
Colon Cancer: From Epidemiology to Prevention

 , , ,
, , ,  ,
,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. Colon Cancer Epidemiology: The Role of Environment
2.1. Modifiable Risk Factors with a Clear Environmental Component
2.1.1. Inactivity, Sedentary Lifestyles and Obesity
2.1.2. Tobacco Consumption
2.1.3. Overconsumption of Red and Processed Meat
2.1.4. Alcohol Consumption
2.1.5. Dietary Fibre and Whole Grains
2.1.6. Dairy Products and Dietary Supplements
2.1.7. Age
3. The Molecular Epidemiology of Colon Cancer: The Role of Genetic Variability
3.1. Risk Factors with a Clear Genetic Component
3.1.1. Hereditary Colorectal Cancer Syndromes
3.1.2. Inflammatory Bowel Disease (IBD)
3.1.3. Personal History of Colonic Adenomas
3.1.4. Comorbidities: History of Diabetes and Other Diseases
3.1.5. Sex
3.1.6. Self-Reported Race/Ethnicity
3.2. Genome Wide Association Studies
4. Intestinal Microbiota Deregulation as a Risk Factor for CRC
4.1. Intestinal Pathogens Linked to CRC
4.2. Human GI Microbiome Inter-Individual Diversity
4.3. Inflammation and Intestinal Dysbiosis
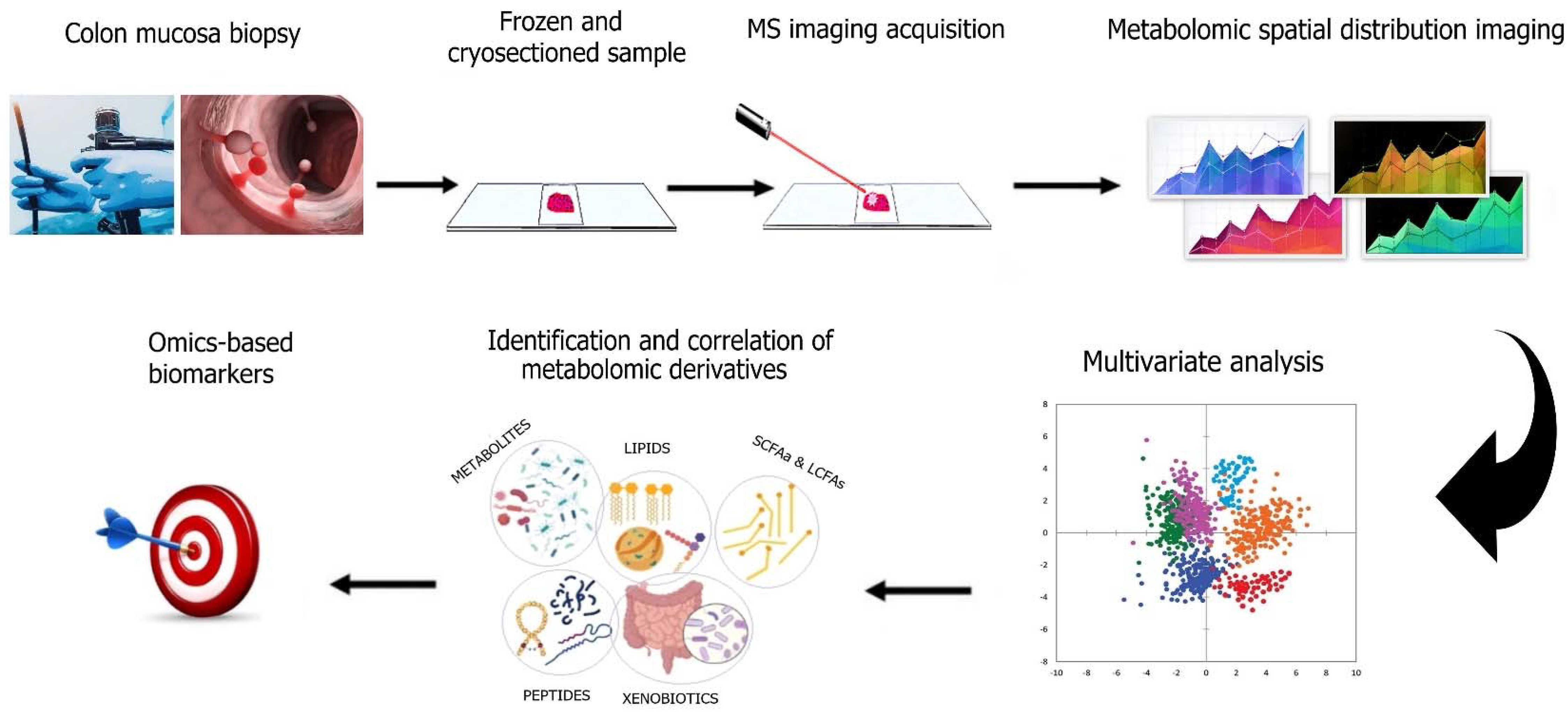
5. Intestinal Metabolomics as an Emerging Way to Study CRC Prevention
6. Epigenetics and Site-Specific Microenvironment
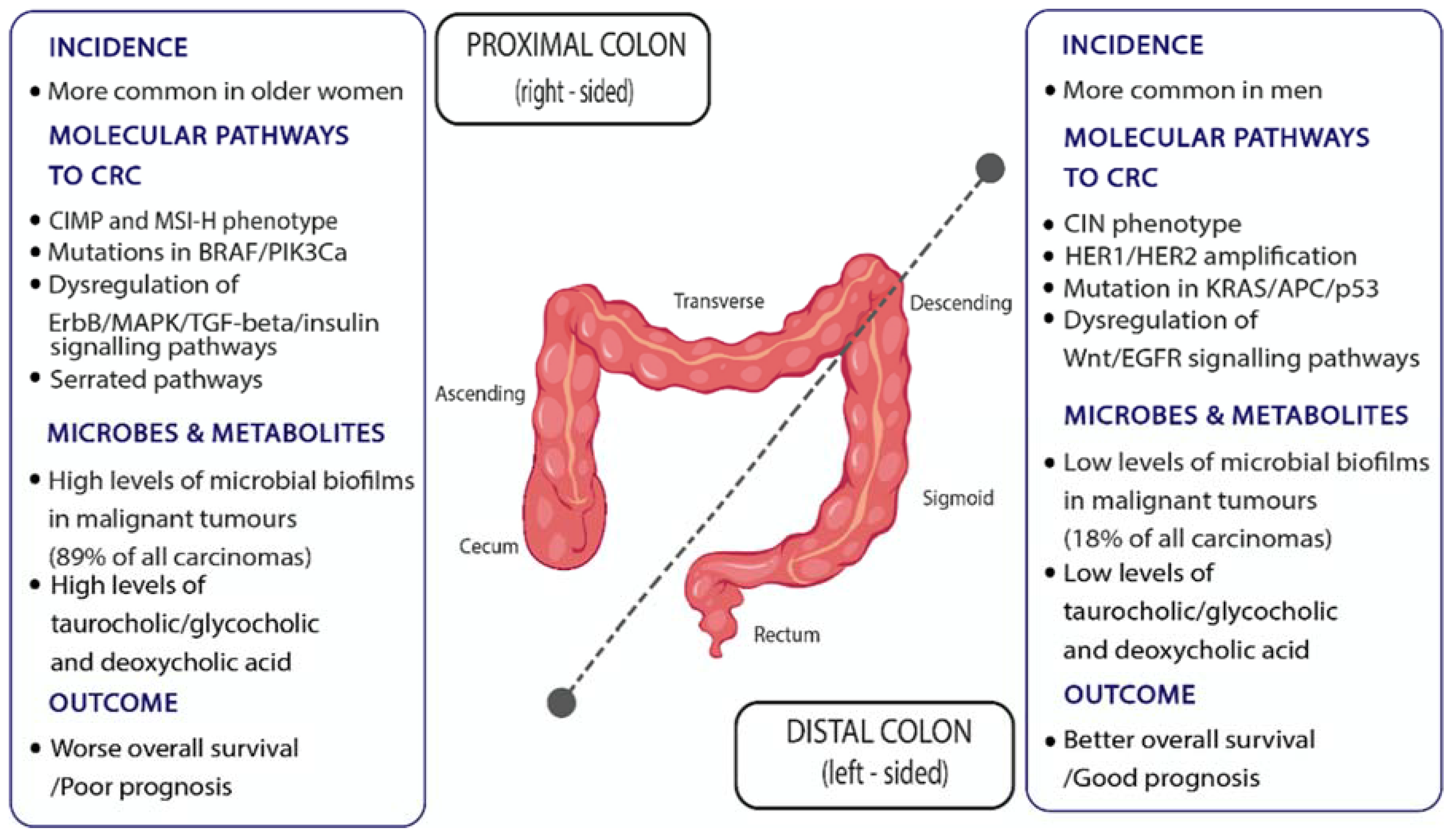
6.1. Intra-Individual (Regional) Differences in Colon Cancer Risk
6.2. Stem Cell Divisions, Regenerative Inflammation, Cell Differentiation, and Colon Cancer Risk
7. Discussion
7.1. Multi-Omics Analysis for Identifying Biomarkers That Can Be Modified by Pre- and Pro-Biotics towards CRC Prevention
7.2. Gut-on-Chip: Towards Experimentation with Personalized Human Gut Tissues
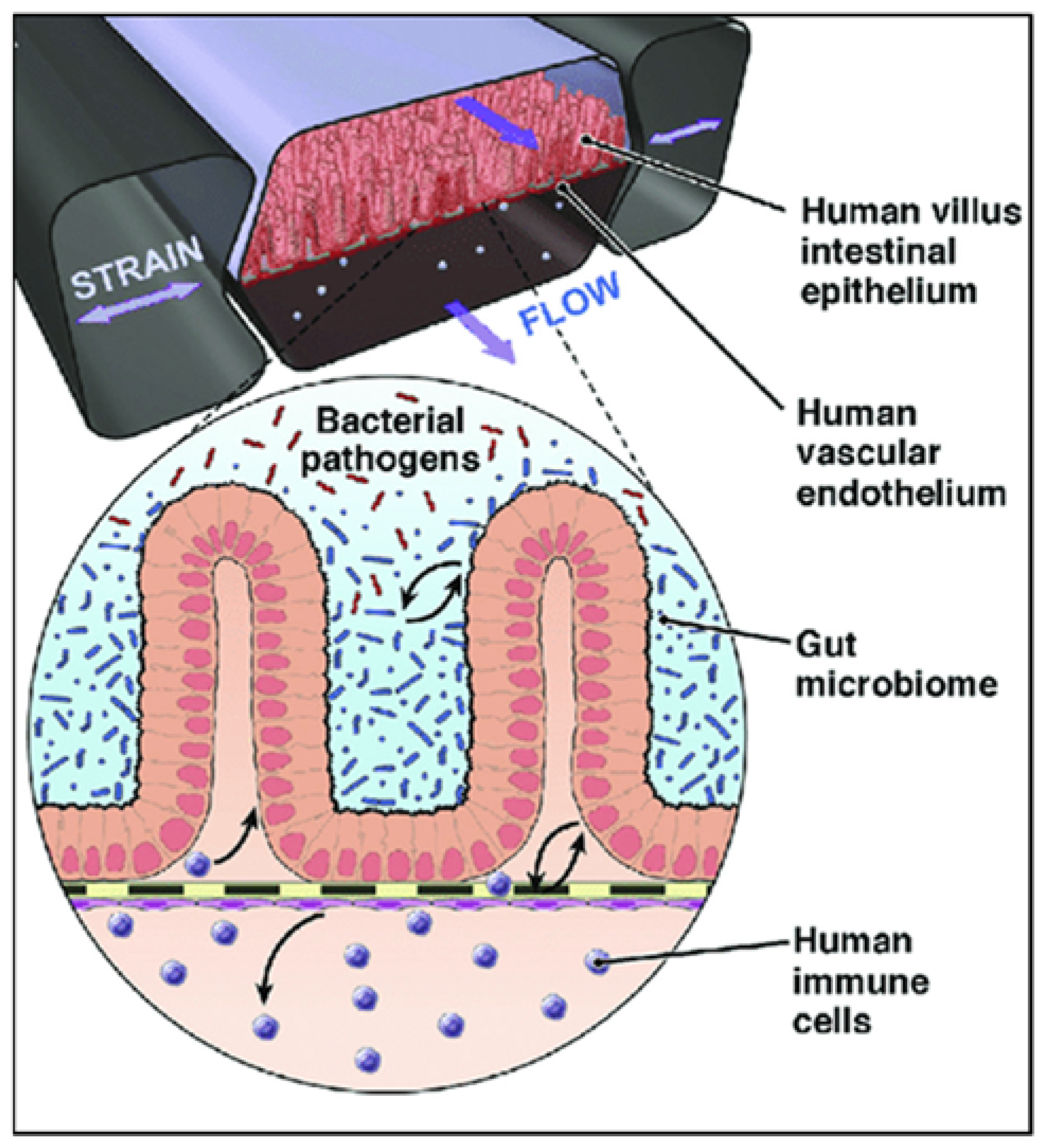
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lai, C.; Shields, P.G. The role of interindividual variation in human carcinogenesis. J. Nutr. 1999, 129, 552S–555S. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Silva, I. Cancer Epidemiology: Principles and Methods; IARC: Lyon, France, 1999. [Google Scholar]
- World Health Organization; Global Cancer Observatory (Globocan). Available online: https://gco.iarc.fr/ (accessed on 1 March 2022).
- Wong, M.C.S.; Huang, J.; Huang, J.L.W.; Pang, T.W.Y.; Choi, P.; Wang, J.; Chiang, J.I.; Jiang, J.Y. Global Prevalence of Colorectal Neoplasia: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2020, 18, 553–561.e510. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Mohandas, K.M. Colorectal cancer in India: Controversies, enigmas and primary prevention. Indian J. Gastroenterol. 2011, 30, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, L.M.; Yabroff, K.R.; Bernstein, A. Climate change and cancer. CA Cancer J. Clin. 2020, 70, 239–244. [Google Scholar] [CrossRef]
- Yang, L.; Cao, C.; Kantor, E.D.; Nguyen, L.H.; Zheng, X.; Park, Y.; Giovannucci, E.L.; Matthews, C.E.; Colditz, G.A.; Cao, Y. Trends in Sedentary Behavior Among the US Population, 2001–2016. JAMA 2019, 321, 1587–1597. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Zheng, C.; Huang, W.Y.; Sheridan, S.; Sit, C.H.; Chen, X.K.; Wong, S.H. COVID-19 Pandemic Brings a Sedentary Lifestyle in Young Adults: A Cross-Sectional and Longitudinal Study. Int. J. Environ. Res. Public Health 2020, 17, 6035. [Google Scholar] [CrossRef]
- Gray, C.L.; Messer, L.C.; Rappazzo, K.M.; Jagai, J.S.; Grabich, S.C.; Lobdell, D.T. The association between physical inactivity and obesity is modified by five domains of environmental quality in U.S. adults: A cross-sectional study. PLoS ONE 2018, 13, e0203301. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couzin-Frankel, J. Obesity doesn’t always mean ill health. Should findings from DNA, animals, and people reframe medical care? Science 2021, 373, 480–483. [Google Scholar] [CrossRef]
- Friedenreich, C.M.; Ryder-Burbidge, C.; McNeil, J. Physical activity, obesity and sedentary behavior in cancer etiology: Epidemiologic evidence and biologic mechanisms. Mol. Oncol. 2021, 15, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Aspects Med. 2019, 69, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Botteri, E.; Iodice, S.; Bagnardi, V.; Raimondi, S.; Lowenfels, A.B.; Maisonneuve, P. Smoking and colorectal cancer: A meta-analysis. JAMA 2008, 300, 2765–2778. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Atkin, W.S.; Morson, B.C.; Cuzick, J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N. Engl. J. Med. 1992, 326, 658–662. [Google Scholar] [CrossRef]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedzwiedzka, E.; Arlukowicz, T.; Przybylowicz, K.E. A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis. Cancers 2021, 13, 2025. [Google Scholar] [CrossRef]
- Aykan, N.F. Red Meat and Colorectal Cancer. Oncol. Rev. 2015, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.L.; Pierre, F.; Corpet, D.E. Processed meat and colorectal cancer: A review of epidemiologic and experimental evidence. Nutr. Cancer 2008, 60, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kusama, K.; Matsukura, N.; Morino, K.; Hasegawa, H.; Sato, S.; Takayama, S.; Sugimura, T. Carcinogenicity in mice of a mutagenic compound, 2-amino-3-methylimidazo[4,5-f]quinoline, from broiled sardine, cooked beef and beef extract. Carcinogenesis 1984, 5, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Corpet, D.E. Red meat and colon cancer: Should we become vegetarians, or can we make meat safer? Meat Sci. 2011, 89, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Myung, S.K.; Lee, J.H. Light Alcohol Drinking and Risk of Cancer: A Meta-Analysis of Cohort Studies. Cancer Res. Treat. 2018, 50, 474–487. [Google Scholar] [CrossRef] [Green Version]
- Salaspuro, M. Microbial metabolism of ethanol and acetaldehyde and clinical consequences. Addict. Biol. 1997, 2, 35–46. [Google Scholar] [CrossRef]
- Amitay, E.L.; Carr, P.R.; Jansen, L.; Roth, W.; Alwers, E.; Herpel, E.; Kloor, M.; Blaker, H.; Chang-Claude, J.; Brenner, H.; et al. Smoking, alcohol consumption and colorectal cancer risk by molecular pathological subtypes and pathways. Br. J. Cancer 2020, 122, 1604–1610. [Google Scholar] [CrossRef] [Green Version]
- Aune, D.; Chan, D.S.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617. [Google Scholar] [CrossRef] [Green Version]
- McRae, M.P. The Benefits of Dietary Fiber Intake on Reducing the Risk of Cancer: An Umbrella Review of Meta-analyses. J. Chiropr. Med. 2018, 17, 90–96. [Google Scholar] [CrossRef]
- Hullings, A.G.; Sinha, R.; Liao, L.M.; Freedman, N.D.; Graubard, B.I.; Loftfield, E. Whole grain and dietary fiber intake and risk of colorectal cancer in the NIH-AARP Diet and Health Study cohort. Am. J. Clin. Nutr. 2020, 112, 603–612. [Google Scholar] [CrossRef]
- Encarnacao, J.C.; Abrantes, A.M.; Pires, A.S.; Botelho, M.F. Revisit dietary fiber on colorectal cancer: Butyrate and its role on prevention and treatment. Cancer Metastasis Rev. 2015, 34, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Thorning, T.K.; Raben, A.; Tholstrup, T.; Soedamah-Muthu, S.S.; Givens, I.; Astrup, A. Milk and dairy products: Good or bad for human health? An assessment of the totality of scientific evidence. Food Nutr. Res. 2016, 60, 32527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampfer, L. Vitamin D and colon cancer. World J. Gastrointest Oncol. 2014, 6, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Thélin, C.S.S.; Sikka, S. Epidemiology of Colorectal Cancer—Incidence, Lifetime Risk Factors Statistics and Temporal Trends. In Screening for Colorectal Cancer with Colonoscopy; IntechOpen Limited: London, UK, 2015. [Google Scholar]
- Peterse, E.F.P.; Meester, R.G.S.; Siegel, R.L.; Chen, J.C.; Dwyer, A.; Ahnen, D.J.; Smith, R.A.; Zauber, A.G.; Lansdorp-Vogelaar, I. The impact of the rising colorectal cancer incidence in young adults on the optimal age to start screening: Microsimulation analysis I to inform the American Cancer Society colorectal cancer screening guideline. Cancer 2018, 124, 2964–2973. [Google Scholar] [CrossRef]
- Vuik, F.E.; Nieuwenburg, S.A.; Bardou, M.; Lansdorp-Vogelaar, I.; Dinis-Ribeiro, M.; Bento, M.J.; Zadnik, V.; Pellise, M.; Esteban, L.; Kaminski, M.F.; et al. Increasing incidence of colorectal cancer in young adults in Europe over the last 25 years. Gut 2019, 68, 1820–1826. [Google Scholar] [CrossRef]
- Frucht, H.L.; Lucas, A. Molecular Genetics of Colorectal Cancer; UpToDate: Waltham, MA, USA, 2020. [Google Scholar]
- Mojarad, E.N.; Kuppen, P.J.; Aghdaei, H.A.; Zali, M.R. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e2073. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e1173. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Available online: https://www.cancer.org/ (accessed on 1 March 2022).
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2019, 247, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Parker, T.W.; Neufeld, K.L. APC controls Wnt-induced beta-catenin destruction complex recruitment in human colonocytes. Sci. Rep. 2020, 10, 2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997, 386, 761–763. [Google Scholar] [CrossRef]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal. Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [Green Version]
- De Jong, A.E.; Morreau, H.; Nagengast, F.M.; Mathus-Vliegen, E.M.; Kleibeuker, J.H.; Griffioen, G.; Cats, A.; Vasen, H.F. Prevalence of adenomas among young individuals at average risk for colorectal cancer. Am. J. Gastroenterol. 2005, 100, 139–143. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Spalinger, M.R.; Manzini, R.; Hering, L.; Riggs, J.B.; Gottier, C.; Lang, S.; Atrott, K.; Fettelschoss, A.; Olomski, F.; Kundig, T.M.; et al. PTPN2 Regulates Inflammasome Activation and Controls Onset of Intestinal Inflammation and Colon Cancer. Cell Rep. 2018, 22, 1835–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.; Su, Y.R.; Petersen, P.; Qi, L.; Kim, A.E.; Figueiredo, J.C.; Lin, Y.; Nan, H.; Sakoda, L.C.; Albanes, D.; et al. Functional informed genome-wide interaction analysis of body mass index, diabetes and colorectal cancer risk. Cancer Med. 2020, 9, 3563–3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abotchie, P.N.; Vernon, S.W.; Du, X.L. Gender differences in colorectal cancer incidence in the United States, 1975–2006. J. Womens Health 2012, 21, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.E.; Paik, H.Y.; Yoon, H.; Lee, J.E.; Kim, N.; Sung, M.K. Sex- and gender-specific disparities in colorectal cancer risk. World J. Gastroenterol. 2015, 21, 5167–5175. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Lim, H.; Moon, A. Sex Differences in Cancer: Epidemiology, Genetics and Therapy. Biomol. Ther. 2018, 26, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Zhang, S.M.; Rexrode, K.M.; Manson, J.E.; Chan, A.T.; Wu, K.; Tworoger, S.S.; Hankinson, S.E.; Fuchs, C.; Gaziano, J.M.; et al. Association between sex hormones and colorectal cancer risk in men and women. Clin. Gastroenterol. Hepatol. 2013, 11, 419–424.e411. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.; Strickler, H.D.; Stanczyk, F.Z.; Xue, X.; Wassertheil-Smoller, S.; Rohan, T.E.; Ho, G.Y.; Anderson, G.L.; Potter, J.D.; Gunter, M.J. A Prospective Evaluation of Endogenous Sex Hormone Levels and Colorectal Cancer Risk in Postmenopausal Women. J. Natl. Cancer Inst. 2015, 107, djv210. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.C.; Caan, B.J.; Prado, C.M.; Cespedes Feliciano, E.M.; Xiao, J.; Kroenke, C.H.; Meyerhardt, J.A. The Association of Abdominal Adiposity With Mortality in Patients With Stage I-III Colorectal Cancer. J. Natl. Cancer Inst. 2020, 112, 377–383. [Google Scholar] [CrossRef]
- World Health Organization. Global Cancer Observatory (Globocan 2020): Cyprus. Available online: https://gco.iarc.fr/today/data/factsheets/populations/196-cyprus-fact-sheets.pdf (accessed on 12 October 2021).
- Onyeaghala, G.; Lintelmann, A.K.; Joshu, C.E.; Lutsey, P.L.; Folsom, A.R.; Robien, K.; Platz, E.A.; Prizment, A.E. Adherence to the World Cancer Research Fund/American Institute for Cancer Research cancer prevention guidelines and colorectal cancer incidence among African Americans and whites: The Atherosclerosis Risk in Communities study. Cancer 2020, 126, 1041–1050. [Google Scholar] [CrossRef]
- Ohri, A.; Robinson, A.; Liu, B.; Bhuket, T.; Wong, R. Updated Assessment of Colorectal Cancer Incidence in the U.S. by Age, Sex, and Race/Ethnicity. Dig. Dis. Sci. 2020, 65, 1838–1849. [Google Scholar] [CrossRef]
- Surveillance, Epidemiology, and End Results Program, Cancer Stat Facts: Colorectal Cancer. Available online: https://seer.cancer.gov/statfacts/html/colorect.html (accessed on 20 February 2022).
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Al-Tassan, N.A.; Whiffin, N.; Hosking, F.J.; Palles, C.; Farrington, S.M.; Dobbins, S.E.; Harris, R.; Gorman, M.; Tenesa, A.; Meyer, B.F.; et al. A new GWAS and meta-analysis with 1000Genomes imputation identifies novel risk variants for colorectal cancer. Sci. Rep. 2015, 5, 10442. [Google Scholar] [CrossRef] [PubMed]
- Hennig, E.E.; Kluska, A.; Piatkowska, M.; Kulecka, M.; Balabas, A.; Zeber-Lubecka, N.; Goryca, K.; Ambrozkiewicz, F.; Karczmarski, J.; Olesinski, T.; et al. GWAS Links New Variant in Long Non-Coding RNA LINC02006 with Colorectal Cancer Susceptibility. Biology 2021, 10, 465. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, C.; Kamatani, Y.; Takahashi, A.; Momozawa, Y.; Leveque, K.; Nagayama, S.; Mimori, K.; Mori, M.; Ishii, H.; Inazawa, J.; et al. GWAS identifies two novel colorectal cancer loci at 16q24.1 and 20q13.12. Carcinogenesis 2018, 39, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Baumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Sassone-Corsi, M.; Raffatellu, M. No vacancy: How beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J. Immunol. 2015, 194, 4081–4087. [Google Scholar] [CrossRef] [Green Version]
- Comelli, E.M.; Simmering, R.; Faure, M.; Donnicola, D.; Mansourian, R.; Rochat, F.; Corthesy-Theulaz, I.; Cherbut, C. Multifaceted transcriptional regulation of the murine intestinal mucus layer by endogenous microbiota. Genomics 2008, 91, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Cerf-Bensussan, N.; Gaboriau-Routhiau, V. The immune system and the gut microbiota: Friends or foes? Nat. Rev. Immunol. 2010, 10, 735–744. [Google Scholar] [CrossRef]
- Natividad, J.M.; Verdu, E.F. Modulation of intestinal barrier by intestinal microbiota: Pathological and therapeutic implications. Pharmacol Res. 2013, 69, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgard, J.E.M.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A.; et al. Colon Cancer-Associated Fusobacterium nucleatum May Originate From the Oral Cavity and Reach Colon Tumors via the Circulatory System. Front. Cell Infect. Microbiol. 2020, 10, 400. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum—symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Garrett, W.S. The gut microbiota and colon cancer. Science 2019, 364, 1133–1135. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clos-Garcia, M.; Garcia, K.; Alonso, C.; Iruarrizaga-Lejarreta, M.; D’Amato, M.; Crespo, A.; Iglesias, A.; Cubiella, J.; Bujanda, L.; Falcon-Perez, J.M. Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer. Cancers 2020, 12, 1142. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Misra, B.B.; Liang, L.; Bi, D.; Weng, W.; Wu, W.; Cai, S.; Qin, H.; Goel, A.; Li, X.; et al. Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics 2019, 9, 4101–4114. [Google Scholar] [CrossRef]
- Murphy, E.C.; Frick, I.M. Gram-positive anaerobic cocci--commensals and opportunistic pathogens. FEMS Microbiol. Rev. 2013, 37, 520–553. [Google Scholar] [CrossRef] [Green Version]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial spp. polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Matsumoto, S. Gut microbiota and colorectal cancer. Genes Environ. 2016, 38, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carra, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, aar7785. [Google Scholar] [CrossRef]
- Cheng, Y.; Ling, Z.; Li, L. The Intestinal Microbiota and Colorectal Cancer. Front. Immunol. 2020, 11, 615056. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum pot.tentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, X.; Wong, C.C.; Tong, L.; Chu, E.S.H.; Ho Szeto, C.; Go, M.Y.Y.; Coker, O.O.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat. Microbiol. 2019, 4, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Ozturk, M.A.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.H.; Christodoulou, D.K.; Pavlidis, N. The Developing Story of Predictive Biomarkers in Colorectal Cancer. J. Pers Med. 2019, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Osman, M.A.; Neoh, H.M.; Ab Mutalib, N.S.; Chin, S.F.; Mazlan, L.; Raja Ali, R.A.; Zakaria, A.D.; Ngiu, C.S.; Ang, M.Y.; Jamal, R. Parvimonas micra, Peptostreptococcus stomatis, Fusobacterium nucleatum and Akkermansia muciniphila as a four-bacteria biomarker panel of colorectal cancer. Sci. Rep. 2021, 11, 2925. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Kabeerdoss, J.; Ferdous, S.; Balamurugan, R.; Mechenro, J.; Vidya, R.; Santhanam, S.; Jana, A.K.; Ramakrishna, B.S. Development of the gut microbiota in southern Indian infants from birth to 6 months: A molecular analysis. J. Nutr. Sci. 2013, 2, e18. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.M.; Sitarik, A.R.; Havstad, S.L.; Fujimura, K.E.; Wegienka, G.; Cassidy-Bushrow, A.E.; Kim, H.; Zoratti, E.M.; Lukacs, N.W.; Boushey, H.A.; et al. Joint effects of pregnancy, sociocultural, and environmental factors on early life gut microbiome structure and diversity. Sci. Rep. 2016, 6, 31775. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Sitarik, A.R.; Woodcroft, K.; Johnson, C.C.; Zoratti, E. Birth Mode, Breastfeeding, Pet Exposure, and Antibiotic Use: Associations With the Gut Microbiome and Sensitization in Children. Curr. Allergy Asthma Rep. 2019, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.E.; Townsend, S.D. Temporal development of the infant gut microbiome. Open Biol. 2019, 9, 190128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selber-Hnatiw, S.; Rukundo, B.; Ahmadi, M.; Akoubi, H.; Al-Bizri, H.; Aliu, A.F.; Ambeaghen, T.U.; Avetisyan, L.; Bahar, I.; Baird, A.; et al. Human Gut Microbiota: Toward an Ecology of Disease. Front. Microbiol. 2017, 8, 1265. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; Wilmes, P. Human Gut Microbiome: Function Matters. Trends Microbiol. 2018, 26, 563–574. [Google Scholar] [CrossRef]
- Neu, J.; Rushing, J. Cesarean versus vaginal delivery: Long-term infant outcomes and the hygiene hypothesis. Clin. Perinatol. 2011, 38, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Guinane, C.M.; Tadrous, A.; Fouhy, F.; Ryan, C.A.; Dempsey, E.M.; Murphy, B.; Andrews, E.; Cotter, P.D.; Stanton, C.; Ross, R.P. Microbial composition of human appendices from patients following appendectomy. mBio 2013, 4, e00366-12. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.B.; Wexler, A.G.; Harding, B.N.; Whitney, J.C.; Bohn, A.J.; Goo, Y.A.; Tran, B.Q.; Barry, N.A.; Zheng, H.; Peterson, S.B.; et al. A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell Host Microbe. 2014, 16, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, B.; Zhang, M.; Rantalainen, M.; Wang, S.; Zhou, H.; Zhang, Y.; Shen, J.; Pang, X.; Zhang, M.; et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 2117–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Pramanik, S. Structural diversity, functional aspects and future therapeutic applications of human gut microbiome. Arch. Microbiol. 2021, 203, 5281–5308. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: A cross-sectional study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Vernocchi, P.; Del Chierico, F.; Putignani, L. Gut Microbiota Profiling: Metabolomics Based Approach to Unravel Compounds Affecting Human Health. Front. Microbiol. 2016, 7, 1144. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Fung, K.Y.; Cosgrove, L.; Lockett, T.; Head, R.; Topping, D.L. A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br. J. Nutr. 2012, 108, 820–831. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Alcoholado, L.; Ramos-Molina, B.; Otero, A.; Laborda-Illanes, A.; Ordonez, R.; Medina, J.A.; Gomez-Millan, J.; Queipo-Ortuno, M.I. The Role of the Gut Microbiome in Colorectal Cancer Development and Therapy Response. Cancers 2020, 12, 1406. [Google Scholar] [CrossRef]
- Tierney, B.T.; Yang, Z.; Luber, J.M.; Beaudin, M.; Wibowo, M.C.; Baek, C.; Mehlenbacher, E.; Patel, C.J.; Kostic, A.D. The Landscape of Genetic Content in the Gut and Oral Human Microbiome. Cell Host Microbe. 2019, 26, 283–295.e288. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saoi, M.; Britz-McKibbin, P. New Advances in Tissue Metabolomics: A Review. Metabolites 2021, 11, 672. [Google Scholar] [CrossRef] [PubMed]
- Ala-Korpela, M.; Kangas, A.J.; Soininen, P. Quantitative high-throughput metabolomics: A new era in epidemiology and genetics. Genome Med. 2012, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirwan, J.A.; Broadhurst, D.I.; Davidson, R.L.; Viant, M.R. Characterising and correcting batch variation in an automated direct infusion mass spectrometry (DIMS) metabolomics workflow. Anal. Bioanal. Chem. 2013, 405, 5147–5157. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Erban, A.; Weber, R.J.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J. Mass appeal: Metabolite identification in mass spec.ctrometry-focused untargeted metabolomics. Metabolomics 2013, 9, 44–66. [Google Scholar] [CrossRef] [Green Version]
- De Preter, V.; Verbeke, K. Metabolomics as a diagnostic tool in gastroenterology. World J. Gastrointest Pharmacol. Ther. 2013, 4, 97–107. [Google Scholar] [CrossRef]
- Ramautar, R.; Somsen, G.W.; de Jong, G.J. CE-MS for metabolomics: Developments and applications in the period 2010-2012. Electrophoresis 2013, 34, 86–98. [Google Scholar] [CrossRef]
- Soga, T.; Ishikawa, T.; Igarashi, S.; Sugawara, K.; Kakazu, Y.; Tomita, M. Analysis of nucleotides by pressure-assisted capillary electrophoresis-mass spectrometry using silanol mask technique. J. Chromatogr. A 2007, 1159, 125–133. [Google Scholar] [CrossRef]
- Barderas, M.G.; Laborde, C.M.; Posada, M.; de la Cuesta, F.; Zubiri, I.; Vivanco, F.; Alvarez-Llamas, G. Metabolomic profiling for identification of novel potential biomarkers in cardiovascular diseases. J. Biomed. Biotechnol. 2011, 2011, 790132. [Google Scholar] [CrossRef] [Green Version]
- Kuehnbaum, N.L.; Britz-McKibbin, P. New advances in separation science for metabolomics: Resolving chemical diversity in a post-genomic era. Chem. Rev. 2013, 113, 2437–2468. [Google Scholar] [CrossRef]
- Dunn, W.B.; Bailey, N.J.; Johnson, H.E. Measuring the metabolome: Current analytical technologies. Analyst 2005, 130, 606–625. [Google Scholar] [CrossRef]
- Van der Greef, J.; Stroobant, P.; van der Heijden, R. The role of analytical sciences in medical systems biology. Curr. Opin. Chem. Biol. 2004, 8, 559–565. [Google Scholar] [CrossRef]
- Buchberger, A.R.; DeLaney, K.; Johnson, J.; Li, L. Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Anal. Chem. 2018, 90, 240–265. [Google Scholar] [CrossRef]
- Porta Siegel, T.; Hamm, G.; Bunch, J.; Cappell, J.; Fletcher, J.S.; Schwamborn, K. Mass Spectrometry Imaging and Integration with Other Imaging Modalities for Greater Molecular Understanding of Biological Tissues. Mol. Imaging Biol. 2018, 20, 888–901. [Google Scholar] [CrossRef] [Green Version]
- Stringer, K.A.; McKay, R.T.; Karnovsky, A.; Quemerais, B.; Lacy, P. Metabolomics and Its Application to Acute Lung Diseases. Front. Immunol. 2016, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Horvath, I.; Barnes, P.J.; Loukides, S.; Sterk, P.J.; Hogman, M.; Olin, A.C.; Amann, A.; Antus, B.; Baraldi, E.; Bikov, A.; et al. A European Respiratory Society technical standard: Exhaled biomarkers in lung disease. Eur. Respir. J. 2017, 49, 1600965. [Google Scholar] [CrossRef] [Green Version]
- Issitt, T.; Wiggins, L.; Veysey, M.; Sweeney, S.T.; Brackenbury, W.J.; Redeker, K. Volatile compounds in human breath: Critical review and meta-analysis. J. Breath Res. 2022, 16, 024001. [Google Scholar] [CrossRef]
- Baxter, B.A.; Parker, K.D.; Nosler, M.J.; Rao, S.; Craig, R.; Seiler, C.; Ryan, E.P. Metabolite profile comparisons between ascending and descending colon tissue in healthy adults. World J. Gastroenterol. 2020, 26, 335–352. [Google Scholar] [CrossRef]
- Brown, D.G.; Rao, S.; Weir, T.L.; O’Malia, J.; Bazan, M.; Brown, R.J.; Ryan, E.P. Metabolomics and metabolic pathway networks from human colorectal cancers, adjacent mucosa, and stool. Cancer Metab. 2016, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Menter, D.G.; Kopetz, S. Right Versus Left Colon Cancer Biology: Integrating the Consensus Molecular Subtypes. J. Natl. Compr. Canc. Netw. 2017, 15, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.M.; Pfau, P.R.; O’Connor, E.S.; King, J.; LoConte, N.; Kennedy, G.; Smith, M.A. Mortality by stage for right- versus left-sided colon cancer: Analysis of surveillance, epidemiology, and end results--Medicare data. J. Clin. Oncol. 2011, 29, 4401–4409. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Pamplona, R.; Cordero, D.; Berenguer, A.; Lejbkowicz, F.; Rennert, H.; Salazar, R.; Biondo, S.; Sanjuan, X.; Pujana, M.A.; Rozek, L.; et al. Gene expression differences between colon and rectum tumors. Clin. Cancer Res. 2011, 17, 7303–7312. [Google Scholar] [CrossRef] [Green Version]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef]
- Omar Al-Hassi, H.; Ng, O.; Brookes, M. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2018, 67, 395. [Google Scholar] [CrossRef]
- Iacopetta, B. Are there two sides to colorectal cancer? Int J. Cancer 2002, 101, 403–408. [Google Scholar] [CrossRef]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef] [Green Version]
- Naughton, S.S.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. Fatty Acid modulation of the endocannabinoid system and the effect on food intake and metabolism. Int. J. Endocrinol. 2013, 2013, 361895. [Google Scholar] [CrossRef] [Green Version]
- Flynn, K.J.; Ruffin, M.T.t.; Turgeon, D.K.; Schloss, P.D. Spatial Variation of the Native Colon Microbiota in Healthy Adults. Cancer Prev. Res. 2018, 11, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Apidianakis, Y.; Rahme, L.G. Drosophila melanogaster as a model for human intestinal infection and pathology. Dis. Model Mech. 2011, 4, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlstein, B.; Spradling, A. The adult Drosophila posterior midgut is maintained by pluripotent stem cells. Nature 2006, 439, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Micchelli, C.A.; Perrimon, N. Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature 2006, 439, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Marianes, A.; Spradling, A.C. Physiological and stem cell compartmentalization within the Drosophila midgut. Elife 2013, 2, e00886. [Google Scholar] [CrossRef] [PubMed]
- Buchon, N.; Osman, D.; David, F.P.; Fang, H.Y.; Boquete, J.P.; Deplancke, B.; Lemaitre, B. Morphological and molecular characterization of adult midgut compartmentalization in Drosophila. Cell Rep. 2013, 3, 1725–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Lazaro, M. The stem cell division theory of cancer. Crit. Rev. Oncol. Hematol. 2018, 123, 95–113. [Google Scholar] [CrossRef]
- Rees, W.D.; Tandun, R.; Yau, E.; Zachos, N.C.; Steiner, T.S. Regenerative Intestinal Stem Cells Induced by Acute and Chronic Injury: The Saving Grace of the Epithelium? Front. Cell Dev. Biol. 2020, 8, 583919. [Google Scholar] [CrossRef]
- Tamamouna, V.; Panagi, M.; Theophanous, A.; Demosthenous, M.; Michail, M.; Papadopoulou, M.; Teloni, S.; Pitsouli, C.; Apidianakis, Y. Evidence of two types of balance between stem cell mitosis and enterocyte nucleus growth in the Drosophila midgut. Development 2020, 147, dev189472. [Google Scholar] [CrossRef]
- Panayidou, S.; Apidianakis, Y. Regenerative inflammation: Lessons from Drosophila intestinal epithelium in health and disease. Pathogens 2013, 2, 209–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Sipos, F.; Leiszter, K.; Tulassay, Z. Effect of ageing on colonic mucosal regeneration. World J. Gastroenterol. 2011, 17, 2981–2986. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Apidianakis, Y.; Pitsouli, C.; Perrimon, N.; Rahme, L. Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 20883–20888. [Google Scholar] [CrossRef] [Green Version]
- Buchon, N.; Broderick, N.A.; Poidevin, M.; Pradervand, S.; Lemaitre, B. Drosophila intestinal response to bacterial infection: Activation of host defense and stem cell proliferation. Cell Host Microbe 2009, 5, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Cronin, S.J.; Nehme, N.T.; Limmer, S.; Liegeois, S.; Pospisilik, J.A.; Schramek, D.; Leibbrandt, A.; Simoes Rde, M.; Gruber, S.; Puc, U.; et al. Genome-wide RNAi screen identifies genes involved in intestinal pathogenic bacterial infection. Science 2009, 325, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Patel, P.H.; Kohlmaier, A.; Grenley, M.O.; McEwen, D.G.; Edgar, B.A. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 2009, 137, 1343–1355. [Google Scholar] [CrossRef] [Green Version]
- Biteau, B.; Hochmuth, C.E.; Jasper, H. JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell 2008, 3, 442–455. [Google Scholar] [CrossRef] [Green Version]
- Lian, T.; Wu, Q.; Hodge, B.A.; Wilson, K.A.; Yu, G.; Yang, M. Drosophila Gut-A Nexus Between Dietary Restriction and Lifespan. Int. J. Mol. Sci. 2018, 19, 3810. [Google Scholar] [CrossRef] [Green Version]
- Beggs, A.D.; Jones, A.; El-Bahrawy, M.; Abulafi, M.; Hodgson, S.V.; Tomlinson, I.P. Whole-genome methylation analysis of benign and malignant colorectal tumours. J. Pathol. 2013, 229, 697–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.C.; Hao, C.Y.; Chiu, Y.S.; Wong, P.; Melnick, J.S.; Brotman, M.; Moretto, J.; Mendes, F.; Smith, A.P.; Bennington, J.L.; et al. Alteration of gene expression in normal-appearing colon mucosa of APC(min) mice and human cancer patients. Cancer Res. 2004, 64, 3694–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Ho, K.S.; Eu, K.W.; Cheah, P.Y. A susceptibility gene set for early onset colorectal cancer that integrates diverse signaling pathways: Implication for tumorigenesis. Clin. Cancer Res. 2007, 13, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiyama, H.; Suzuki, K.; Maeda, T.; Koizumi, K.; Miyaki, Y.; Okada, S.; Kawamura, Y.J.; Samuelsson, J.K.; Alonso, S.; Konishi, F.; et al. DNA demethylation in normal colon tissue predicts predisposition to multiple cancers. Oncogene 2012, 31, 5029–5037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaz, A.M.; Wong, C.J.; Dzieciatkowski, S.; Luo, Y.; Schoen, R.E.; Grady, W.M. Patterns of DNA methylation in the normal colon vary by anatomical location, gender, and age. Epigenetics 2014, 9, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, P.; Chan, A.T.; Nishihara, R.; Fuchs, C.S.; Beck, A.H.; Giovannucci, E.; Ogino, S. Etiologic field effect: Reappraisal of the field effect concept in cancer predisposition and progression. Mod. Pathol. 2015, 28, 14–29. [Google Scholar] [CrossRef] [Green Version]
- Markou, P.; Apidianakis, Y. Pathogenesis of intestinal Pseudomonas aeruginosa infection in patients with cancer. Front. Cell Infect. Microbiol. 2014, 3, 115. [Google Scholar] [CrossRef]
- Panagi, M.; Georgila, K.; Eliopoulos, A.G.; Apidianakis, Y. Constructing personalized longitudinal holo’omes of colon cancer-prone humans and their modeling in flies and mice. Oncotarget 2019, 10, 4224–4246. [Google Scholar] [CrossRef] [Green Version]
- Ellis, S.R.; Bruinen, A.L.; Heeren, R.M. A critical evaluation of the current state-of-the-art in quantitative imaging mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 1275–1289. [Google Scholar] [CrossRef]
- Kertesz, V.; Cahill, J.F. Spatially resolved absolute quantitation in thin tissue by mass spectrometry. Anal. Bioanal. Chem. 2021, 413, 2619–2636. [Google Scholar] [CrossRef]
- Hinsch, A.; Buchholz, M.; Odinga, S.; Borkowski, C.; Koop, C.; Izbicki, J.R.; Wurlitzer, M.; Krech, T.; Wilczak, W.; Steurer, S.; et al. MALDI imaging mass spectrometry reveals multiple clinically relevant masses in colorectal cancer using large-scale tissue microarrays. J. Mass Spectrom 2017, 52, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Umbreit, C.; Kruger, T.; Pelzel, D.; Ernst, G.; Kniemeyer, O.; Guntinas-Lichius, O.; Berndt, A.; von Eggeling, F. Identification of Proteomic Markers in Head and Neck Cancer Using MALDI-MS Imaging, LC-MS/MS, and Immunohistochemistry. Proteomics Clin. Appl. 2019, 13, e1700173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, L.M.; Clench, M.R. Mass spectrometry imaging tools in oncology. Biomark Med. 2015, 9, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Yamamoto, E.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.O.; Sugai, T.; An, B.; et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014, 74, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Bailey, M.; Christoforidou, Z.; Lewis, M.C. The evolutionary basis for differences between the immune systems of man, mouse, pig and ruminants. Vet. Immunol. Immunopathol. 2013, 152, 13–19. [Google Scholar] [CrossRef]
- Basson, M.D. Effects of repetitive deformation on intestinal epithelial cells. Inflammopharmacology 2007, 15, 109–114. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Biragyn, A.; Ferrucci, L. Gut dysbiosis: A potential link between increased cancer risk in ageing and inflammaging. Lancet Oncol. 2018, 19, e295–e304. [Google Scholar] [CrossRef]
- Brugmann, S.A.; Wells, J.M. Building additional complexity to in vitro-derived intestinal tissues. Stem Cell Res. Ther. 2013, 4 (Suppl. 1), S1. [Google Scholar] [CrossRef] [Green Version]
- Burke, C.M.; Darling, A.E. A method for high precision sequencing of near full-length 16S rRNA genes on an Illumina MiSeq. PeerJ 2016, 4, e2492. [Google Scholar] [CrossRef] [PubMed]
- Elsea, S.H.; Lucas, R.E. The mousetrap: What we can learn when the mouse model does not mimic the human disease. ILAR J. 2002, 43, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gayer, C.P.; Basson, M.D. The effects of mechanical forces on intestinal physiology and pathology. Cell Signal. 2009, 21, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassart, A.; Malarde, V.; Gobaa, S.; Sartori-Rupp, A.; Kerns, J.; Karalis, K.; Marteyn, B.; Sansonetti, P.; Sauvonnet, N. Bioengineered Human Organ-on-Chip Reveals Intestinal Microenvironment and Mechanical Forces Impacting Shigella Infection. Cell Host Microbe. 2019, 26, 565. [Google Scholar] [CrossRef]
- Halldorsson, S.; Lucumi, E.; Gomez-Sjoberg, R.; Fleming, R.M.T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab. Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, J.; Choi, J.H.; Bahinski, A.; Ingber, D.E. Co-culture of Living Microbiome with Microengineered Human Intestinal Villi in a Gut-on-a-Chip Microfluidic Device. J. Vis. Exp. 2016, 114, e54344. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Singhal, R.; Shah, Y.M. Oxygen battle in the gut: Hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J. Biol. Chem. 2020, 295, 10493–10505. [Google Scholar] [CrossRef]
- Van Berlo, D.; Van de Steeg, E.; Amirabadi, H.E.; Masereeuw, R. The potential of multi-organ-on-chip models for assessment of drug disposition as alternative to animal testing. Curr. Opin. Toxicol. 2021, 27, 8–17. [Google Scholar] [CrossRef]
- Bein, A.; Shin, W.; Jalili-Firoozinezhad, S.; Park, M.H.; Sontheimer-Phelps, A.; Tovaglieri, A.; Chalkiadaki, A.; Kim, H.J.; Ingber, D.E. Microfluidic Organ-on-a-Chip Models of Human Intestine. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]


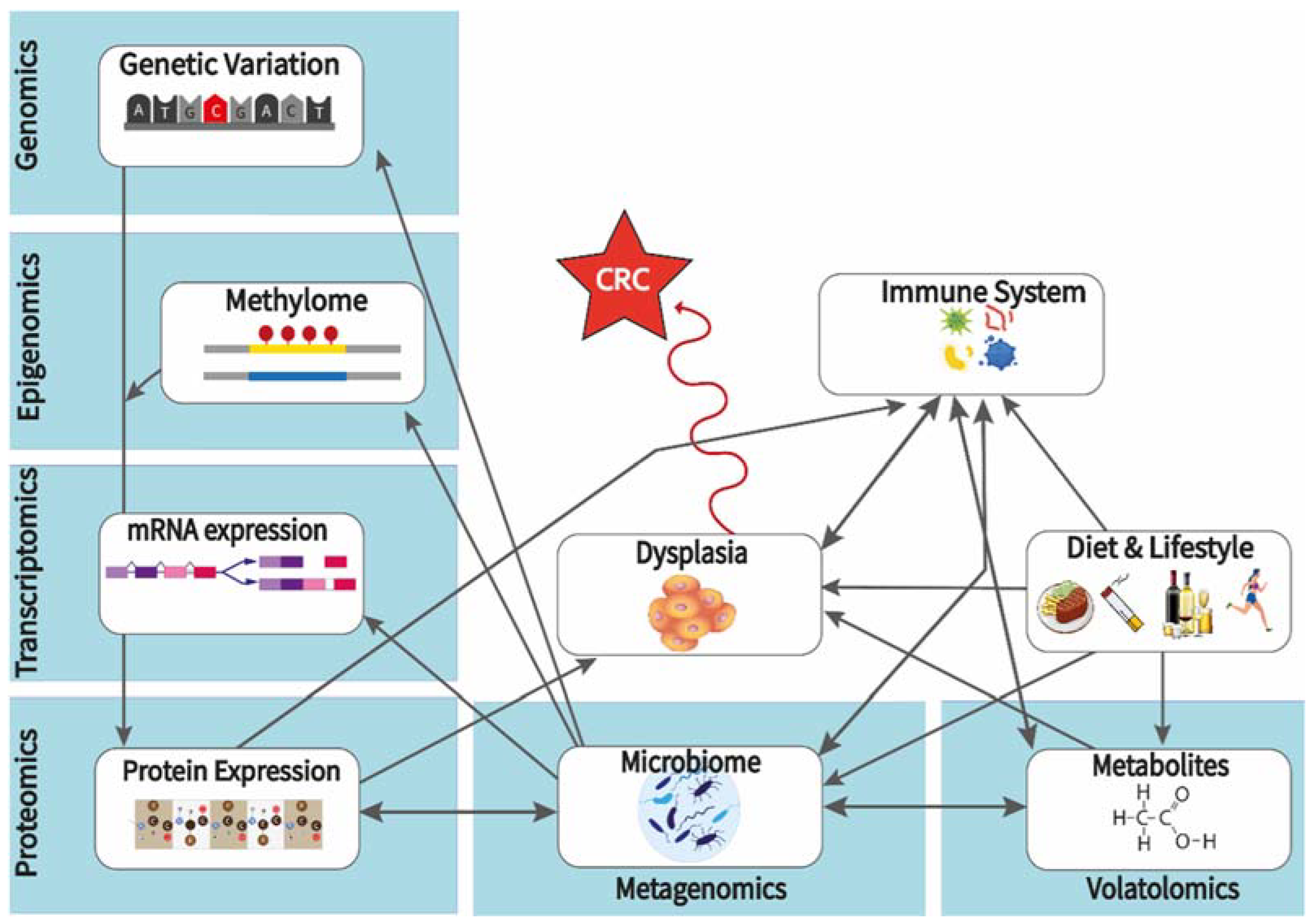{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
Healthy
Individuals | CRC Patients | Advanced Adenoma & Removed Polyp patients | IBD Patients | ||
|---|---|---|---|---|---|
| Microbiota | Phylum level | Firmicutes ↑ | Proteobacteria ↑ | Firmicutes ↓ | |
| Actinobacteria ↑ | Fusobacteria ↑ | Proteobacteria ↑ | |||
| Lentisphaerae ↑ | |||||
| Genus level | Escherichia – Shigella ↑ | Clostridium ↑ | |||
| Parvimonas ↑ | |||||
| Fusobacterium ↑ | |||||
| Porphyromonas ↑ | |||||
| Staphylococcus ↑ | |||||
| Pepto-streptococcus ↑ | |||||
| Peptococcus ↑ | |||||
| OTUs | Firmicutes ↑ | Gamma-proteobacteria ↑ | Adlercreutzia ↑ | F. prausnitzii ↓ | |
| Clostridiales ↑ | Enterobacteriaceae ↑ | B. adolescentis ↓ | |||
| Clostridia ↑ | Fusobacteriales ↑ | D. invisus ↓ | |||
| Lachnospiraceae ↑ | Erysipelotrichaceae ↑ | Clostridium defficil (cluster XIVa) ↓ | |||
| Ruminococcaceae ↑ | Lachnospiraceae ↓ | R. gnavus ↑ | |||
| Selenomonadales ↑ | Lactobacilli ↑ | ||||
| Negativicutes ↑ | Adherent-invasive E. coli ↑ | ||||
| Faecalibacterium ↑ | Adherent-invasive Campylobacter concisus ↑ | ||||
| Enterotoxigenic B. fragilis (ETBF) ↑ | |||||
| B. vulgatus ↑ | |||||
| Fusobacterium varium ↑ | |||||
| Klebssiella pneumonie ↑ | |||||
| Roseburia hominis ↓ | |||||
| Faecalibacterium ↓ | |||||
| Mycobacterium anium paratuberculosis ↑ | |||||
| Metabolic Method | Breadth of Compounds | Detection Sensitivity | Quantitative Accuracy | Sample Material | Sample Preparation |
|---|---|---|---|---|---|
| NMR | Biomolecules, including metabolites | μΜ to mM | Yes | Biofluids and tissues | Minimal |
| GC-MS | Thermally stable volatiles (fatty & organic acids, steroids, di-glycerides, sugars, sugar alcohols) | <μM | Yes | Biofluids and tissues | Multiple steps/ Chemical derivatization |
| LC-MS & UHPLC-MS | Polar & non-polar metabolites, ribonucleotides, amino acids, amines, sugars, organic acids | pM to nM | Yes | Biofluids and tissues | Minimal |
| CE-MS | Polar metabolites (wider spectrum than LC/MS), ionic compounds | nM | Yes | Biofluids and tissues | Minimal |
| MALDI MSI | Metabolites, lipids, peptides, glycans, proteins, drugs, drug metabolites | 0.5 μm to 100 μm depending on instrumentation | No | Biological tissue sections | Minimal or multi-step |
| DESI MSI | Metabolites, peptides | ~50μm spatial resolution | Semi-quantitative | Biological tissue sections | No |
| SIMS I & EASI MSI | Metabolites, peptides | nm to mm sample surface resolution | Yes | Biological tissue sections | Minimal |
| Healthy Individuals | CRC Patients | Advanced Adenoma & Removed Polyp Patients | IBD Patients | Overweight/Obese Individuals | |
|---|---|---|---|---|---|
| Metabolites | Sugars (maltose, fructose, iditol, glycerol, sedoheptulose) ↑ | Polyamines (cadaverine, putrescine, 1,4-Butanediamine) ↑ | Triacyloglycerol ↑ | Methylamine, trimethylamine ↓ | Trimethylamine N-oxide (TMAO) ↑ {asc} |
| Sugar alcohols ↑ | Amino acids (Pro, Glu, Phe, Ala, Lys, 5-oxo-Pro, Val, Leu, Orn) ↑ | 2-arachidonoylglycerol ↓ | SCFAs (Acetate, butyrate) ↓ | Endocannabinoids (linoleoylethanolamine, oleoylethanolamine) ↓ {asc} | |
| Amines (galactosamine) ↑ | Cholesteryl esters (ChoE) ↑ | 3-phosphoglycerate ↓ | Amino acids: Ala, Iso, Leu, Lys, Val ↑ [faecal matter] | Chenodeoxycholate ↑ | |
| Organic and fatty acids (octadecanoic acid, hexadecenoic acid, benzenepropanoic acid, linoleic acid, oleic acid) ↑ | Sphingomyelin classes ↑ | 6-phosphoglyconate ↓ | Amino acids: Ala, Cho, Glu, Iso, Leu, Val ↓ [colon mucosa tissue] | Cholate ↑ [desc} | |
| Mannitol ↑ | Glycerophosphatidylcholine ↑ | 1-dihomo-linoleuylglycerol ↓ {asc} | Amino acids: Arg, Lys ↑ [faecal matter] | Taurodeoxycholate ↑ {asc} | |
| Poly- and monounsaturated fatty acids ↑ | Aspartate ↓ {asc} | Taurine ↑ | 3-hydroxybutyrate (BHBA) ↑ | ||
| Deoxycholic acid ↑ | Glycerophosphorycholine (GPC) ↓ {asc} | Cadaverine ↑ | 2-arachidonoyglycerol ↑ | ||
| Glutarate ↓ {desc} | Indole ↑ | Long chain fatty acids ↑ {desc} | |||
| 2-hydroxyarachidate ↓ {desc} | Anti-oxidants ↑ | Heptadecanoic acid (margarate) ↑ {desc} | |||
| Myoinositol ↑ | |||||
| Betaine ↑ | |||||
| Glycerophosphorylcholine ↑ | |||||
| Lactate, formate, glutamate ↓ | |||||
| Succinate ↓ | |||||
| Phenolic compounds ↑ | |||||
| Glycerophospoglycine ↑ | |||||
| Glucose ↑ | |||||
| Metabolic Pathways | MAsp metabolism ↑ | Asp metabolism ↑ | SCFA synthesis ↓ | ||
| Ala metabolism ↑ | Ammonia recycling ↑ | Amino acid biosynthesis ↓ | |||
| Protein biosynthesis ↑ | Protein biosynthesis ↑ | ||||
| Glu-Ala cycle ↑ | Trp metabolism ↑ | ||||
| Selenoamino acid metabolism ↑ | |||||
| Mitochondrial electron transport chain ↑ | |||||
| Ammonia recycling ↑ | |||||
| Glutamate metabolism ↑ | |||||
| Urea cycle ↑ | |||||
| Citric acid cycle ↑ | |||||
| Methionine metabolism ↑ | |||||
| Galactose metabolism ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsaounou, K.; Nicolaou, E.; Vogazianos, P.; Brown, C.; Stavrou, M.; Teloni, S.; Hatzis, P.; Agapiou, A.; Fragkou, E.; Tsiaoussis, G.; et al. Colon Cancer: From Epidemiology to Prevention. Metabolites 2022, 12, 499. https://doi.org/10.3390/metabo12060499
Katsaounou K, Nicolaou E, Vogazianos P, Brown C, Stavrou M, Teloni S, Hatzis P, Agapiou A, Fragkou E, Tsiaoussis G, et al. Colon Cancer: From Epidemiology to Prevention. Metabolites. 2022; 12(6):499. https://doi.org/10.3390/metabo12060499
Chicago/Turabian StyleKatsaounou, Kyriaki, Elpiniki Nicolaou, Paris Vogazianos, Cameron Brown, Marios Stavrou, Savvas Teloni, Pantelis Hatzis, Agapios Agapiou, Elisavet Fragkou, Georgios Tsiaoussis, and et al. 2022. "Colon Cancer: From Epidemiology to Prevention" Metabolites 12, no. 6: 499. https://doi.org/10.3390/metabo12060499
APA StyleKatsaounou, K., Nicolaou, E., Vogazianos, P., Brown, C., Stavrou, M., Teloni, S., Hatzis, P., Agapiou, A., Fragkou, E., Tsiaoussis, G., Potamitis, G., Zaravinos, A., Andreou, C., Antoniades, A., Shiammas, C., & Apidianakis, Y. (2022). Colon Cancer: From Epidemiology to Prevention. Metabolites, 12(6), 499. https://doi.org/10.3390/metabo12060499