
Assessments of Ceanothanes Triterpenes as Cholinesterase Inhibitors: An Investigation of Potential Agents with Novel Inspiration for Drug Treatment of Neurodegenerative Diseases

 , , , , , and
, , , , , and
Abstract
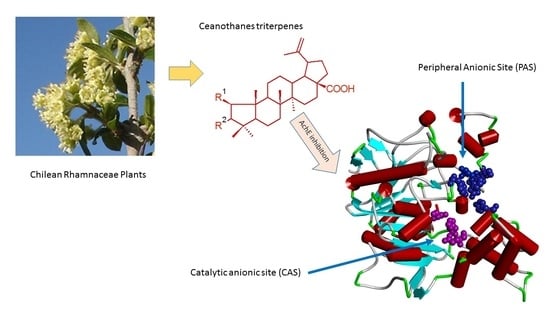
:
1. Introduction
2. Results and Discussion
2.1. Structural Elucidation of Ceanothane Triterpenes
2.2. Enzyme Inhibition and Kinetics Assays
2.3. Docking Studies
2.4. Propidium Iodide Displacement Assay
3. Materials and Methods
3.1. Equipment and General Experimental Procedures
3.2. Plant Materials
3.3. Extraction and Isolation
3.4. Spectroscopic Data
3.5. Preparation of 3-Oxo-Ceanothic Acid 6
3.6. Preparation of 3-Acetoxyceanothic Acid 7
3.7. In Vitro AChE/BChE Inhibitory Activity Assay
3.8. Kinetic Characterization of AChE Inhibition
3.9. Propidium Displacement Assay
3.10. In Silico Assays
3.10.1. Ligand Construction
3.10.2. Molecular Docking
3.10.3. MD Protocol
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr. Opin. Pharmacol. 2005, 5, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-H.; Wu, J.; Liu, H.-L.; Zhao, J.-H.; Liu, K.-T.; Chuang, C.-K.; Lin, H.-Y.; Tsai, W.-B.; Ho, Y. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011, 18, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisaru, D.; Sternfeld, M.; Eldor, A.; Glick, D.; Soreq, H. Structural roles of acetylcholinesterase variants in biology and pathology. Eur. J. Biochem. 1999, 264, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, F.J.; Aldunate, R.; Inestrosa, N.C. Peripheral binding site is involved in the neurotrophic activity of acetylcholinesterase. Neuroreport 1999, 10, 3621–3625. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Ramirez, G.; De Ferrari, G.V.; Inestrosa, N.C. Acetylcholinesterase induces the expression of the b-amyloid precursor protein in glia and activates glial cells in culture. Neurobiol. Dis. 2003, 14, 447–457. [Google Scholar] [CrossRef]
- Inestrosa, N.; Alvarez, A.; Dinamarca, M.; Perez-Acle, T.; Colombres, M. Acetylcholinesterase-Amyloid-b-peptide Interaction: Effect of Congo Red and the Role of the Wnt Pathway. Curr. Alzheimer Res. 2005, 2, 301–306. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S.W. Identification of a structural site on acetylcholinesterase that promotes neurite outgrowth and binds laminin-1 and collagen IV. Biochem. Biophys. Res. Commun. 2004, 319, 448–455. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef]
- Miao, Y.; He, N.; Zhu, J.-J. History and New Developments of Assays for Cholinesterase Activity and Inhibition. Chem. Rev. 2010, 110, 5216–5234. [Google Scholar] [CrossRef]
- Colovic, M.B.; KRstic, D.Z.; Lazarevi’c-Pasti, T.D.; Bondzic, A.M.; Vasic, V.S. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tougu, V. Acetylcholinesterase: Mechanism of Catalysis and Inhibition. Curr. Med. Chem.-Cent. Nerv. Syst. Agents 2001, 1, 155–170. [Google Scholar] [CrossRef]
- Ogura, H.; Kosasa, T.; Kuriya, Y.; Yamanishi, Y. Comparison of inhibitory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butyrylcholinesterase in vitro. Methods Find. Exp. Clin. Pharmacol. 2000, 22, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and safety of donezepil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. Clin. Interv. Aging 2008, 3, 211–225. [Google Scholar]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ’wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139 (Suppl. 2), 237–252. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, A.; Cağlar, P.; Dirmenci, T.; Gören, N.; Topçu, G. Un nuevo diterpenoide de isopimarano con actividad inhibidora de la acetilcolinesterasa de Nepeta sorgerae, una especie endémica de la montaña Nemrut. Comun. Prod. Nat. 2012, 7, 693–696. [Google Scholar]
- Houghton, P.J.; Ren, Y.; Howes, M.J. Acetylcholinesterase inhibitors from plants and fungi. Nat. Prod. Rep. 2006, 23, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.Y.; Park, S.Y. Terpenoids as potential anti-Alzheimer’s disease therapeutics. Molecules 2012, 17, 3524–3538. [Google Scholar] [CrossRef] [PubMed]
- Gurovic, M.S.; Castro, M.J.; Richmond, V.; Faraoni, M.B.; Maier, M.S.; Murray, A.P. Triterpenoids with acetylcholinesterase inhibition from Chuquiraga erinacea D. Don. subsp. erinacea (Asteraceae). Planta Med. 2010, 76, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, J.; Kříž, Z.; Kuča, K.; Jun, D.; Koča, J. Acetylcholinesterases—The structural similarities and differences. J. Enzym. Inhib. Med. Chem. 2007, 22, 417–424. [Google Scholar] [CrossRef]
- Guo, S.; Duan, J.; Tang, Y.; Qian, Y.; Zhao, J.; Qian, D. Triterpenoids from fruits of Ziziphus jujube var. spinose. Biochem. Syst. Ecol. 2011, 39, 880–882. [Google Scholar]
- Quiroz, S.; Cespedes, C.L.; Alderete, J.B.; Alarcón, J. Ceanothane and oleanane-type triterpenes from Talguenea quinquenervia have insecticidal activity against Cydia pomonella, Tenebrio molitor and Drosophila melanogaster. Ind. Crops Prod. 2015, 74, 759–766. [Google Scholar] [CrossRef]
- Lee, S.M.; Min, B.S.; Lee, C.G.; Kim, K.S.; Kho, Y.H. Cytotoxic triterpenoids from the fruits of Zizyphus jujube. Planta Med. 2003, 69, 1051–1054. [Google Scholar]
- Li, X.C.; ElSohly, H.N.; Nimrod, A.C.; Clark, A.M. Antifungal jujubogenin saponins from Colubrina retusa. J. Nat. Prod. 1999, 62, 674–677. [Google Scholar] [CrossRef]
- Quiroz-Carreño, S.; Cespedes-Acuña, C.L.; Seigler, D.S.; Alarcón-Enos, J. Identification of structurally diverse alkaloids in Talguenea quinquinervia (gill. Et hook) by liquid chromatography/electrospray ionisation tandem mass spectroscopy and insecticidal activity. Phytochem. Anal. 2019, 30, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-S.; Chen, w.C.; Huang, C.-F.; Sy, Y. Preparation and cytotoxic effects of ceanothic acid derivatives. J. Nat. Prod. 1998, 61, 1343–1347. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharm. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S.W. The peripheral anionic site of acetylcholinesterase: Structure, Functions and Potential Role in Rational Drug Design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. Β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Sauvaître, T.; Barlier, M.; Herlem, D.; Gresh, N.; Chiaroni, A.; Guenard, D.; Guillou, C. New Potent Acetylcholinesterase Inhibitors in the Tetracyclic Triterpene. J. Med. Chem. 2007, 50, 5311–5323. [Google Scholar] [CrossRef]
- Eubanks, L.M.; Rogers, C.J.; Koob, G.F.; Olson, A.J.; Dickerson, T.J.; Janda, K.D. A Molecular Link between the Active Component of Marijuana and Alzheimer’s Disease Pathology. Mol. Pharm. 2006, 3, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Coelho, T.; Mota, T.; Serra, B.A.; Camara, A.L.; De Andrades, A.M. Naturally Occurring Acetylcholinesterase Inhibitors and Their Potential Use for Alzheimer’s Disease Therapy. Front. Pharmacol. 2018, 9, 1102. [Google Scholar]
- Alarcón, J.; Cespedes, C.L. Chemical Constituents and Biological Activities of South American Rhamnaceae. Phytochem. Rev. 2015, 14, 389–401. [Google Scholar] [CrossRef]
- Cardoso-Lopes, E.M.; Maier, J.A.; Rogerido da Silva, M.; Regasini, L.O.; Simote, S.Y.; Peporine, N.; Rubens, J.; da Silva, V.; Marx, M.C. Alkaloids from stems of Esenbeckia leiocarpa Engl. (Rutaceae) as Potential Treatment for Alzheimer Disease. Molecules 2010, 15, 9205–9213. [Google Scholar] [CrossRef] [Green Version]
- Murray, A.P.; Faraoni, M.B.; Castro, M.J.; Alza, N.P.; Cavallaro, V. Natural AChE Inhibitors from Plants and their Contribution to Alzheimer’s Disease Therapy. Curr. Neuropharmacol. 2013, 11, 388–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, V.; Ayi, K.; Lu, Z.; Liby, K.; Sporn, M.; Kain, K. Synthetic oleanane triterpenoids enhance blood brain barrier integrity and improve survival in experimental cerebral malaria. Malar. J. 2017, 16, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.I.; Lin, S.G.; Chen, X.; Zhou, Z.W.; Liang, J.; Duan, W.; Chowbay, B.; Wen, J.Y.; Chan, E.; Cao, J.; et al. Transport of Cryptotanshinone, a Major Active Triterpenoid in Salvia Miltiorrhiza Bunge Widely Used in the Treatment of Stroke and Alzheimer’s Disease, Across the Blood-Brain Barrier. Curr. Drug Metab. 2007, 8, 365–377. [Google Scholar] [CrossRef]
- Galdeano, C.; Coquelle, N.; Cieslikiewicz-Bouet, M.; Bartolin, M.; Perez, B.; Clos, M.V.; Israel, S.; Jean, L.; Colletier, J.; Renard, P.; et al. Increasing polarity in tacrine and huprine derivatives: Potent anticholinesterase agents for the treatment of Myasthenia Gravis. Molecules 2018, 23, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.; Goodsell, D.; Halliday, R.; Huey, R.; Hart, W.; Belew, R.; Olson, A. Automated docking using a Lamarckian Genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS Biomolecular Solvation Software Suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid Specific Protein Backbone Parameters Trained against Quantum Mechanics Surface in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; Darden, T.A.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Berendsen, H.J.C.; Postman, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 ± SEM, µM | Ki | MolDock | ||||
|---|---|---|---|---|---|---|---|
| M | AChE * | AChE hb a,* | BuChE | Inhibition Type | µM | Score | |
| 1 | 452.7 | 0.184 ± 0.0007 | 0.215 ± 0.005 | >500 | acompetitive | 0.046 | −109.891 |
| 2 | 470.7 | 0.150 ± 0.0009 | 0.173 ± 0.006 | >500 | acompetitive | 0.028 | −144.641 |
| 3 | 486.7 | 0.125 ± 0.0004 | 0.146 ± 0.003 | >500 | competitive | 0.055 | −145.850 |
| 4 | 454.6 | 0.188 ± 0.0005 | 0.219 ± 0.004 | >500 | acompetitive | 0.022 | −107.504 |
| 5 | 472.7 | 0.155 ± 0.0001 | 0.181 ± 0.0007 | >500 | acompetitive | 0.056 | −141.850 |
| 6 | 484.7 | 0.172 ± 0.0012 | 0.199 ± 0.009 | >500 | acompetitive | 0.047 | −112.846 |
| 7 | 528.7 | 0.179 ± 0.0001 | 0.207 ± 0.006 | >500 | competitive | 0.050 | −121.490 |
| Galantamine | 287.4 | 0.046 ± 0.0005 | 0.040 ± 0.0004 | 0.739 ± 0.012 | competitive | 0.045 | −184.840 |
| Compound | IC50 µM | % Displacement of Propidium Iodide | |
|---|---|---|---|
| 0.75 µM | 1.5 µM | ||
| 1 | 0.184 ± 0.0007 | 19.7 ± 0.8 | 26.3 ± 1.3 |
| 2 | 0.150 ± 0.0009 | 17. 4 ± 0.7 | 23.2 ± 0.8 |
| 3 | 0.125 ± 0.0004 | 34.2 ± 0.6 | 45.6 ± 1.1 |
| 4 | 0.188 ± 0.0005 | 20.1 ± 1.0 | 26.7 ± 1.3 |
| 5 | 0.155 ± 0.0001 | 14.5 ± 0.3 | 19.3 ± 1.5 |
| 6 | 0.172 ± 0.0012 | 24.3 ± 0.7 | 40.5 ± 1.5 |
| 7 | 0.179 ± 0.0001 | 24. 1 ± 0.3 | 32.1 ± 1.3 |
| Donepezil | 0.012 ± 0.007 | 73.2 ± 1.9 | 84.6 ± 3.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Nuñez, E.; Quiroz-Carreño, S.; Pastene-Navarrete, E.; Seigler, D.S.; Céspedes-Acuña, C.; Martínez Valenzuela, I.; Oppliger Muñoz, M.; Salas-Burgos, A.; Alarcón-Enos, J. Assessments of Ceanothanes Triterpenes as Cholinesterase Inhibitors: An Investigation of Potential Agents with Novel Inspiration for Drug Treatment of Neurodegenerative Diseases. Metabolites 2022, 12, 668. https://doi.org/10.3390/metabo12070668
Muñoz-Nuñez E, Quiroz-Carreño S, Pastene-Navarrete E, Seigler DS, Céspedes-Acuña C, Martínez Valenzuela I, Oppliger Muñoz M, Salas-Burgos A, Alarcón-Enos J. Assessments of Ceanothanes Triterpenes as Cholinesterase Inhibitors: An Investigation of Potential Agents with Novel Inspiration for Drug Treatment of Neurodegenerative Diseases. Metabolites. 2022; 12(7):668. https://doi.org/10.3390/metabo12070668
Chicago/Turabian StyleMuñoz-Nuñez, Evelyn, Soledad Quiroz-Carreño, Edgar Pastene-Navarrete, David S. Seigler, Carlos Céspedes-Acuña, Ignacio Martínez Valenzuela, Martina Oppliger Muñoz, Alexis Salas-Burgos, and Julio Alarcón-Enos. 2022. "Assessments of Ceanothanes Triterpenes as Cholinesterase Inhibitors: An Investigation of Potential Agents with Novel Inspiration for Drug Treatment of Neurodegenerative Diseases" Metabolites 12, no. 7: 668. https://doi.org/10.3390/metabo12070668
APA StyleMuñoz-Nuñez, E., Quiroz-Carreño, S., Pastene-Navarrete, E., Seigler, D. S., Céspedes-Acuña, C., Martínez Valenzuela, I., Oppliger Muñoz, M., Salas-Burgos, A., & Alarcón-Enos, J. (2022). Assessments of Ceanothanes Triterpenes as Cholinesterase Inhibitors: An Investigation of Potential Agents with Novel Inspiration for Drug Treatment of Neurodegenerative Diseases. Metabolites, 12(7), 668. https://doi.org/10.3390/metabo12070668