodialysis patients. J. Am. Soc. Nephrol. 2015, 26, 484–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradi, M.; Mahmoudi, M.; Saedisomeolia, A.; Zahirihashemi, R.; Koohdani, F. The effect of weight loss on HDL subfractions and LCAT activity in two genotypes of APOA-II -265T>C polymorphism. Nutr. J. 2017, 16, 34. [Google Scholar] [CrossRef]
- Brown, J.; Trivedi, S.; Kwok, F.; Rowczenio, D.; Thomas, L.; Varikatt, W. Novel apolipoprotein AII mutation associated renal amyloidosis and fibrillary/immunotactoid cardiomyopathy. Pathology 2019, 51, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Chang, C.C.; Chen, H.W.; Lin, C.Y.; Chen, J.S. Serum ApoA4 levels predicted the progression of renal impairment in T2DM. Eur. J Clin. Investig. 2018, 48, e12937. [Google Scholar] [CrossRef] [PubMed]
- Bringans, S.; Ito, J.; Casey, T.; Thomas, S.; Peters, K.; Crossett, B.; Coleman, O.; Ebhardt, H.A.; Pennington, S.R.; Lipscombe, R. A robust multiplex immunoaffinity mass spectrometry assay (PromarkerD) for clinical prediction of diabetic kidney disease. Clin. Proteom. 2020, 17, 37. [Google Scholar] [CrossRef]
- Boes, E.; Fliser, D.; Ritz, E.; Konig, P.; Lhotta, K.; Mann, J.F.; Muller, G.A.; Neyer, U.; Riegel, W.; Riegler, P.; et al. Apolipoprotein A-IV predicts progression of chronic kidney disease: The mild to moderate kidney disease study. J. Am. Soc. Nephrol. 2006, 17, 528–536. [Google Scholar] [CrossRef]
- Shao, B.; Mathew, A.V.; Thornock, C.; Pennathur, S.; Michigan Kidney Translational Core, C.I.G. Altered HDL proteome predicts incident CVD in chronic kidney disease patients. J. Lipid Res. 2021, 62, 100135. [Google Scholar] [CrossRef]
- Perampalam, P.; Hassan, H.M.; Lilly, G.E.; Passos, D.T.; Torchia, J.; Kiser, P.K.; Bozovic, A.; Kulasingam, V.; Dick, F.A. Disrupting the DREAM transcriptional repressor complex induces apolipoprotein overexpression and systemic amyloidosis in mice. J. Clin. Investig. 2021, 131, e140903. [Google Scholar] [CrossRef]
- Li, X.; Wang, F.; Xu, M.; Howles, P.; Tso, P. ApoA-IV improves insulin sensitivity and glucose uptake in mouse adipocytes via PI3K-Akt Signaling. Sci. Rep. 2017, 7, 41289. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Cho, Y.I.; Kim, S.Y.; Yoon, Y.E.; Kim, K.S.; Hong, S.J.; Han, W.K. TNF-alpha-induced Inflammation Stimulates Apolipoprotein-A4 via Activation of TNFR2 and NF-kappaB Signaling in Kidney Tubular Cells. Sci. Rep. 2017, 7, 8856. [Google Scholar] [CrossRef] [Green Version]
- Brahm, A.J.; Hegele, R.A. Chylomicronaemia--current diagnosis and future therapies. Nat. Rev. Endocrinol. 2015, 11, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Hyun, Y.J.; Jang, Y.; Chae, J.S.; Kim, J.Y.; Paik, J.K.; Kim, S.Y.; Yang, J.Y.; Ordovas, J.M.; Ko, Y.G.; Lee, J.H. Association of apolipoprotein A5 concentration with serum insulin and triglyceride levels and coronary artery disease in Korean men. Atherosclerosis 2009, 205, 568–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Hayashi, T.; Adachi, M.; Taira, T.; Hattori, H. Marked decrease of apolipoprotein A-V in both diabetic and nondiabetic patients with end-stage renal disease. Metabolism 2007, 56, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Hishida, A.; Wakai, K.; Naito, M.; Suma, S.; Sasakabe, T.; Hamajima, N.; Hosono, S.; Horita, M.; Turin, T.C.; Suzuki, S.; et al. Polymorphisms of genes involved in lipid metabolism and risk of chronic kidney disease in Japanese—Cross-sectional data from the J-MICC study. Lipids Health Dis. 2014, 13, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouatou, S.; Ajjemami, M.; Charoute, H.; Sefri, H.; Ghalim, N.; Rhaissi, H.; Benrahma, H.; Barakat, A.; Rouba, H. Association of APOA5 rs662799 and rs3135506 polymorphisms with arterial hypertension in Moroccan patients. Lipids Health Dis. 2014, 13, 60. [Google Scholar] [CrossRef] [Green Version]
- de Luis, D.; Izaola, O.; Primo, D. APOA-5 Genetic Variant rs662799: Role in Lipid Changes and Insulin Resistance after a Mediterranean Diet in Caucasian Obese Subjects. Dis. Markers 2021, 2021, 1257145. [Google Scholar] [CrossRef]
- Hussain, M.M.; Bakillah, A.; Nayak, N.; Shelness, G.S. Amino acids 430-570 in apolipoprotein B are critical for its binding to microsomal triglyceride transfer protein. J. Biol. Chem. 1998, 273, 25612–25615. [Google Scholar] [CrossRef] [Green Version]
- Sniderman, A.D.; Thanassoulis, G.; Glavinovic, T.; Navar, A.M.; Pencina, M.; Catapano, A.; Ference, B.A. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019, 4, 1287–1295. [Google Scholar] [CrossRef]
- Hussain, M.M.; Fatma, S.; Pan, X.; Iqbal, J. Intestinal lipoprotein assembly. Curr. Opin. Lipidol. 2005, 16, 281–285. [Google Scholar] [CrossRef]
- Kwon, S.; Kim, D.K.; Oh, K.H.; Joo, K.W.; Lim, C.S.; Kim, Y.S.; Han, S.S. Apolipoprotein B is a risk factor for end-stage renal disease. Clin. Kidney J. 2021, 14, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Goek, O.N.; Kottgen, A.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Astor, B.C. Association of apolipoprotein A1 and B with kidney function and chronic kidney disease in two multiethnic population samples. Nephrol. Dial. Transpl. 2012, 27, 2839–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, S.; Gunnarsson, I.; Jacobson, S.H. Impact of the apolipoprotein B/apolipoprotein A-I ratio on renal outcome in immunoglobulin A nephropathy. Scand. J. Urol. Nephrol. 2012, 46, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shi, S.; Zhao, X.J.; Wang, J.K.; Liu, Z.W.; Liu, F.Q.; Zhu, L.; Zhu, S.M.; Zhang, Y.; Pan, S. Association Between the Lipid Profile and Renal Dysfunction in the Heart Failure Patients. Kidney Blood Press. Res. 2019, 44, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xie, Y.; Ma, X.; Gu, L.; Li, H.; Li, X.; Guo, G.; Zhang, X. Preoperative apolipoprotein B/A1 ratio is an independent prognostic factor in metastatic renal cell carcinoma. Urol. Oncol. 2019, 37, 184.e9–184.e17. [Google Scholar] [CrossRef]
- Tseng, C.H. Lipid abnormalities associated with urinary albumin excretion rate in Taiwanese type 2 diabetic patients. Kidney Int. 2005, 67, 1547–1553. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.C.; Tseng, C.H. Dyslipidemia, kidney disease, and cardiovascular disease in diabetic patients. Rev. Diabet Stud. 2013, 10, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Mazidi, M.; Webb, R.J.; Lip, G.Y.H.; Kengne, A.P.; Banach, M.; Davies, I.G. Discordance between LDL-C and Apolipoprotein B Levels and Its Association with Renal Dysfunction: Insights from a Population-Based Study. J. Clin. Med. 2022, 11, 313. [Google Scholar] [CrossRef]
- Lim, A. Diabetic nephropathy—complications and treatment. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 361–381. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Wang, S.; Zhao, H.; Yu, M.; Deng, X.; Jiang, Y.; Cao, Y.; Li, P.; Niu, W. Susceptibility of ApoB and PCSK9 Genetic Polymorphisms to Diabetic Kidney Disease Among Chinese Diabetic Patients. Front. Med. 2021, 8, 659188. [Google Scholar] [CrossRef]
- Krzystanek, M.; Pedersen, T.X.; Bartels, E.D.; Kjaehr, J.; Straarup, E.M.; Nielsen, L.B. Expression of apolipoprotein B in the kidney attenuates renal lipid accumulation. J. Biol. Chem. 2010, 285, 10583–10590. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.M.; Rava, P.; Walsh, M.; Rana, M.; Iqbal, J. Multiple functions of microsomal triglyceride transfer protein. Nutr. Metab. 2012, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straarup, E.M.; Fisker, N.; Hedtjarn, M.; Lindholm, M.W.; Rosenbohm, C.; Aarup, V.; Hansen, H.F.; Orum, H.; Hansen, J.B.; Koch, T. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic. Acids Res. 2010, 38, 7100–7111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Nagothu, K.; Ranganathan, G.; Ali, S.M.; Shank, B.; Gokden, N.; Ayyadevara, S.; Megyesi, J.; Olivecrona, G.; Chugh, S.S.; et al. Reduced kidney lipoprotein lipase and renal tubule triglyceride accumulation in cisplatin-mediated acute kidney injury. Am. J. Physiol. Renal. Physiol. 2012, 303, F437–F448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jong, M.C.; Hofker, M.H.; Havekes, L.M. Role of ApoCs in lipoprotein metabolism: Functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Fuior, E.V.; Gafencu, A.V. Apolipoprotein C1: Its Pleiotropic Effects in Lipid Metabolism and Beyond. Int. J. Mol. Sci. 2019, 20, 5939. [Google Scholar] [CrossRef] [Green Version]
- Pillois, X.; Gautier, T.; Bouillet, B.; Pais de Barros, J.P.; Jeannin, A.; Verges, B.; Bonnet, J.; Lagrost, L. Constitutive inhibition of plasma CETP by apolipoprotein C1 is blunted in dyslipidemic patients with coronary artery disease. J. Lipid Res. 2012, 53, 1200–1209. [Google Scholar] [CrossRef] [Green Version]
- Dautin, G.; Soltani, Z.; Ducloux, D.; Gautier, T.; Pais de Barros, J.P.; Gambert, P.; Lagrost, L.; Masson, D. Hemodialysis reduces plasma apolipoprotein C-I concentration making VLDL a better substrate for lipoprotein lipase. Kidney Int. 2007, 72, 871–878. [Google Scholar] [CrossRef]
- Raaijmakers, A.; Corveleyn, A.; Devriendt, K.; van Tienoven, T.P.; Allegaert, K.; Van Dyck, M.; van den Heuvel, L.; Kuypers, D.; Claes, K.; Mekahli, D.; et al. Criteria for HNF1B analysis in patients with congenital abnormalities of kidney and urinary tract. Nephrol. Dial. Transpl. 2015, 30, 835–842. [Google Scholar] [CrossRef] [Green Version]
- Kaysen, G.A. Lipid and lipoprotein metabolism in chronic kidney disease. J. Ren Nutr. 2009, 19, 73–77. [Google Scholar] [CrossRef]
- Bus, P.; Pierneef, L.; Bor, R.; Wolterbeek, R.; van Es, L.A.; Rensen, P.C.; de Heer, E.; Havekes, L.M.; Bruijn, J.A.; Berbee, J.F.; et al. Apolipoprotein C-I plays a role in the pathogenesis of glomerulosclerosis. J. Pathol. 2017, 241, 589–599. [Google Scholar] [CrossRef]
- Westerterp, M.; Berbee, J.F.; Pires, N.M.; van Mierlo, G.J.; Kleemann, R.; Romijn, J.A.; Havekes, L.M.; Rensen, P.C. Apolipoprotein C-I is crucially involved in lipopolysaccharide-induced atherosclerosis development in apolipoprotein E-knockout mice. Circulation 2007, 116, 2173–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Miao, C.; Hou, C.; Wang, Z.; Liu, B. Apolipoprotein C1 (APOC1): A Novel Diagnostic and Prognostic Biomarker for Clear Cell Renal Cell Carcinoma. Front. Oncol. 2020, 10, 1436. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Wu, L.W.; Zeng, L.H.; Zhang, Z.Y.; Wang, W.; Zhang, C.; Lin, N.M. ApoC1 promotes the metastasis of clear cell renal cell carcinoma via activation of STAT3. Oncogene 2020, 39, 6203–6217. [Google Scholar] [CrossRef]
- Yamamura, T.; Sudo, H.; Ishikawa, K.; Yamamoto, A. Familial type I hyperlipoproteinemia caused by apolipoprotein C-II deficiency. Atherosclerosis 1979, 34, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Peng, D. The emerging role of apolipoprotein C-III: Beyond effects on triglyceride metabolism. Lipids Health Dis. 2016, 15, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacevic, L.; Lu, H.; Caruso, J.A.; Govil-Dalela, T.; Thomas, R.; Lakshmanan, Y. Marked increase in urinary excretion of apolipoproteins in children with nephrolithiasis associated with hypercalciuria. Pediatr. Nephrol. 2017, 32, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Drayna, D.; Fielding, C.; McLean, J.; Baer, B.; Castro, G.; Chen, E.; Comstock, L.; Henzel, W.; Kohr, W.; Rhee, L.; et al. Cloning and expression of human apolipoprotein D cDNA. J. Biol. Chem. 1986, 261, 16535–16539. [Google Scholar] [CrossRef]
- Muffat, J.; Walker, D.W. Apolipoprotein D: An overview of its role in aging and age-related diseases. Cell Cycle 2010, 9, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Vaca, F.; Pownall, H.J. Disulfide linked dimers of apolipoprotein D in urine. Electrophoresis 1993, 14, 1086–1087. [Google Scholar] [CrossRef]
- DeWan, A.T.; Arnett, D.K.; Atwood, L.D.; Province, M.A.; Lewis, C.E.; Hunt, S.C.; Eckfeldt, J. A genome scan for renal function among hypertensives: The HyperGEN study. Am. J. Hum. Genet. 2001, 68, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Blue, M.L.; Williams, D.L.; Zucker, S.; Khan, S.A.; Blum, C.B. Apolipoprotein E synthesis in human kidney, adrenal gland, and liver. Proc. Natl. Acad. Sci. USA 1983, 80, 283–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, C.; Nie, W.; Tang, D.; Yi, L.; Mei, C. Apolipoprotein E gene variants on the risk of end stage renal disease. PLoS ONE 2013, 8, e83367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, M.; Segerer, S.; Dantas, M.; Brown, P.A.; Hudkins, K.L.; Goodpaster, T.; Kirk, E.; LeBoeuf, R.C.; Alpers, C.E. Renal injury in apolipoprotein E-deficient mice. Lab. Investig. 2002, 82, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Bruneval, P.; Bariety, J.; Belair, M.F.; Mandet, C.; Heudes, D.; Nicoletti, A. Mesangial expansion associated with glomerular endothelial cell activation and macrophage recruitment is developing in hyperlipidaemic apoE null mice. Nephrol. Dial. Transpl. 2002, 17, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Cambruzzi, E.; Pegas, K.L. Pathogenesis, histopathologic findings and treatment modalities of lipoprotein glomerulopathy: A review. J. Bras. Nefrol. 2019, 41, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Magistroni, R.; Bertolotti, M.; Furci, L.; Fano, R.A.; Leonelli, M.; Pisciotta, L.; Pellegrini, E.; Calabresi, L.; Bertolini, S.; Calandra, S. Lipoprotein glomerulopathy associated with a mutation in apolipoprotein e. Clin. Med. Insights Case Rep. 2013, 6, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Shing, C.M.; Fassett, R.G.; Peake, J.M.; Coombes, J.S. Voluntary exercise decreases atherosclerosis in nephrectomised ApoE knockout mice. PLoS ONE 2015, 10, e0120287. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Ushiogi, Y.; Yokoyama, H.; Hara, S.; Matsunaga, A.; Muso, E.; Saito, T. A case of apolipoprotein E Toyonaka and homozygous apolipoprotein E2/2 showing non-immune membranous nephropathy-like glomerular lesions with foamy changes. CEN Case Rep. 2019, 8, 106–111. [Google Scholar] [CrossRef]
- Saito, T.; Matsunaga, A.; Fukunaga, M.; Nagahama, K.; Hara, S.; Muso, E. Apolipoprotein E-related glomerular disorders. Kidney Int. 2020, 97, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Morton, R.E.; Liu, Y.; Izem, L. ApoF knockdown increases cholesteryl ester transfer to LDL and impairs cholesterol clearance in fat-fed hamsters. J. Lipid Res. 2019, 60, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Izem, L.; Morton, R.E. Identification of a hormone response element that mediates suppression of APOF by LXR and PPARalpha agonists. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158583. [Google Scholar] [CrossRef] [PubMed]
- Lagor, W.R.; Fields, D.W.; Khetarpal, S.A.; Kumaravel, A.; Lin, W.; Weintraub, N.; Wu, K.; Hamm-Alvarez, S.F.; Drazul-Schrader, D.; de la Llera-Moya, M.; et al. The effects of apolipoprotein F deficiency on high density lipoprotein cholesterol metabolism in mice. PLoS ONE 2012, 7, e31616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serdyuk, A.P.; Morton, R.E. Lipid transfer inhibitor protein activity deficiency in normolipidemic uremic patients on continuous ambulatory peritoneal dialysis. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L. The proximal tubule is the primary target of injury and progression of kidney disease: Role of the glomerulotubular junction. Am. J. Physiol. Renal. Physiol. 2016, 311, F145–F161. [Google Scholar] [CrossRef] [PubMed]
- Provost, P.R.; Boucher, E.; Tremblay, Y. Apolipoprotein A-I, A-II, C-II, and H expression in the developing lung and sex difference in surfactant lipids. J. Endocrinol. 2009, 200, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Norden, A.G.; Lapsley, M.; Lee, P.J.; Pusey, C.D.; Scheinman, S.J.; Tam, F.W.; Thakker, R.V.; Unwin, R.J.; Wrong, O. Glomerular protein sieving and implications for renal failure in Fanconi syndrome. Kidney Int. 2001, 60, 1885–1892. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, M.; Chen, H.Y.; Rong, J.; Dufresne, L.; Yao, J.; Guo, X.; Tsai, M.Y.; Tsimikas, S.; Post, W.S.; Vasan, R.S.; et al. Genome-Wide Association Study Highlights APOH as a Novel Locus for Lipoprotein(a) Levels-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 458–464. [Google Scholar]
- de Silva, H.V.; Stuart, W.D.; Duvic, C.R.; Wetterau, J.R.; Ray, M.J.; Ferguson, D.G.; Albers, H.W.; Smith, W.R.; Harmony, J.A. A 70-kDa apolipoprotein designated ApoJ is a marker for subclasses of human plasma high density lipoproteins. J. Biol. Chem. 1990, 265, 13240–13247. [Google Scholar] [CrossRef]
- Navab, M.; Hama-Levy, S.; Van Lenten, B.J.; Fonarow, G.C.; Cardinez, C.J.; Castellani, L.W.; Brennan, M.L.; Lusis, A.J.; Fogelman, A.M.; La Du, B.N. Mildly oxidized LDL induces an increased apolipoprotein J/paraoxonase ratio. J. Clin. Investig. 1997, 99, 2005–2019. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Kawasaki, S.; Adachi, W.; Kinoshita, S. Apolipoprotein J expression in human ocular surface epithelium. Investig. Ophthalmol. Vis. Sci. 1996, 37, 2285–2292. [Google Scholar]
- de Retana, S.F.; Marazuela, P.; Sole, M.; Colell, G.; Bonaterra, A.; Sanchez-Quesada, J.L.; Montaner, J.; Maspoch, D.; Cano-Sarabia, M.; Hernandez-Guillamon, M. Peripheral administration of human recombinant ApoJ/clusterin modulates brain beta-amyloid levels in APP23 mice. Alzheimers Res. Ther. 2019, 11, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, M.E.; Girton, R.; Finkel, D.; Chmielewski, D.; Barrie, A., 3rd; Witte, D.P.; Zhu, G.; Bissler, J.J.; Harmony, J.A.; Aronow, B.J. Apolipoprotein J/clusterin prevents a progressive glomerulopathy of aging. Mol. Cell Biol. 2002, 22, 1893–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, M.E.; Silkensen, J. Clusterin and the kidney. Exp. Nephrol. 1995, 3, 9–14. [Google Scholar] [PubMed]
- Silkensen, J.R.; Schwochau, G.B.; Rosenberg, M.E. The role of clusterin in tissue injury. Biochem. Cell Biol. 1994, 72, 483–488. [Google Scholar] [CrossRef]
- Myung, J.; Schmal, C.; Hong, S.; Tsukizawa, Y.; Rose, P.; Zhang, Y.; Holtzman, M.J.; De Schutter, E.; Herzel, H.; Bordyugov, G.; et al. The choroid plexus is an important circadian clock component. Nat. Commun. 2018, 9, 1062. [Google Scholar] [CrossRef]
- Heo, J.Y.; Kim, J.E.; Dan, Y.; Kim, Y.W.; Kim, J.Y.; Cho, K.H.; Bae, Y.K.; Im, S.S.; Liu, K.H.; Song, I.H.; et al. Clusterin deficiency induces lipid accumulation and tissue damage in kidney. J. Endocrinol. 2018, 237, 175–191. [Google Scholar] [CrossRef] [Green Version]
- Pant, J.; Giovinazzo, J.A.; Tuka, L.S.; Pena, D.; Raper, J.; Thomson, R. Apolipoproteins L1-6 share key cation channel-regulating residues but have different membrane insertion and ion conductance properties. J. Biol. Chem. 2021, 297, 100951. [Google Scholar] [CrossRef]
- Pollak, M.R.; Genovese, G.; Friedman, D.J. APOL1 and kidney disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 179–182. [Google Scholar] [CrossRef]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef] [Green Version]
- Madhavan, S.M.; O’Toole, J.F.; Konieczkowski, M.; Ganesan, S.; Bruggeman, L.A.; Sedor, J.R. APOL1 localization in normal kidney and nondiabetic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 2119–2128. [Google Scholar] [CrossRef] [Green Version]
- Duchateau, P.N.; Pullinger, C.R.; Orellana, R.E.; Kunitake, S.T.; Naya-Vigne, J.; O’Connor, P.M.; Malloy, M.J.; Kane, J.P. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J. Biol. Chem. 1997, 272, 25576–25582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchateau, P.N.; Pullinger, C.R.; Cho, M.H.; Eng, C.; Kane, J.P. Apolipoprotein L gene family: Tissue-specific expression, splicing, promoter regions; discovery of a new gene. J. Lipid. Res. 2001, 42, 620–630. [Google Scholar] [CrossRef]
- Page, N.M.; Butlin, D.J.; Lomthaisong, K.; Lowry, P.J. The human apolipoprotein L gene cluster: Identification, classification, and sites of distribution. Genomics 2001, 74, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; Kowalewska, J.; McCune, T. Transplantation With APOL1 Risk Variant Kidney May Be Associated With Lifetime Risk for Recurrence of Focal Segmental Glomerulosclerosis: A Case Report and Review of Literature. Transpl. Proc. 2019, 51, 3077–3079. [Google Scholar] [CrossRef]
- Zee, J.; McNulty, M.T.; Hodgin, J.B.; Zhdanova, O.; Hingorani, S.; Jefferson, J.A.; Gibson, K.L.; Trachtman, H.; Fornoni, A.; Dell, K.M.; et al. APOL1 genotype-associated morphologic changes among patients with focal segmental glomerulosclerosis. Pediatr. Nephrol. 2021, 36, 2747–2757. [Google Scholar] [CrossRef]
- Olabisi, O.A.; Zhang, J.Y.; VerPlank, L.; Zahler, N.; DiBartolo, S., 3rd; Heneghan, J.F.; Schlondorff, J.S.; Suh, J.H.; Yan, P.; Alper, S.L.; et al. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc. Natl. Acad. Sci. USA 2016, 113, 830–837. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.; Schmitz, J.; Fischer, K.; Granado, D.; Groh, A.C.; Krausel, V.; Luttgenau, S.M.; Amelung, T.M.; Pavenstadt, H.; Weide, T. Evolution of Renal-Disease Factor APOL1 Results in Cis and Trans Orientations at the Endoplasmic Reticulum That Both Show Cytotoxic Effects. Mol. Biol. Evol. 2021, 38, 4962–4976. [Google Scholar] [CrossRef]
- Bisgaard, L.S.; Christoffersen, C. The apoM/S1P Complex-A Mediator in Kidney Biology and Disease? Front. Med. 2021, 8, 754490. [Google Scholar] [CrossRef]
- Wolfrum, C.; Poy, M.N.; Stoffel, M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat. Med. 2005, 11, 418–422. [Google Scholar] [CrossRef]
- Christoffersen, C.; Benn, M.; Christensen, P.M.; Gordts, P.; Roebroek, A.J.M.; Frikke-Schmidt, R.; Tybjaerg-Hansen, A.; Dahlback, B.; Nielsen, L.B. The plasma concentration of HDL-associated apoM is influenced by LDL receptor-mediated clearance of apoB-containing particles. J. Lipid Res. 2012, 53, 2198–2204. [Google Scholar] [CrossRef] [Green Version]
- Faber, K.; Hvidberg, V.; Moestrup, S.K.; Dahlback, B.; Nielsen, L.B. Megalin is a receptor for apolipoprotein M, and kidney-specific megalin-deficiency confers urinary excretion of apolipoprotein M. Mol. Endocrinol. 2006, 20, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, W.; Wu, Y.; Zhang, X.; Lv, K.; Zhang, Y.; Li, Z.; Liang, F.; Dai, C.; Wang, L.; Gao, J.; et al. Deletion of ApoM gene induces apoptosis in mouse kidney via mitochondrial and endoplasmic reticulum stress pathways. Biochem. Biophys. Res. Commun. 2018, 505, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Blaho, V.A.; Galvani, S.; Engelbrecht, E.; Liu, C.; Swendeman, S.L.; Kono, M.; Proia, R.L.; Steinman, L.; Han, M.H.; Hla, T. HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 2015, 523, 342–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christoffersen, C.; Bartels, E.D.; Aarup, A.; Nielsen, L.B.; Pedersen, T.X. ApoB and apoM—New aspects of lipoprotein biology in uremia-induced atherosclerosis. Eur. J. Pharmacol. 2017, 816, 154–160. [Google Scholar] [CrossRef]
- Brinck, J.W.; Thomas, A.; Brulhart-Meynet, M.C.; Lauer, E.; Frej, C.; Dahlback, B.; Stenvinkel, P.; James, R.W.; Frias, M.A. High-density lipoprotein from end-stage renal disease patients exhibits superior cardioprotection and increase in sphingosine-1-phosphate. Eur. J. Clin. Investig. 2018, 48, e12866. [Google Scholar] [CrossRef] [Green Version]
- Svarrer, E.M.; Andersen, H.O.; Helvind, M.; Slagman, M.C.; Navis, G.; Dullaart, R.P.; Dahlback, B.; Nielsen, L.B. Urinary apolipoprotein M as a biomarker of acute kidney injury in children undergoing heart surgery. Biomark. Med. 2016, 10, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Kimak, E.; Bylina, J.; Solski, J.; Halabis, M.; Baranowicz-Gaszczyk, I.; Ksiazek, A. Association between lipids, lipoproteins composition of HDL particles and triglyceride-rich lipoproteins, and LCAT and CETP activity in post-renal transplant patients. Cell Biochem. Biophys. 2013, 67, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Piedrahita, J.A.; Zhang, S.H.; Hagaman, J.R.; Oliver, P.M.; Maeda, N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc. Natl. Acad. Sci. USA 1992, 89, 4471–4475. [Google Scholar] [CrossRef] [Green Version]
- Fazio, S.; Linton, M.F. Mouse models of hyperlipidemia and atherosclerosis. Front. Biosci. 2001, 6, D515–D525. [Google Scholar] [CrossRef]
- Kon, V.; Linton, M.F.; Fazio, S. Atherosclerosis in chronic kidney disease: The role of macrophages. Nat. Rev. Nephrol. 2011, 7, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Taleb, A.; Witztum, J.L.; Tsimikas, S. Oxidized phospholipids on apoB-100-containing lipoproteins: A biomarker predicting cardiovascular disease and cardiovascular events. Biomark. Med. 2011, 5, 673–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.W.; Qu, J.; Black, D.D.; Tso, P. Regulation of intestinal lipid metabolism: Current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Dummer, P.D.; Limou, S.; Rosenberg, A.Z.; Heymann, J.; Nelson, G.; Winkler, C.A.; Kopp, J.B. APOL1 Kidney Disease Risk Variants: An Evolving Landscape. Semin. Nephrol. 2015, 35, 222–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasembeli, A.N.; Duarte, R.; Ramsay, M.; Mosiane, P.; Dickens, C.; Dix-Peek, T.; Limou, S.; Sezgin, E.; Nelson, G.W.; Fogo, A.B.; et al. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South Africans. J. Am. Soc. Nephrol. 2015, 26, 2882–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruggeman, L.A.; O’Toole, J.F.; Sedor, J.R. APOL1 polymorphisms and kidney disease: Loss-of-function or gain-of-function? Am. J. Physiol. Renal. Physiol. 2019, 316, F1–F8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrensch, F.; Crouchet, E.; Ligat, G.; Zeisel, M.B.; Keck, Z.Y.; Foung, S.K.H.; Schuster, C.; Baumert, T.F. Hepatitis C Virus (HCV)-Apolipoprotein Interactions and Immune Evasion and Their Impact on HCV Vaccine Design. Front. Immunol. 2018, 9, 1436. [Google Scholar] [CrossRef]
- Pol, S.; Parlati, L.; Jadoul, M. Hepatitis C virus and the kidney. Nat. Rev. Nephrol. 2019, 15, 73–86. [Google Scholar] [CrossRef]
- Pan, X.; Bradfield, C.A.; Hussain, M.M. Global and hepatocyte-specific ablation of Bmal1 induces hyperlipidaemia and enhances atherosclerosis. Nat. Commun. 2016, 7, 13011. [Google Scholar] [CrossRef]
- Pan, X.; Jiang, X.C.; Hussain, M.M. Impaired cholesterol metabolism and enhanced atherosclerosis in clock mutant mice. Circulation 2013, 128, 1758–1769. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Hussain, M.M. Bmal1 regulates production of larger lipoproteins by modulating cAMP-responsive element-binding protein H and apolipoprotein AIV. Hepatology 2021. [Google Scholar] [CrossRef]
- Rao, S.; Walters, K.B.; Wilson, L.; Chen, B.; Bolisetty, S.; Graves, D.; Barnes, S.; Agarwal, A.; Kabarowski, J.H. Early lipid changes in acute kidney injury using SWATH lipidomics coupled with MALDI tissue imaging. Am. J. Physiol. Renal. Physiol. 2016, 310, F1136–F1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeZwaan-McCabe, D.; Sheldon, R.D.; Gorecki, M.C.; Guo, D.F.; Gansemer, E.R.; Kaufman, R.J.; Rahmouni, K.; Gillum, M.P.; Taylor, E.B.; Teesch, L.M.; et al. ER Stress Inhibits Liver Fatty Acid Oxidation while Unmitigated Stress Leads to Anorexia-Induced Lipolysis and Both Liver and Kidney Steatosis. Cell Rep. 2017, 19, 1794–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, A.; Rudnitskaya, A.; Chariyavilaskul, P.; Dhaun, N.; Melville, V.; Goddard, J.; Webb, D.J.; Pitt, A.R.; Spickett, C.M. Top-down lipidomics of low density lipoprotein reveal altered lipid profiles in advanced chronic kidney disease. J. Lipid Res. 2015, 56, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonacina, F.; Coe, D.; Wang, G.; Longhi, M.P.; Baragetti, A.; Moregola, A.; Garlaschelli, K.; Uboldi, P.; Pellegatta, F.; Grigore, L.; et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat. Commun. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Roula, D.; Theiler, A.; Luschnig, P.; Sturm, G.J.; Tomazic, P.V.; Marsche, G.; Heinemann, A.; Sturm, E.M. Apolipoprotein A-IV acts as an endogenous anti-inflammatory protein and is reduced in treatment-naive allergic patients and allergen-challenged mice. Allergy 2020, 75, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisvert, W.A.; Black, A.S.; Curtiss, L.K. ApoA1 reduces free cholesterol accumulation in atherosclerotic lesions of ApoE-deficient mice transplanted with ApoE-expressing macrophages. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Duan, H.; Mazzone, T. Apolipoprotein E-dependent cholesterol efflux from macrophages: Kinetic study and divergent mechanisms for endogenous versus exogenous apolipoprotein E. J. Lipid Res. 1999, 40, 1618–1627. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Apolipoprotein | Lipoprotein | Main Tissue |
|---|---|---|
| ApoA-I | HDL, VLDL, CM | Liver, small intestine, kidney, macrophages |
| ApoA-II | HDL, VLDL, CM, | Liver, stomach, small intestine, tongue, skin |
| ApoA-IV | HDL, CM, | Intestine, liver, kidney |
| ApoA-V | HDL, VLDL, CM, | Liver |
| ApoB 48 | CM, IDL/CM remnants | Intestine, |
| ApoB100 | Lp(a), IDL, LDL, VLDL | Liver |
| ApoC-I | HDL, IDL, VLDL, CM | Liver, brain |
| ApoC-II | HDL, IDL, VLDL, CM | Liver, brain |
| ApoC-III | HDL, IDL/CM remnants, VLDL, CM | Liver, intestine |
| ApoD | HDL | Brain, kidney, muscle |
| ApoE | HDL, IDL/CM remnants, VLDL, CM | Liver, kidney, lung, skin |
| ApoF | HDL, LDL | Liver, kidney, brain |
| ApoH | HDL | Liver, kidney, lung |
| ApoJ | HDL | Brain, liver, kidney, lung, |
| ApoL-1 | HDL, LDL | Liver, pancreas, kidney, brain, |
| ApoL-2 | HDL | Liver, kidney, lung, brain |
| ApoM | HDL, LDL, VLDL, CM | Intestine, liver, kidney |
| Apolipoprotein | Physiological Functions | Pathophysiology |
|---|---|---|
| ApoA-I | main structural protein in HDL, Cholesterol transport | CVD, CKD, FSGS [91,92,93,94,95,97,98] |
| ApoA-II | main structural protein in HDL, Cholesterol transport | CVD, T2D, DKD, Kidney stones, ESRD [102,103,104,105,106,107] |
| ApoA-IV | may increase triacylglycerol secretion | DKD, CKD [105,111,115,116,117] |
| ApoA-V | enhances triacylglycerol uptake | ESRD, CKD, T2D nephropathy [119,120,121,122,123] |
| ApoB 48 | remove excessive triglycerides | albuminuria, gomerulosclerosis, ESRD [94,128,129,130,131,132,133,134] |
| ApoB100 | binds to LDL receptor, remove excessive triglycerides | Albuminuria [132,133] |
| ApoC-I | activates LCAT | CKD, diabetic nephropathy, glomerulosclerosis, renal cancer [142,147,148,149] |
| ApoC-II | activates lipoprotein lipase | poorly defined dieases |
| ApoC-III | inhibits lipoprotein lipase, controls triacylglycerol turnover | renal insufficiency in T2D, kidney stone formation [59,153] |
| ApoD | associated with LCAT, progesterone binding | poorly defined dieases |
| ApoE | binds to LDL receptor, remove excessive triglycerides | nondiabetic ESRD, nephrotic syndrome, diabetic nephropathy, ESRD [162,163,164,165,166] |
| ApoF | inhibits CETP activity | uremia [170] |
| ApoH | binding to phospholipids | antiphospholipid Syndrome-related kidney dieases [173,174] |
| ApoJ | cholesterol clearance | acute and chronic renal disease, polycystic kidney disease, ischemic renal tissues, lupus-like nephritis [179,180,181,182,183] |
| ApoL-1 | encodes a secreted HDL, bind to ApoA1, efflux of cholesterol | Renal failure, FSGS, Glomerulonephritis and HIV-related KD [186,187,190,191,192,193] |
| ApoM | main structural protein in HDL, transports S1P | CKD [202,203] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X. The Roles of Fatty Acids and Apolipoproteins in the Kidneys. Metabolites 2022, 12, 462. https://doi.org/10.3390/metabo12050462
Pan X. The Roles of Fatty Acids and Apolipoproteins in the Kidneys. Metabolites. 2022; 12(5):462. https://doi.org/10.3390/metabo12050462
Chicago/Turabian StylePan, Xiaoyue. 2022. "The Roles of Fatty Acids and Apolipoproteins in the Kidneys" Metabolites 12, no. 5: 462. https://doi.org/10.3390/metabo12050462
APA StylePan, X. (2022). The Roles of Fatty Acids and Apolipoproteins in the Kidneys. Metabolites, 12(5), 462. https://doi.org/10.3390/metabo12050462