
Metabolomic and Transcriptomic Profiling Identified Significant Genes in Thymic Epithelial Tumor

Abstract
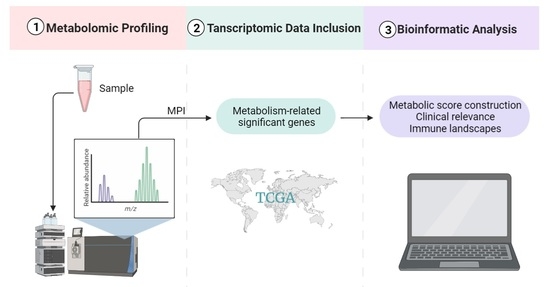
:
1. Introduction
2. Results
2.1. Differential Metabolism between TCTs and ANTs
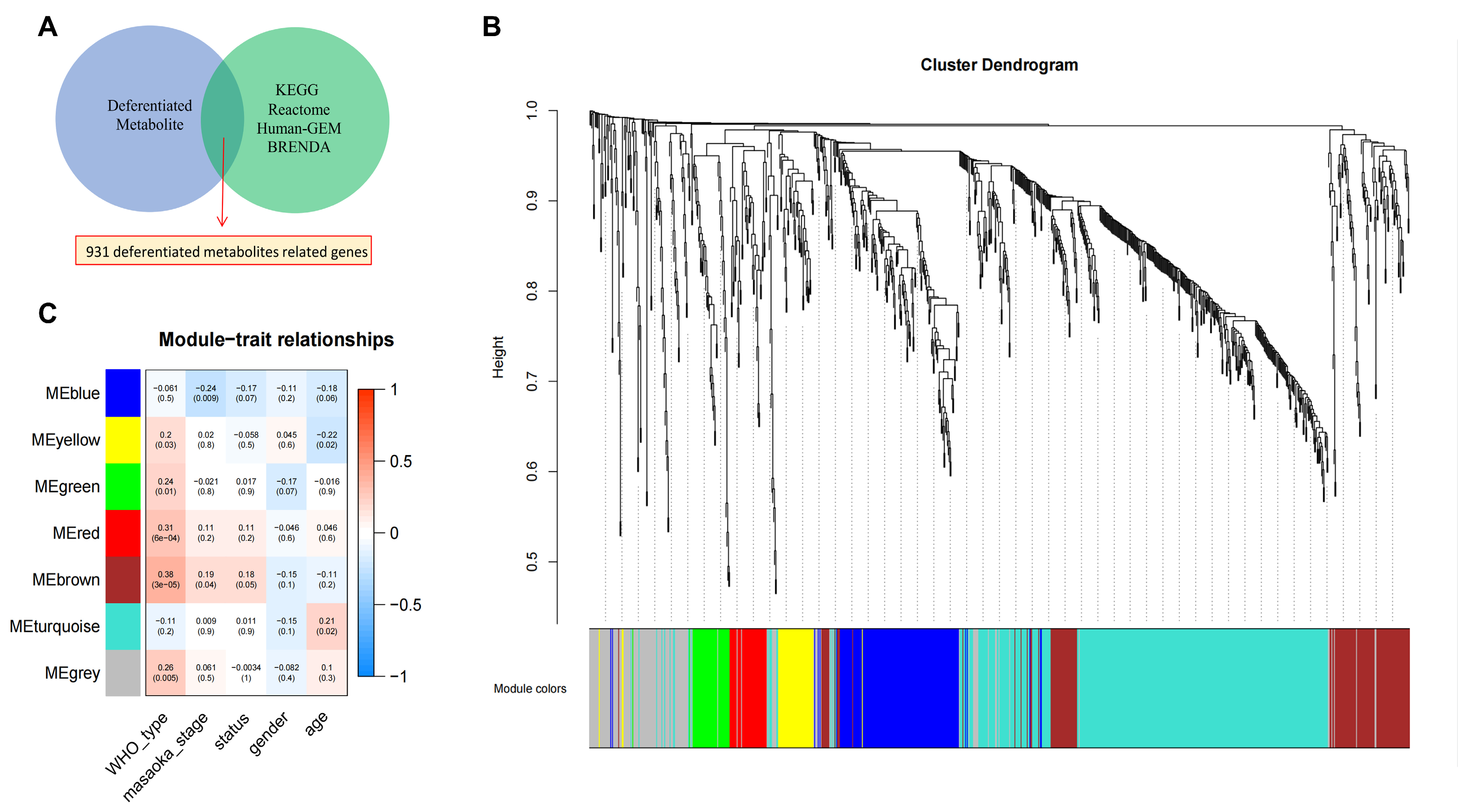
2.2. Identification of Differential Metabolism-Related Genes in TETs with Prognostic Relevance
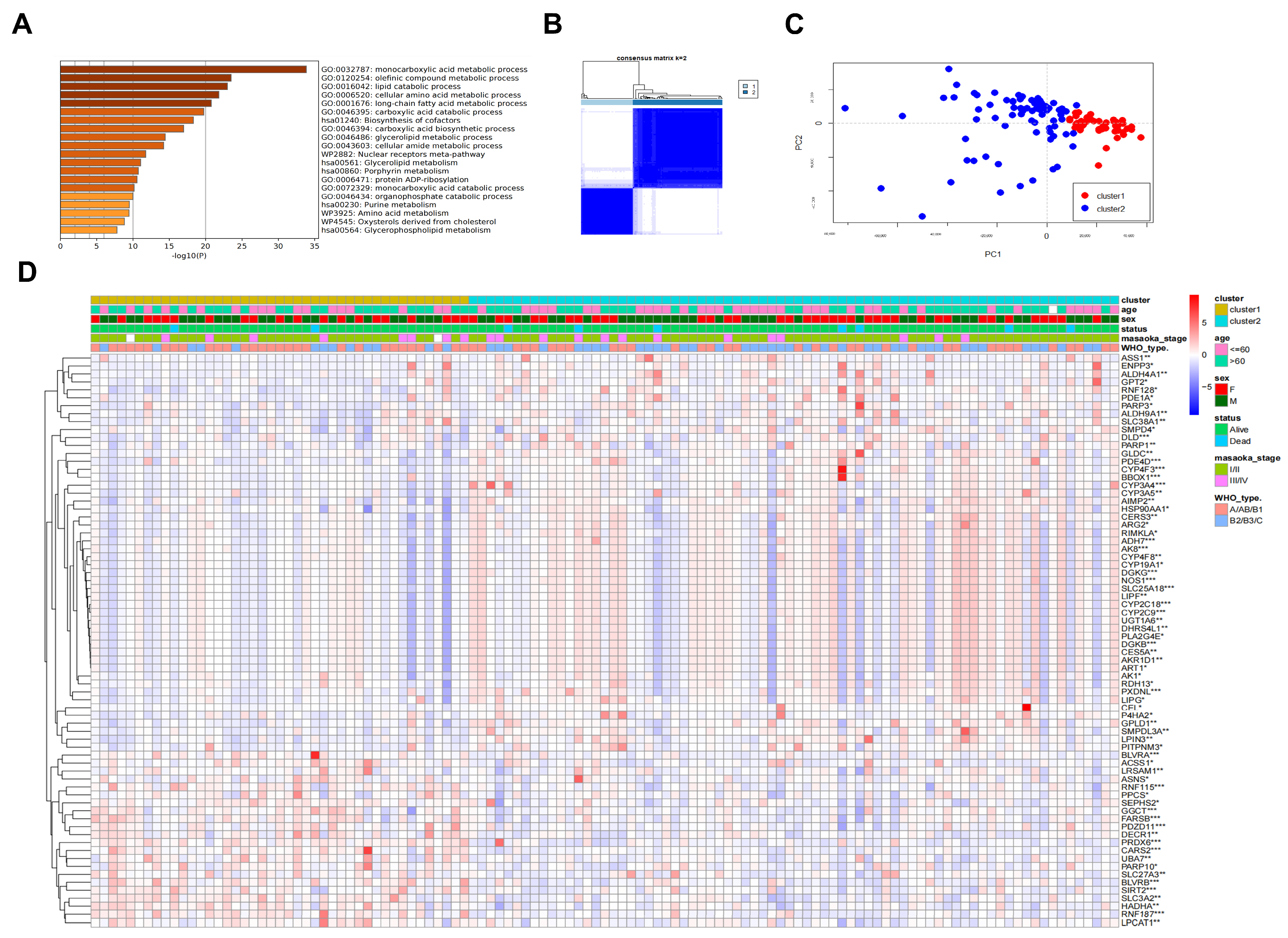
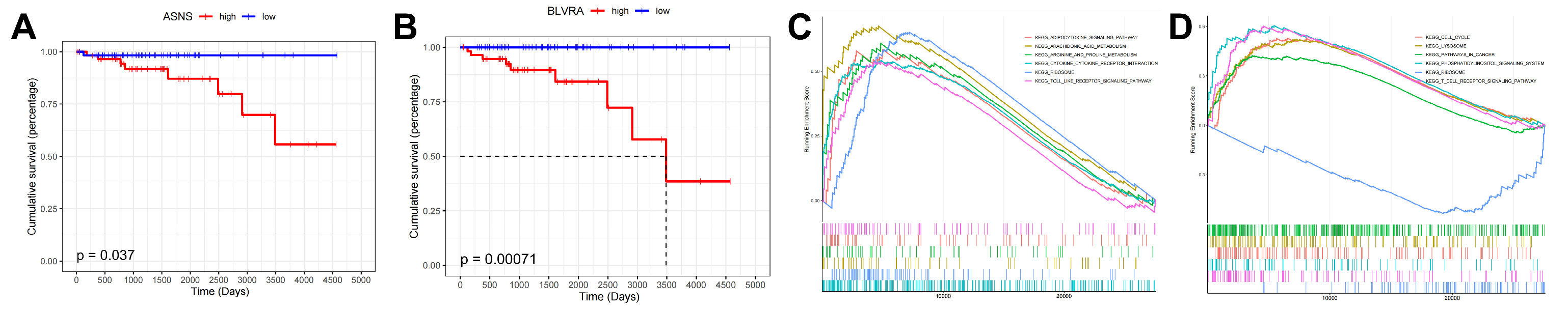
2.3. Prognosis-Associated Metabolic Features Mediated by TP-MRGs
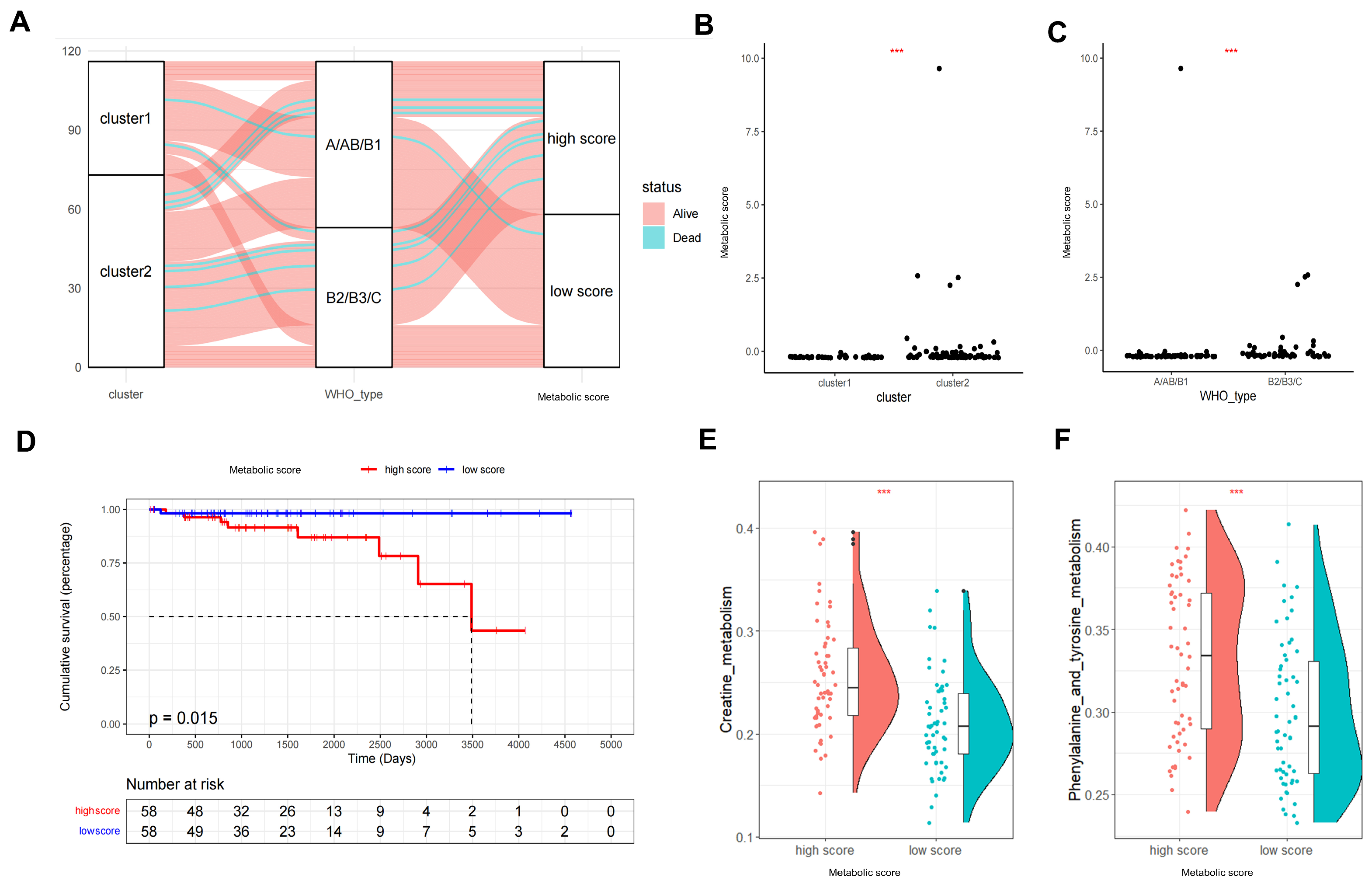
2.4. Metabolic Score Construction and Its Clinical Relevance
2.5. Metabolic Patterns Characterized by Distinct Immune Landscapes
2.6. Expression, Prognosis, and GSEA of MSGs in TETs
3. Discussion
4. Materials and Methods
4.1. Metabolomics Sample Collection and Preparation
4.2. LC-MS Analysis and Data Processing
4.3. Transcriptomic Data Collection and Processing
4.4. WGCNA Analysis
4.5. Consensus Clustering for TP-MRGs
4.6. Construction of the Metabolic Score
4.7. Gene Set Variation Analysis and Gene Set Enrichment Analysis
4.8. Tumor-Infiltrating Immune Cell Evaluation and ESTIMATE
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marx, A.; Chan, J.K.; Coindre, J.M.; Detterbeck, F.; Girard, N.; Harris, N.L.; Jaffe, E.S.; Kurrer, M.O.; Marom, E.M.; Moreira, A.L.; et al. The 2015 World Health Organization Classification of Tumors of the Thymus: Continuity and Changes. J. Thorac. Oncol. 2015, 10, 1383–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorsetti, M.; Leo, F.; Trama, A.; D’Angelillo, R.; Serpico, D.; Macerelli, M.; Zucali, P.; Gatta, G.; Garassino, M.C. Thymoma and thymic carcinomas. Crit. Rev. Oncol./Hematol. 2016, 99, 332–350. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Riely, G.J.; Akerley, W.; Borghaei, H.; Chang, A.C.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Govindan, R.; et al. National Comprehensive Cancer Network. Thymomas and thymic carcinomas: Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2013, 11, 562–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Zhang, Y. Outcomes after surgical resection of thymic carcinoma: A study from a single tertiary referral centre. Eur. J. Surg. Oncol. 2014, 40, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shi, J.; Fan, L.; Hu, D.; Yang, J.; Zhao, H. Surgical treatment of thymoma: An 11-year experience with 761 patients. Eur. J. Cardiothorac. Surg. 2016, 49, 1144–1149. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.W.; Palma, D.A.; Camidge, D.R.; Jones, B.L.; Robin, T.P.; Sher, D.J.; Koshy, M.; Kavanagh, B.D.; Gaspar, L.E.; Rusthoven, C.G. The Impact of Postoperative Radiotherapy for Thymoma and Thymic Carcinoma. J. Thorac. Oncol. 2017, 12, 734–744. [Google Scholar] [CrossRef] [Green Version]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies-Challenges and Emerging Directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Bae, M.K.; Byun, C.S.; Lee, C.Y.; Lee, J.G.; Park, I.K.; Kim, D.J.; Yang, W.I.; Chung, K.Y. Clinical outcomes and prognosis of recurrent thymoma management. J. Thorac. Oncol. 2012, 7, 1304–1314. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Marx, A.; Chen, W.H.; Yong, J.; Puppe, B.; Stroebel, P.; Mueller-Hermelink, H.K. New WHO histologic classification predicts prognosis of thymic epithelial tumors: A clinicopathologic study of 200 thymoma cases from China. Cancer 2002, 95, 420–429. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Y.; Wang, W.; Chen, H.; Ling, T.; Yang, R.; Wang, Y.; Duan, C.; Liu, Y.; Guo, X.; et al. Identification and Characterization of Robust Hepatocellular Carcinoma Prognostic Subtypes Based on an Integrative Metabolite-Protein Interaction Network. Adv. Sci. 2021, 8, e2100311. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef] [PubMed]
- Placzek, S.; Schomburg, I.; Chang, A.; Jeske, L.; Ulbrich, M.; Tillack, J.; Schomburg, D. BRENDA in 2017: New perspectives and new tools in BRENDA. Nucleic Acids Res. 2017, 45, D380–D388. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Richards, N.G.; Schuster, S.M. Mechanistic issues in asparagine synthetase catalysis. Adv. Enzymol. Relat. Areas Mol. Biol. 1998, 72, 145–198. [Google Scholar] [CrossRef]
- Shen, Y.; Li, M.; Xiong, Y.; Gui, S.; Bai, J.; Zhang, Y.; Li, C. Proteomics Analysis Identified ASNS as a Novel Biomarker for Predicting Recurrence of Skull Base Chordoma. Front. Oncol. 2021, 11, 698497. [Google Scholar] [CrossRef]
- Mao, H.; Xu, Y.; Zhang, Z.; Sun, G.; Wang, Z.; Qiao, D.; Yin, X.; Liu, S.; Bo, P. Biliverdin Reductase A (BLVRA) Promotes Colorectal Cancer Cell Progression by Activating the Wnt/beta-Catenin Signaling Pathway. Cancer Manag. Res. 2020, 12, 2697–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, E.; Liu, S.; Zhang, Z.; Zhang, R.; Huang, D.; Gao, T.; Zhang, T.; Xu, G. Therapeutic Potential of Glutamine Pathway in Lung Cancer. Front. Oncol. 2021, 11, 835141. [Google Scholar] [CrossRef]
- Alwahsh, M.; Knitsch, R.; Marchan, R.; Lambert, J.; Hoerner, C.; Zhang, X.; Schalke, B.; Lee, D.-H.; Bulut, E.; Graeter, T.; et al. Metabolic Profiling of Thymic Epithelial Tumors Hints to a Strong Warburg Effect, Glutaminolysis and Precarious Redox Homeostasis as Potential Therapeutic Targets. Cancers 2022, 14, 1564. [Google Scholar] [CrossRef]
- Wu, H.; Wang, L.; Zhan, X.; Wang, B.; Wu, J.; Zhou, A. A UPLC-Q-TOF/MS-based plasma metabolomics approach reveals the mechanism of Compound Kushen Injection-based intervention against non-small cell lung cancer in Lewis tumor-bearing mice. Phytomedicine 2020, 76, 153259. [Google Scholar] [CrossRef]
- Locke, M.; Ghazaly, E.; Freitas, M.O.; Mitsinga, M.; Lattanzio, L.; Lo Nigro, C.; Nagano, A.; Wang, J.; Chelala, C.; Szlosarek, P.; et al. Inhibition of the Polyamine Synthesis Pathway Is Synthetically Lethal with Loss of Argininosuccinate Synthase 1. Cell Rep. 2016, 16, 1604–1613. [Google Scholar] [CrossRef] [Green Version]
- Delage, B.; Fennell, D.A.; Nicholson, L.; McNeish, I.; Lemoine, N.R.; Crook, T.; Szlosarek, P.W. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 2010, 126, 2762–2772. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Prusinkiewicz, M.A.; Gameiro, S.F.; Ghasemi, F.; Dodge, M.J.; Zeng, P.Y.F.; Maekebay, H.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Survival-Associated Metabolic Genes in Human Papillomavirus-Positive Head and Neck Cancers. Cancers 2020, 12, 253. [Google Scholar] [CrossRef] [Green Version]
- Milman, H.A.; Cooney, D.A. The distribution of L-asparagine synthetase in the principal organs of several mammalian and avian species. Biochem. J. 1974, 142, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; He, X.; Zheng, Y.; Feng, W.; Xia, X.; Yu, X.; Lin, Z. Down-regulation of asparagine synthetase induces cell cycle arrest and inhibits cell proliferation of breast cancer. Chem. Biol. Drug Des. 2014, 84, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Sircar, K.; Huang, H.; Hu, L.; Cogdell, D.; Dhillon, J.; Tzelepi, V.; Efstathiou, E.; Koumakpayi, I.H.; Saad, F.; Luo, D.; et al. Integrative molecular profiling reveals asparagine synthetase is a target in castration-resistant prostate cancer. Am. J. Pathol. 2012, 180, 895–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Dong, L.W.; Tan, Y.X.; Zhang, J.; Pan, Y.F.; Yang, C.; Li, M.H.; Ding, Z.W.; Liu, L.J.; Jiang, T.Y.; et al. Asparagine synthetase is an independent predictor of surgical survival and a potential therapeutic target in hepatocellular carcinoma. Br. J. Cancer 2013, 109, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Kubickova, K.N.; Subhanova, I.; Konickova, R.; Matousova, L.; Urbanek, P.; Parobkova, H.; Kupec, M.; Pudil, J.; Vitek, L. Predictive role BLVRA mRNA expression in hepatocellular cancer. Ann. Hepatol. 2016, 15, 881–887. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Miralem, T.; Maines, M.D. Biliverdin reductase: A target for cancer therapy? Front. Pharmacol. 2015, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Song, S.; Yi, Z.; Zhang, M.; Li, J.; Yang, F.; Yin, H.; Yu, X.; Guan, C.; Liu, Y.; et al. HGF induces EMT in non-small-cell lung cancer through the hBVR pathway. Eur. J. Pharmacol. 2017, 811, 180–190. [Google Scholar] [CrossRef]
- Liu, M.; Liu, Y.; Feng, H.; Jing, Y.; Zhao, S.; Yang, S.; Zhang, N.; Jin, S.; Li, Y.; Weng, M.; et al. Clinical Significance of Screening Differential Metabolites in Ovarian Cancer Tissue and Ascites by LC/MS. Front. Pharmacol. 2021, 12, 701487. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhu, J.; Ma, X.; Zhang, L.; Wu, Y.; Zhu, W.; Xing, Y.; Jia, Y.; Wang, Y. Transcriptomic and Metabolomic Profiling in Helicobacter pylori-Induced Gastric Cancer Identified Prognosis- and Immunotherapy-Relevant Gene Signatures. Front. Cell Dev. Biol. 2021, 9, 769409. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene Expression Profiling in Breast Cancer: Understanding the Molecular Basis of Histologic Grade To Improve Prognosis. JNCI J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Li, M.; Zhou, R.; Zhang, J.; Sun, H.; Shi, M.; Bin, J.; Liao, Y.; Rao, J.; Liao, W. Tumor Microenvironment Characterization in Gastric Cancer Identifies Prognostic and Immunotherapeutically Relevant Gene Signatures. Cancer Immunol. Res. 2019, 7, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Factors | Variables | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|---|
| p-Value | HR (95% CI) | p-Value | HR (95% CI) | ||
| WHO type | A/AB/B1 vs. B2/B3/C | 0.83 | 0.86 (0.21–3.5) | 0.65 | 0.66 (0.11–4) |
| Masaoka stage | I/II vs. III/IV | 0.54 | 1.6 (0.34–8) | 0.31 | 2.7 (0.4–18) |
| Sex | Male vs. Female | 0.53 | 0.66 (0.18–2.5) | 0.62 | 1.5 (0.31–7.2) |
| Age | ≤60 vs. >60 (years old) | 0.17 | 3 (0.63–15) | 0.14 | 4 (0.63–25) |
| ASNS | Expression (high vs. low) | 0.071 | 0.15 (0.018–1.2) | 0.23 | 0.25 (0.027–2.4) |
| BLVRA | Expression (high vs. low) | 1 | 1.3 × 10−9 (0–Inf) | 1 | 5.2 × 10−10 (0–Inf) |
| Age, Years | |
|---|---|
| Median (range) | 53.5 (32–80) |
| Sex (%) | |
| Female | 14 (43.8) |
| Male | 18 (56.2) |
| WHO histological type (%) | |
| A | 2 (6.3) |
| AB | 12 (37.5) |
| B1 | 1 (3.1) |
| B2 | 4 (12.5) |
| B3 | 3 (9.3) |
| C | 8 (25.0) |
| NA | 2 (6.3) |
| Myasthenia gravis (%) | |
| (+) | 10 (31.3) |
| (−) | 20 (62.5) |
| NA | 2 (6.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, E.; Zhou, Y.; Liu, S.; Zhang, Z.; Zhang, R.; Huang, D.; Gao, T.; Zhang, T.; Xu, G. Metabolomic and Transcriptomic Profiling Identified Significant Genes in Thymic Epithelial Tumor. Metabolites 2022, 12, 567. https://doi.org/10.3390/metabo12060567
Tang E, Zhou Y, Liu S, Zhang Z, Zhang R, Huang D, Gao T, Zhang T, Xu G. Metabolomic and Transcriptomic Profiling Identified Significant Genes in Thymic Epithelial Tumor. Metabolites. 2022; 12(6):567. https://doi.org/10.3390/metabo12060567
Chicago/Turabian StyleTang, Enyu, Yang Zhou, Siyang Liu, Zhiming Zhang, Rixin Zhang, Dejing Huang, Tong Gao, Tianze Zhang, and Guangquan Xu. 2022. "Metabolomic and Transcriptomic Profiling Identified Significant Genes in Thymic Epithelial Tumor" Metabolites 12, no. 6: 567. https://doi.org/10.3390/metabo12060567
APA StyleTang, E., Zhou, Y., Liu, S., Zhang, Z., Zhang, R., Huang, D., Gao, T., Zhang, T., & Xu, G. (2022). Metabolomic and Transcriptomic Profiling Identified Significant Genes in Thymic Epithelial Tumor. Metabolites, 12(6), 567. https://doi.org/10.3390/metabo12060567