
Succinate as a New Actor in Pluripotency and Early Development?


{kind=link}
{kind=link}
Abstract
:1. Introduction
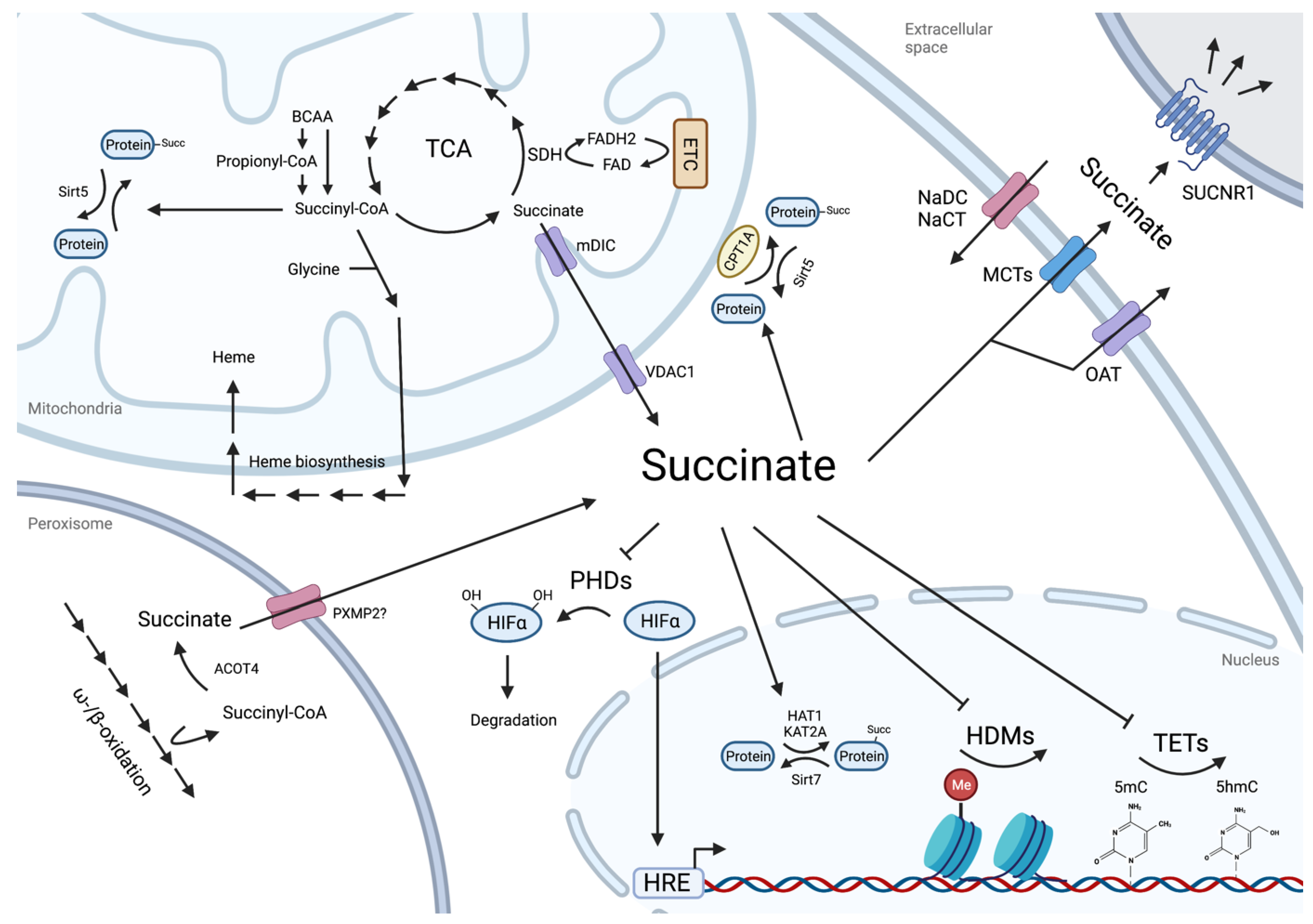
2. Succinate as Metabolic Cross-Roads
3. Regulatory Roles of Intracellular Succinate
3.1. Protein Lysine Residue Succinylation
3.2. Succinate as an Epigenetic Landscape Remodeler
4. Succinate as a Paracrine Effector
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Evans, M.J.; Kaufman, M.H. Establishment in Culture of Pluripotential Cells from Mouse Embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a Pluripotent Cell Line from Early Mouse Embryos Cultured in Medium Conditioned by Teratocarcinoma Stem Cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, J.; Smith, A. Naive and Primed Pluripotent States. Cell Stem Cell 2009, 4, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, L.; Ayyash, M.; Novershtern, N.; Hanna, J.H. Dynamic Stem Cell States: Naive to Primed Pluripotency in Rodents and Humans. Nat. Rev. Mol. Cell Biol. 2016, 17, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Rebuzzini, P.; Zuccotti, M.; Garagna, S. Building Pluripotency Identity in the Early Embryo and Derived Stem Cells. Cells 2021, 10, 2049. [Google Scholar] [CrossRef] [PubMed]
- Brons, I.G.M.; Smithers, L.E.; Trotter, M.W.B.; Rugg-Gunn, P.; Sun, B.; Chuva de Sousa Lopes, S.M.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A.; et al. Derivation of Pluripotent Epiblast Stem Cells from Mammalian Embryos. Nature 2007, 448, 191–195. [Google Scholar] [CrossRef]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D.G. New Cell Lines from Mouse Epiblast Share Defining Features with Human Embryonic Stem Cells. Nature 2007, 448, 196–199. [Google Scholar] [CrossRef]
- Kinoshita, M.; Barber, M.; Mansfield, W.; Cui, Y.; Spindlow, D.; Stirparo, G.G.; Dietmann, S.; Nichols, J.; Smith, A. Capture of Mouse and Human Stem Cells with Features of Formative Pluripotency. Cell Stem Cell 2020. [Google Scholar] [CrossRef]
- Du, P.; Pirouz, M.; Choi, J.; Huebner, A.J.; Clement, K.; Meissner, A.; Hochedlinger, K.; Gregory, R.I. An Intermediate Pluripotent State Controlled by MicroRNAs Is Required for the Naive-to-Primed Stem Cell Transition. Cell Stem Cell 2018, 22, 851–864.e5. [Google Scholar] [CrossRef] [Green Version]
- Bulut-Karslioglu, A.; Biechele, S.; Jin, H.; MacRae, T.A.; Hejna, M.; Gertsenstein, M.; Song, J.S.; Ramalho-Santos, M. Inhibition of MTOR Induces a Paused Pluripotent State. Nature 2016, 540, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Hussein, A.M.; Wang, Y.; Mathieu, J.; Margaretha, L.; Song, C.; Jones, D.C.; Cavanaugh, C.; Miklas, J.W.; Mahen, E.; Showalter, M.R.; et al. Metabolic Control over MTOR-Dependent Diapause-like State. Dev. Cell 2020, 52, 236–250.e7. [Google Scholar] [CrossRef] [PubMed]
- Macfarlan, T.S.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.M.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic Stem Cell Potency Fluctuates with Endogenous Retrovirus Activity. Nature 2012, 487, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percharde, M.; Lin, C.J.; Yin, Y.; Guan, J.; Peixoto, G.A.; Bulut-Karslioglu, A.; Biechele, S.; Huang, B.; Shen, X.; Ramalho-Santos, M. A LINE1-Nucleolin Partnership Regulates Early Development and ESC Identity. Cell 2018, 174, 391–405.e19. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Ocampo, A.; Belmonte, J.C.I. Cellular Metabolism and Induced Pluripotency. Cell 2016, 166, 1371–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, J.; Ruohola-Baker, H. Metabolic Remodeling during the Loss and Acquisition of Pluripotency. Development 2017, 144, 541–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsogtbaatar, E.; Landin, C.; Minter-Dykhouse, K.; Folmes, C.D.L. Energy Metabolism Regulates Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α Induced Switch from Bivalent to Exclusively Glycolytic Metabolism during ESC-to-EpiSC/HESC Transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The Metabolome Regulates the Epigenetic Landscape during Naive-to-Primed Human Embryonic Stem Cell Transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef]
- Sone, M.; Morone, N.; Nakamura, T.; Tanaka, A.; Okita, K.; Woltjen, K.; Nakagawa, M.; Heuser, J.E.; Yamada, Y.; Yamanaka, S.; et al. Hybrid Cellular Metabolism Coordinated by Zic3 and Esrrb Synergistically Enhances Induction of Naive Pluripotency. Cell Metab. 2017, 25, 1103–1117.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ratanasirintrawoot, S.; Chandrasekaran, S.; Wu, Z.; Ficarro, S.B.; Yu, C.; Ross, C.A.; Cacchiarelli, D.; Xia, Q.; Seligson, M.; et al. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell Stem Cell 2016, 19, 66–80. [Google Scholar] [CrossRef] [Green Version]
- Tobias, I.C.; Isaac, R.R.; Dierolf, J.G.; Khazaee, R.; Cumming, R.C.; Betts, D.H. Metabolic Plasticity during Transition to Naïve-like Pluripotency in Canine Embryo-Derived Stem Cells. Stem Cell Res. 2018, 30, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic Regulation in Pluripotent Stem Cells during Reprogramming and Self-Renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-Ketoglutarate Maintains the Pluripotency of Embryonic Stem Cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TeSlaa, T.; Chaikovsky, A.C.; Lipchina, I.; Escobar, S.L.; Hochedlinger, K.; Huang, J.; Graeber, T.G.; Braas, D.; Teitell, M.A. α-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016, 24, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics – Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- Ehnes, D.D.; Hussein, A.M.; Ware, C.B.; Mathieu, J.; Ruohola-Baker, H. Combinatorial Metabolism Drives the Naive to Primed Pluripotent Chromatin Landscape. Exp. Cell Res. 2020, 389, 111913. [Google Scholar] [CrossRef]
- Arnold, P.K.; Jackson, B.T.; Paras, K.I.; Brunner, J.S.; Hart, M.L.; Newsom, O.J.; Alibeckoff, S.P.; Endress, J.; Drill, E.; Sullivan, L.B.; et al. A Non-Canonical Tricarboxylic Acid Cycle Underlies Cellular Identity. Nature 2022, 603, 477–481. [Google Scholar] [CrossRef]
- Atamna, H. Heme, Iron, and the Mitochondrial Decay of Ageing. Ageing Res. Rev. 2004, 3, 303–318. [Google Scholar] [CrossRef]
- Bowers, M.A.; Aicher, L.D.; Davis, H.A.; Woods, J.S. Quantitative Determination of Porphyrins in Rat and Human Urine and Evaluation of Urinary Porphyrin Profiles during Mercury and Lead Exposures. J. Lab. Clin. Med. 1992, 120, 272–281. [Google Scholar] [CrossRef]
- Daniell, W.E.; Stockbridge, H.L.; Labbe, R.F.; Woods, J.S.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.R.; Ellefson, R.D.; Moore, M.R.; Pierach, C.A.; et al. Environmental Chemical Exposures and Disturbances of Heme Synthesis. Environ. Health Perspect. 1997, 105, 37–53. [Google Scholar] [PubMed] [Green Version]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.-C.; Gouya, L.; et al. Acute Intermittent Porphyria Causes Hepatic Mitochondrial Energetic Failure in a Mouse Model. Int. J. Biochem. Cell Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westin, M.A.K.; Hunt, M.C.; Alexson, S.E.H. The Identification of a Succinyl-CoA Thioesterase Suggests a Novel Pathway for Succinate Production in Peroxisomes. J. Biol. Chem. 2005, 280, 38125–38132. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, X.; Yao, H.; Chen, X.; Shang, L.; Li, P.; Cui, X.; Zeng, J. Peroxisome-Generated Succinate Induces Lipid Accumulation and Oxidative Stress in the Kidneys of Diabetic Mice. J. Biol. Chem. 2022, 298, 101660. [Google Scholar] [CrossRef]
- Martinez-Val, A.; Lynch, C.J.; Calvo, I.; Ximénez-Embún, P.; Garcia, F.; Zarzuela, E.; Serrano, M.; Munoz, J. Dissection of Two Routes to Naïve Pluripotency Using Different Kinase Inhibitors. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Boroviak, T.; Loos, R.; Lombard, P.; Okahara, J.; Behr, R.; Sasaki, E.; Nichols, J.; Smith, A.; Bertone, P. Lineage-Specific Profiling Delineates the Emergence and Progression of Naive Pluripotency in Mammalian Embryogenesis. Dev. Cell 2015, 35, 366–382. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Prezioso, G.; Quagliariello, E.; Klingenberg, M. Kinetic Study of the Dicarboxylate Carrier in Rat Liver Mitochondria. Eur. J. Biochem. 1971, 22, 66–74. [Google Scholar] [CrossRef]
- Hodge, T.; Colombini, M. Regulation of Metabolite Flux through Voltage-Gating of VDAC Channels. J. Membr. Biol. 1997, 157, 271–279. [Google Scholar] [CrossRef]
- Andrienko, T.N.; Pasdois, P.; Pereira, G.C.; Ovens, M.J.; Halestrap, A.P. The Role of Succinate and ROS in Reperfusion Injury - A Critical Appraisal. J. Mol. Cell. Cardiol. 2017, 110, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.; Bozi, L.H.M.; Yaghi, O.K.; Mills, E.L.; Xiao, H.; Nicholson, H.E.; Paschini, M.; Paulo, J.A.; Garrity, R.; Laznik-Bogoslavski, D.; et al. PH-Gated Succinate Secretion Regulates Muscle Remodeling in Response to Exercise. Cell 2020, 183, 62–75.e17. [Google Scholar] [CrossRef]
- Anzai, N.; Kanai, Y.; Endou, H. Organic Anion Transporter Family: Current Knowledge. J. Pharmacol. Sci. 2006, 100, 411–426. [Google Scholar] [CrossRef] [Green Version]
- Pajor, A.M. Sodium-Coupled Dicarboxylate and Citrate Transporters from the SLC13 Family. Pflugers Arch. 2014, 466, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Khamaysi, A.; Anbtawee-Jomaa, S.; Fremder, M.; Eini-Rider, H.; Shimshilashvili, L.; Aharon, S.; Aizenshtein, E.; Shlomi, T.; Noguchi, A.; Springer, D.; et al. Systemic Succinate Homeostasis and Local Succinate Signaling Affect Blood Pressure and Modify Risks for Calcium Oxalate Lithogenesis. J. Am. Soc. Nephrol. 2019, 30, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Detraux, D.; Kuppers, D.; Wang, Y.; Cavanaugh, C.; Sidhu, S.; Levy, S.; Robitaille, A.M.; Ferreccio, A.; Bottorff, T.; et al. Folliculin Regulates MTORC1/2 and WNT Pathways in Early Human Pluripotency. Nat. Commun. 2019, 10, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, B.; Ueda, M.; Sabri, S.; Brumbaugh, J.; Huebner, A.J.; Sahakyan, A.; Clement, K.; Clowers, K.J.; Erickson, A.R.; Shioda, K.; et al. Reduced MEK Inhibition Preserves Genomic Stability in Naive Human Embryonic Stem Cells. Nat. Methods 2018, 15, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Grow, E.J.; Flynn, R.A.; Chavez, S.L.; Bayless, N.L.; Wossidlo, M.; Wesche, D.J.; Martin, L.; Ware, C.B.; Blish, C.A.; Chang, H.Y.; et al. Intrinsic Retroviral Reactivation in Human Preimplantation Embryos and Pluripotent Cells. Nature 2015, 522, 221–246. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Okamoto, I.; Sasaki, K.; Yabuta, Y.; Iwatani, C.; Tsuchiya, H.; Seita, Y.; Nakamura, S.; Yamamoto, T.; Saitou, M. A Developmental Coordinate of Pluripotency among Mice, Monkeys and Humans. Nature 2016, 537, 57–62. [Google Scholar] [CrossRef]
- Yang, M.; Yu, H.; Yu, X.; Liang, S.; Hu, Y.; Luo, Y.; Izsvák, Z.; Sun, C.; Wang, J. Chemical-Induced Chromatin Remodeling Reprograms Mouse ESCs to Totipotent-like Stem Cells. Cell Stem Cell 2022, 29, 400–418.e13. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of Lysine Succinylation as a New Post-Translational Modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef]
- Sreedhar, A.; Wiese, E.K.; Hitosugi, T. Enzymatic and Metabolic Regulation of Lysine Succinylation. Genes Dis. 2020, 7, 166–171. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.; Lee, J.-H.; Li, X.; et al. KAT2A Coupled with the α-KGDH Complex Acts as a Histone H3 Succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurmi, K.; Hitosugi, S.; Wiese, E.K.; Boakye-Agyeman, F.; Gonsalves, W.I.; Lou, Z.; Karnitz, L.M.; Goetz, M.P.; Hitosugi, T. Carnitine Palmitoyltransferase 1A Has a Lysine Succinyltransferase Activity. Cell Rep. 2018, 22, 1365–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Yuan, Y.; Yuan, H.; Wang, J.; Yun, H.; Geng, Y.; Zhao, M.; Li, L.; Weng, Y.; Liu, Z.; et al. Histone Acetyltransferase 1 Is a Succinyltransferase for Histones and Non-histones and Promotes Tumorigenesis. EMBO Rep. 2021, 22, e50967. [Google Scholar] [CrossRef] [PubMed]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J. Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism. iScience 2018, 2, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Shi, L.; Yang, S.; Yan, R.; Zhang, D.; Yang, J.; He, L.; Li, W.; Yi, X.; Sun, L.; et al. SIRT7 Is a Histone Desuccinylase That Functionally Links to Chromatin Compaction and Genome Stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef] [Green Version]
- Gut, P.; Matilainen, S.; Meyer, J.G.; Pällijeff, P.; Richard, J.; Carroll, C.J.; Euro, L.; Jackson, C.B.; Isohanni, P.; Minassian, B.A.; et al. SUCLA2 Mutations Cause Global Protein Succinylation Contributing to the Pathomechanism of a Hereditary Mitochondrial Disease. Nat. Commun. 2020, 11, 5927. [Google Scholar] [CrossRef]
- Guo, Z.; Pan, F.; Peng, L.; Tian, S.; Jiao, J.; Liao, L.; Lu, C.; Zhai, G.; Wu, Z.; Dong, H.; et al. Systematic Proteome and Lysine Succinylome Analysis Reveals Enhanced Cell Migration by Hyposuccinylation in Esophageal Squamous Cell Carcinoma. Mol. Cell. Proteomics 2021, 20, 100053. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.M.; Skinner, M.E.; et al. SIRT5-Mediated Lysine Desuccinylation Impacts Diverse Metabolic Pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 Regulates the Mitochondrial Lysine Succinylome and Metabolic Networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [Green Version]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-Assisted Proteomics Identifies Succinylation and SIRT5 as Important Regulators of Cardiac Function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Lu, J.; Maringer, K.V.; Sims-Lucas, S.; Prochownik, E.V.; Gibson, B.W.; Goetzman, E.S. Lysine Desuccinylase SIRT5 Binds to Cardiolipin and Regulates the Electron Transport Chain. J. Biol. Chem. 2017, 292, 10239–10249. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Peasley, K.D.; Cargill, K.R.; Maringer, K.V.; Bharathi, S.S.; Mukherjee, E.; Zhang, Y.; Holtz, A.; Basisty, N.; Yagobian, S.D.; et al. Sirtuin 5 Regulates Proximal Tubule Fatty Acid Oxidation to Protect against AKI. J. Am. Soc. Nephrol. 2019, 30, 2384–2398. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Q.-b.; Bu, C.; Wang, D.; Yu, K.; Gan, Z.; Chang, J.; Cheng, Z.; Liu, Z. Quantitative Dynamics of Proteome, Acetylome, and Succinylome during Stem-Cell Differentiation into Hepatocyte-like Cells. J. Proteome Res. 2018, 17, 2491–2498. [Google Scholar] [CrossRef]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-Inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an Intermediate in Metabolism, Signal Transduction, ROS, Hypoxia, and Tumorigenesis. Biochim. Biophys. Acta-Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Cervera, A.M.; Bayley, J.P.; Devilee, P.; McCreath, K.J. Inhibition of Succinate Dehydrogenase Dysregulates Histone Modification in Mammalian Cells. Mol. Cancer 2009, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-Dependent Histone and DNA Demethylases by Fumarate and Succinate That Are Accumulated in Mutations of FH and SDH Tumor Suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [Green Version]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I.; Jahromi, M.S.; Xekouki, P.; et al. Succinate Dehydrogenase Mutation Underlies Global Epigenomic Divergence in Gastrointestinal Stromal Tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Xie, W. Rebooting the Epigenomes during Mammalian Early Embryogenesis. Stem Cell Rep. 2020, 15, 1158–1175. [Google Scholar] [CrossRef]
- Morin, A.; Goncalves, J.; Moog, S.; Castro-Vega, L.-J.; Job, S.; Buffet, A.; Fontenille, M.-J.; Woszczyk, J.; Gimenez-Roqueplo, A.-P.; Letouzé, E.; et al. TET-Mediated Hypermethylation Primes SDH-Deficient Cells for HIF2α-Driven Mesenchymal Transition. Cell Rep. 2020, 30, 4551–4566.e7. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.; Chang, W.Y.; Hunkapiller, J.; Cagney, G.; Garcha, K.; Torchia, J.; Krogan, N.J.; Reiter, J.F.; Stanford, W.L. Polycomb-like 2 Associates with PRC2 and Regulates Transcriptional Networks during Mouse Embryonic Stem Cell Self-Renewal and Differentiation. Cell Stem Cell 2010, 6, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landeira, D.; Sauer, S.; Poot, R.; Dvorkina, M.; Mazzarella, L.; Jørgensen, H.F.; Pereira, C.F.; Leleu, M.; Piccolo, F.M.; Spivakov, M.; et al. Jarid2 Is a PRC2 Component in Embryonic Stem Cells Required for Multi-Lineage Differentiation and Recruitment of PRC1 and RNA Polymerase II to Developmental Regulators. Nat. Cell Biol. 2010, 12, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Moody, J.D.; Levy, S.; Mathieu, J.; Xing, Y.; Kim, W.; Dong, C.; Tempel, W.; Robitaille, A.M.; Dang, L.T.; Ferreccio, A.; et al. First Critical Repressive H3K27me3 Marks in Embryonic Stem Cells Identified Using Designed Protein Inhibitor. Proc. Natl. Acad. Sci. USA 2017, 114, 10125–10130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ain, R.; Seshagiri, P.B. Succinate and Malate Improve Development of Hamster Eight-Cell Embryos in Vitro: Confirmation of Viability by Embryo Transfer. Mol. Reprod. Dev. 1997, 47, 440–447. [Google Scholar] [CrossRef]
- Ispada, J.; da Fonseca Junior, A.M.; de Lima, C.B.; Dos Santos, E.C.; Fontes, P.K.; Nogueira, M.F.G.; da Silva, V.L.; Almeida, F.N.; Leite, S.d.C.; Chitwood, J.L.; et al. Tricarboxylic Acid Cycle Metabolites as Mediators of DNA Methylation Reprogramming in Bovine Preimplantation Embryos. Int. J. Mol. Sci. 2020, 21, 6868. [Google Scholar] [CrossRef]
- Wang, X.-H.; Xu, S.; Zhou, X.-Y.; Zhao, R.; Lin, Y.; Cao, J.; Zang, W.-D.; Tao, H.; Xu, W.; Li, M.-Q.; et al. Low Chorionic Villous Succinate Accumulation Associates with Recurrent Spontaneous Abortion Risk. Nat. Commun. 2021, 12, 3428. [Google Scholar] [CrossRef]
- Atallah, R.; Gindlhuber, J.; Platzer, W.; Bärnthaler, T.; Tatzl, E.; Toller, W.; Strutz, J.; Rittchen, S.; Luschnig, P.; Birner-Gruenberger, R.; et al. Sucnr1 Is Expressed in Human Placenta and Mediates Angiogenesis: Significance in Gestational Diabetes. Int. J. Mol. Sci. 2021, 22, 12048. [Google Scholar] [CrossRef]
- Selak, M.A.; Durán, R.V.; Gottlieb, E. Redox Stress Is Not Essential for the Pseudo-Hypoxic Phenotype of Succinate Dehydrogenase Deficient Cells. Biochim. Biophys. Acta-Bioenerg. 2006, 1757, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Kluckova, K.; Tennant, D.A. Metabolic Implications of Hypoxia and Pseudohypoxia in Pheochromocytoma and Paraganglioma. Cell Tissue Res. 2018, 372, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, J.; Zhou, W.; Xing, Y.; Sperber, H.; Ferreccio, A.; Agoston, Z.; Kuppusamy, K.T.; Moon, R.T.; Ruohola-Baker, H. Hypoxia-Inducible Factors Have Distinct and Stage-Specific Roles during Reprogramming of Human Cells to Pluripotency. Cell Stem Cell 2014, 14, 592–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Miao, F.J.P.; Lin, D.C.H.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.-L.; Tian, H.; Ling, L. Citric Acid Cycle Intermediates as Ligands for Orphan G-Protein-Coupled Receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Cho, S.W.; Saxena, D.; Li, X. Multifaceted Actions of Succinate as a Signaling Transmitter Vary with Its Cellular Locations. Endocrinol. Metab. 2020, 35, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Woo, S.H.; Choi, D.H.; Cho, E.-H. Succinate Causes α-SMA Production through GPR91 Activation in Hepatic Stellate Cells. Biochem. Biophys. Res. Commun. 2015, 463, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Sapieha, P.; Sirinyan, M.; Hamel, D.; Zaniolo, K.; Joyal, J.S.; Cho, J.H.; Honoré, J.C.; Kermorvant-Duchemin, E.; Varma, D.R.; Tremblay, S.; et al. The Succinate Receptor GPR91 in Neurons Has a Major Role in Retinal Angiogenesis. Nat. Med. 2008, 14, 1067–1076. [Google Scholar] [CrossRef]
- Gilissen, J.; Geubelle, P.; Dupuis, N.; Laschet, C.; Pirotte, B.; Hanson, J. Forskolin-Free CAMP Assay for Gi-Coupled Receptors. Biochem. Pharmacol. 2015, 98, 381–391. [Google Scholar] [CrossRef]
- Robben, J.H.; Fenton, R.A.; Vargas, S.L.; Schweer, H.; Peti-Peterdi, J.; Deen, P.M.T.; Milligan, G. Localization of the Succinate Receptor in the Distal Nephron and Its Signaling in Polarized MDCK Cells. Kidney Int. 2009, 76, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Li, T.; Du, S.; Chen, Y.; Wang, S.; Xiong, F.; Wu, Q. The MAPK Signaling Pathway Mediates the GPR91-Dependent Release of VEGF from RGC-5 Cells. Int. J. Mol. Med. 2015, 36, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Correa, P.R.A.V.; Kruglov, E.A.; Thompson, M.; Leite, M.F.; Dranoff, J.A.; Nathanson, M.H. Succinate Is a Paracrine Signal for Liver Damage. J. Hepatol. 2007, 47, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, C.J.; Andrade, V.L.; Gomes, E.R.M.; Alves, M.N.M.; Ladeira, M.S.; Pinheiro, A.C.N.; Gomes, D.A.; Almeida, A.P.; Goes, A.M.; Resende, R.R.; et al. Succinate Modulates Ca(2+) Transient and Cardiomyocyte Viability through PKA-Dependent Pathway. Cell Calcium 2010, 47, 37–46. [Google Scholar] [CrossRef]
- Aguiar, C.J.; Rocha-Franco, J.A.; Sousa, P.A.; Santos, A.K.; Ladeira, M.; Rocha-Resende, C.; Ladeira, L.O.; Resende, R.R.; Botoni, F.A.; Melo, M.B.; et al. Succinate Causes Pathological Cardiomyocyte Hypertrophy through GPR91 Activation. Cell Commun. Signal. 2014, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate Receptor GPR91 Provides a Direct Link between High Glucose Levels and Renin Release in Murine and Rabbit Kidney. J. Clin. Investig. 2008, 118. [Google Scholar] [CrossRef] [Green Version]
- Mossa, A.; Velasquez-Flores, M.; Cammisotto, P.G.; Campeau, L. Receptor GPR91 Contributes to Voiding Function and Detrusor Relaxation Mediated by Succinate. Neurourol. Urodyn. 2021, 40, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Vicario, N.; Costa, A.S.H.; Kwok, C.K.; Leonardi, T.; Booty, L.M.; Bicci, I.; Balzarotti, B.; Volpe, G.; et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell 2018, 22, 355–368.e13. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-T.; Li, L.-Z.; Yang, Y.-L.; Yin, X.; Liu, Q.; Zhang, L.; Liu, K.; Liu, B.; Li, J.; Qi, L.-W. Succinate Induces Aberrant Mitochondrial Fission in Cardiomyocytes through GPR91 Signaling. Cell Death Dis. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detraux, D.; Caruso, M.; Feller, L.; Fransolet, M.; Meurant, S.; Mathieu, J.; Arnould, T.; Renard, P. A Critical Role for Heme Synthesis and Succinate in the Regulation of Pluripotent States Transitions. bioRxiv 2022. [Google Scholar] [CrossRef]
- Khan, S.A.; Park, K.-m.; Fischer, L.A.; Dong, C.; Lungjangwa, T.; Jimenez, M.; Casalena, D.; Chew, B.; Dietmann, S.; Auld, D.S.; et al. Probing the Signaling Requirements for Naive Human Pluripotency by High-Throughput Chemical Screening. Cell Rep. 2021, 35, 109233. [Google Scholar] [CrossRef]
- Lanner, F.; Rossant, J. The Role of FGF/Erk Signaling in Pluripotent Cells. Development 2010, 137, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Alonso, R.; Bustos, F.; Williams, C.A.C.; Findlay, G.M. Protein Kinases in Pluripotency-Beyond the Usual Suspects. J. Mol. Biol. 2017, 429, 1504–1520. [Google Scholar] [CrossRef] [Green Version]
- Beltran-Povea, A.; Caballano-Infantes, E.; Salguero-Aranda, C.; Martín, F.; Soria, B.; Bedoya, F.J.; Tejedo, J.R.; Cahuana, G.M. Role of Nitric Oxide in the Maintenance of Pluripotency and Regulation of the Hypoxia Response in Stem Cells. World J. Stem Cells 2015, 7, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Caballano-Infantes, E.; Cahuana, G.M.; Bedoya, F.J.; Salguero-Aranda, C.; Tejedo, J.R. The Role of Nitric Oxide in Stem Cell Biology. Antioxidants 2022, 11, 497. [Google Scholar] [CrossRef] [PubMed]
- Tejedo, J.R.; Tapia-Limonchi, R.; Mora-Castilla, S.; Cahuana, G.M.; Hmadcha, A.; Martin, F.; Bedoya, F.J.; Soria, B. Low Concentrations of Nitric Oxide Delay the Differentiation of Embryonic Stem Cells and Promote Their Survival. Cell Death Dis. 2010, 1, e80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tranguch, S.; Steuerwald, N.; Huet-Hudson, Y.M. Nitric Oxide Synthase Production and Nitric Oxide Regulation of Preimplantation Embryo Development. Biol. Reprod. 2003, 68, 1538–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.W.; Li, C.Y. Regulation of HIF-1α Stability through S-Nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.-Y.; Ellent, D.P.; Lee, S.; Goldsby, J.; Ko, B.-S.; Matijevic, N.; Huang, J.-C.; Wu, K.K. Cyclooxygenase-2-Derived Prostaglandin E2 Protects Mouse Embryonic Stem Cells from Apoptosis. Stem Cells 2007, 25, 1096–1103. [Google Scholar] [CrossRef]
- Kim, Y.H.; Han, H.J. High-Glucose-Induced Prostaglandin E(2) and Peroxisome Proliferator-Activated Receptor Delta Promote Mouse Embryonic Stem Cell Proliferation. Stem Cells 2008, 26, 745–755. [Google Scholar] [CrossRef]
- Yun, S.P.; Lee, M.Y.; Ryu, J.M.; Han, H.J. Interaction between PGE2 and EGF Receptor through MAPKs in Mouse Embryonic Stem Cell Proliferation. Cell. Mol. Life Sci. 2009, 66, 1603–1616. [Google Scholar] [CrossRef]
- Yun, S.P.; Ryu, J.M.; Park, J.H.; Kim, M.O.; Lee, J.H.; Han, H.J. Prostaglandin E₂ Maintains Mouse ESC Undifferentiated State through Regulation of Connexin31, Connexin43 and Connexin45 Expression: Involvement of Glycogen Synthase Kinase 3β/β-Catenin. Biol. Cell 2012, 104, 378–396. [Google Scholar] [CrossRef]
- Zhang, B.; He, L.; Liu, Y.; Zhang, J.; Zeng, Q.; Wang, S.; Fan, Z.; Fang, F.; Chen, L.; Lv, Y.; et al. Prostaglandin E 2 Is Required for BMP4-Induced Mesoderm Differentiation of Human Embryonic Stem Cells. Stem Cell Rep. 2018, 10, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Ware, C.B.; Nelson, A.M.; Mecham, B.; Hesson, J.; Zhou, W.; Jonlin, E.C.; Jimenez-Caliani, A.J.; Deng, X.; Cavanaugh, C.; Cook, S.; et al. Derivation of Naive Human Embryonic Stem Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 4484–4489. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.-S.; Göke, J.; Ng, J.-H.; Lu, X.; Gonzales, K.A.U.; Tan, C.-P.; Tng, W.-Q.; Hong, Z.-Z.; Lim, Y.-S.; Ng, H.-H. Induction of a Human Pluripotent State with Distinct Regulatory Circuitry That Resembles Preimplantation Epiblast. Cell Stem Cell 2013, 13, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Gafni, O.; Weinberger, L.; Mansour, A.A.; Manor, Y.S.; Chomsky, E.; Ben-Yosef, D.; Kalma, Y.; Viukov, S.; Maza, I.; Zviran, A.; et al. Derivation of Novel Human Ground State Naive Pluripotent Stem Cells. Nature 2013, 504, 282–286. [Google Scholar] [CrossRef]
- Takashima, Y.; Guo, G.; Loos, R.; Nichols, J.; Ficz, G.; Krueger, F.; Oxley, D.; Santos, F.; Clarke, J.; Mansfield, W.; et al. Resetting Transcription Factor Control Circuitry toward Ground-State Pluripotency in Human. Cell 2014, 158, 1254–1269. [Google Scholar] [CrossRef] [Green Version]
- Theunissen, T.W.; Powell, B.E.; Wang, H.; Mitalipova, M.; Faddah, D.A.; Reddy, J.; Fan, Z.P.; Maetzel, D.; Ganz, K.; Shi, L.; et al. Systematic Identification of Culture Conditions for Induction and Maintenance of Naive Human Pluripotency. Cell Stem Cell 2014, 15, 471–487. [Google Scholar] [CrossRef] [Green Version]
- Veenvliet, J.V.; Lenne, P.F.; Turner, D.A.; Nachman, I.; Trivedi, V. Sculpting with Stem Cells: How Models of Embryo Development Take Shape. Development 2021, 148. [Google Scholar] [CrossRef]
- Van den Brink, S.C.; van Oudenaarden, A. 3D Gastruloids: A Novel Frontier in Stem Cell-Based in Vitro Modeling of Mammalian Gastrulation. Trends Cell Biol. 2021, 31, 747–759. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Detraux, D.; Renard, P. Succinate as a New Actor in Pluripotency and Early Development? Metabolites 2022, 12, 651. https://doi.org/10.3390/metabo12070651
Detraux D, Renard P. Succinate as a New Actor in Pluripotency and Early Development? Metabolites. 2022; 12(7):651. https://doi.org/10.3390/metabo12070651
Chicago/Turabian StyleDetraux, Damien, and Patricia Renard. 2022. "Succinate as a New Actor in Pluripotency and Early Development?" Metabolites 12, no. 7: 651. https://doi.org/10.3390/metabo12070651