
Imputation of Missing Values for Multi-Biospecimen Metabolomics Studies: Bias and Effects on Statistical Validity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
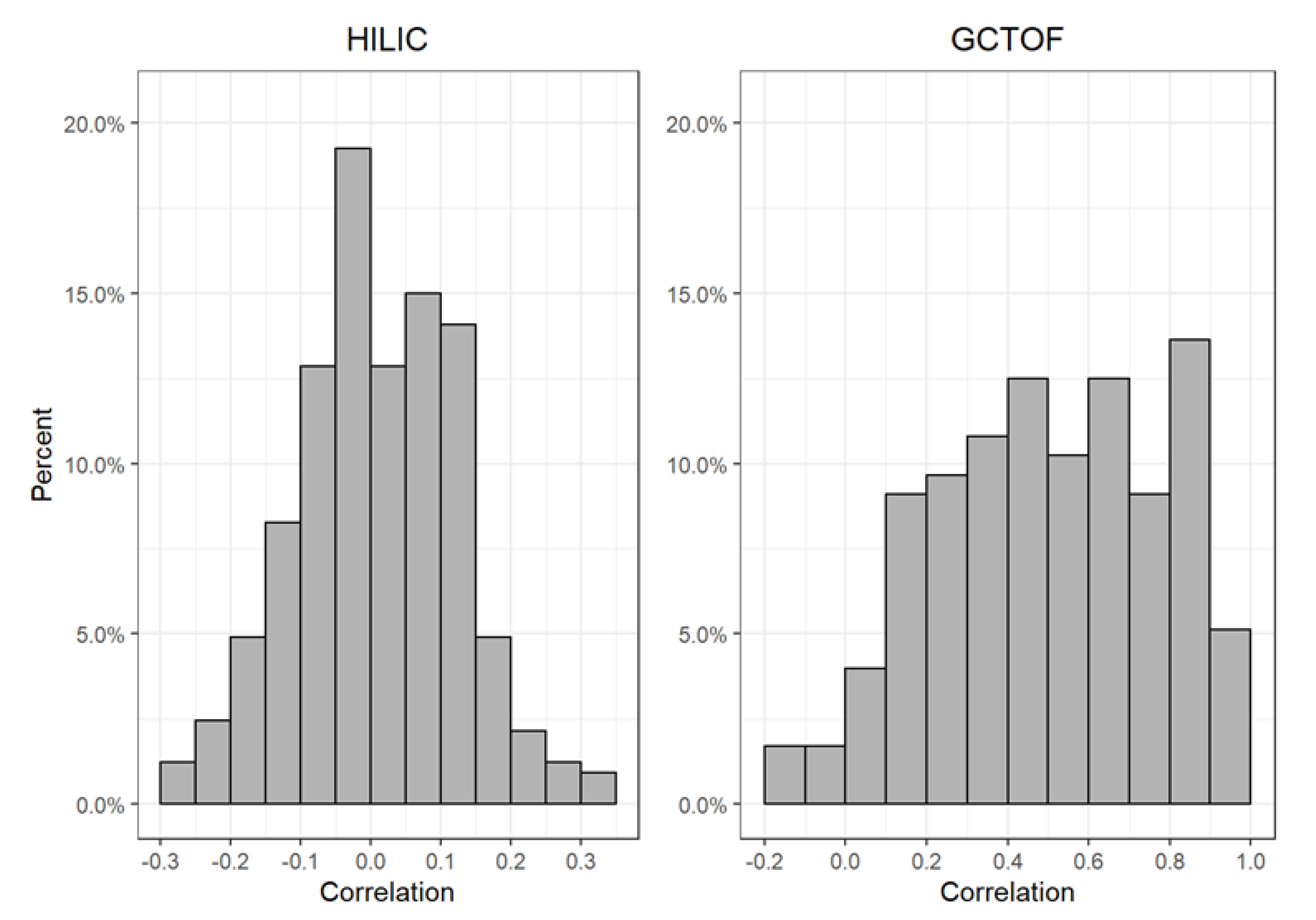
2.1. Data Sets
2.2. Comparison of the True vs. Imputed Between-Biospecimen Correlations
Combined Versus Separate Imputation Approaches
2.3. Performance of the Imputation Methods
2.3.1. GCTOF Data Set
2.3.2. HILIC Data Set
2.4. Bias in the Between-Biospecimen Correlation
2.5. Effects on Statistical Significance Tests
2.5.1. Sensitivity
2.5.2. Specificity
2.5.3. Accuracy
2.6. Effects of Bias in the Between-Biospecimen Correlation Estimates on Statistical Significance Tests
3. Discussion
4. Materials and Methods
4.1. Data Sets
4.1.1. GCTOF
4.1.2. HILIC
4.2. Simulating Missingness
4.3. Metrics
4.3.1. Sensitivity (True Positive Rate)
4.3.2. Specificity (True Negative Rate)
4.3.3. Accuracy (True Discovery)
4.4. Combined vs. Separate Imputation
4.5. Imputation Methods
4.5.1. Expectation-Maximization with Bootstrap Method
4.5.2. Random Forest Method
4.5.3. K-Nearest Neighbor Method
4.5.4. Quantile Regression Method
4.5.5. Half-Minimum Method
4.5.6. Software
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, K.; Mall, C.; Taylor, S.L.; Hitchcock, S.; Zhang, C.; Wettersten, H.I.; Jones, A.D.; Chapman, A.; Weiss, R.H. Mealtime, temporal, and daily variability of the human urinary and plasma metabolomes in a tightly controlled environment. PLoS ONE 2014, 9, e86223. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Taylor, S.L.; Ganti, S.; Guo, L.; Osier, M.V.; Weiss, R.H. Urine metabolomic analysis identifies potential biomarkers and pathogenic pathways in kidney cancer. Omics A J. Integr. Biol. 2011, 15, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Trott, J.F.; Gao, G.; Chapman, A.; Weiss, R.H. Plasma metabolites and lipids associate with kidney function and kidney volume in hypertensive ADPKD patients early in the disease course. BMC Nephrol. 2019, 20, 66. [Google Scholar] [CrossRef] [PubMed]
- Clough, T.; Key, M.; Ott, I.; Ragg, S.; Schadow, G.; Vitek, O. Protein quantification in label-free LC-MS experiments. J. Proteome Res. 2009, 8, 5275–5287. [Google Scholar] [CrossRef]
- Betts, K.; Sawyer, K. Use of Metabolomics to Advance Research on Environmental Exposures and the Human Exposome: Workshop in Brief; Board on Life Sciences; Division on Earth and Life Studies; National Academies of Science, Engineering, and Medicine: Washington, DC, USA, 2016. [Google Scholar]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Lankadurai, B.P.; Nagato, E.G.; Simpson, M.J. Environmental metabolomics: An emerging approach to study organism responses to environmental stressors. Environ. Rev. 2013, 21, 180–205. [Google Scholar] [CrossRef]
- Dai, Y.; Huo, X.; Chen, S.; Faas, M.M.; Xu, X. Early-life exposure to widespread environmental toxicants and maternal-fetal health risk: A focus on metabolomic biomarkers. Sci. Total Environ. 2020, 739, 139626. [Google Scholar] [CrossRef]
- Ganti, S.; Taylor, S.L.; Abu Aboud, O.; Yang, J.; Evans, C.; Osier, M.V.; Alexander, D.C.; Kim, K.; Weiss, R.H. Kidney Tumor Biomarkers Revealed by Simultaneous Multiple Matrix Metabolomics Analysis. Cancer Res. 2012, 72, 3471–3479. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Wang, X.-H.; Huang, Z.-Z.; Lin, L.; Gao, Y.; Zhu, E.-Y.; Xing, J.-C.; Zheng, J.-X.; Hang, W. A study of human bladder cancer by serum and urine metabonomics. Chin. J. Anal. Chem. 2012, 40, 1322–1328. [Google Scholar] [CrossRef]
- De Paepe, E.; Van Meulebroek, L.; Rombouts, C.; Huysman, S.; Verplanken, K.; Lapauw, B.; Wauters, J.; Hemeryck, L.Y.; Vanhaecke, L. A validated multi-matrix platform for metabolomic fingerprinting of human urine, feces and plasma using ultra-high performance liquid chromatography coupled to hybrid orbitrap high-resolution mass spectrometry. Anal. Chim. Acta 2018, 1033, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, K.; Nishiumi, S.; Kitamoto-Matsuda, J.; Fujita, T.; Morimoto, K.; Yamashita, D.; Saito, M.; Otsuki, N.; Irino, Y.; Shinohara, M.; et al. Serum and tissue metabolomics of head and neck cancer. Cancer Genom. Proteom. 2013, 11, 233–238. [Google Scholar]
- Jordan, K.W.; Adkins, C.B.; Su, L.; Halpern, E.F.; Mark, E.J.; Christiani, D.C.; Cheng, L.L. Comparison of squamous cell carcinoma and adenocarcinoma of the lung by metabolomic analysis of tissue-serum pairs. Lung Cancer 2010, 68, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Austdal, M.; Skråstad, R.B.; Gundersen, A.S.; Austgulen, R.; Iversen, A.-C.; Bathen, T.F. Metabolomic Biomarkers in Serum and Urine in Women with Preeclampsia. PLoS ONE 2014, 9, e91923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.L.; Ruhaak, L.R.; Kelly, K.; Weiss, R.H.; Kim, K. Effects of imputation on correlation: Implications for analysis of mass spectrometry data from multiple biological matrices. Brief. Bioinform. 2017, 18, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.L.; Ruhaak, L.R.; Weiss, R.H.; Kelly, K.; Kim, K. Multivariate two-part statistics for analysis of correlated mass spectrometry data from multiple biological specimens. Bioinformatics 2017, 33, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrydziuszko, O.; Viant, M.R. Missing values in mass spectrometry based metabolomics: An undervalued step in the data processing pipeline. Metabolomics 2012, 8, S161–S174. [Google Scholar] [CrossRef]
- Wang, X.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. A hybrid approach to protein differential expression in mass spectrometry-based proteomics. Bioinformatics 2012, 28, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Webb-Robertson, B.J.; Wiberg, H.K.; Matzke, M.M.; Brown, J.N.; Wang, J.; McDermott, J.E.; Smith, R.D.; Rodland, K.D.; Metz, T.O.; Pounds, J.G.; et al. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Proteome Res. 2015, 14, 1993–2001. [Google Scholar] [CrossRef] [Green Version]
- Ruben, D. Inference and missing data. Biometrika 1976, 63, 581–592. [Google Scholar] [CrossRef]
- Greenlees, J.S.; Reece, W.S.; Zieschang, K.D. Imputation of Missing Values When the Probability of Response Depends on the Variable Being Imputed. J. Am. Stat. Assoc. 1982, 77, 251–261. [Google Scholar] [CrossRef]
- Do, K.T.; Wahl, S.; Raffler, J.; Molnos, S.; Laimighofer, M.; Adamski, J.; Suhre, K.; Strauch, K.; Peters, A.; Gieger, C.; et al. Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies. Metabolomics 2018, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efron, B. Missing Data, Imputation, and the Bootstrap. J. Am. Stat. Assoc. 1994, 89, 463–475. [Google Scholar] [CrossRef]
- Dempster, A.; Laird, N.; Rubin, D. Maximum Likelihood Estimation from Incomplete Data via the EM Algorithm. J. R. Stat. Soc. B 1977, 39, 1–22. [Google Scholar]
- Lazar, C.; Gatto, L.; Ferro, M.; Bruley, C.; Burger, T. Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. J. Proteome Res. 2016, 15, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.F.; Rueda, M. New imputation methods for missing data using quantiles. J. Comput. Appl. Math. 2009, 232, 305–317. [Google Scholar] [CrossRef]
- Lee, M.; Rahbar, M.H.; Brown, M.; Gensler, L.; Weisman, M.; Diekman, L.; Reveille, J.D. A multiple imputation method based on weighted quantile regression models for longitudinal censored biomarker data with missing values at early visits. BMC Med. Res. Methodol. 2018, 18, 8. [Google Scholar] [CrossRef] [Green Version]
- Lazar, C. QRILC: A Quantile Regression Approach for the Imputation of Left-Censored Missing Data in Quantitative Proteomics; R Package: Madison, WI, USA, 2021. [Google Scholar]
- Stekhoven, D.J.; Buhlmann, P. MissForest—non-parametric missing value imputation for mixed-type data. Bioinformatics 2012, 28, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Ampong, I.; Zimmerman, K.D.; Nathanielsz, P.W.; Cox, L.A.; Olivier, M. Optimization of Imputation Strategies for High-Resolution Gas Chromatography–Mass Spectrometry (HR GC–MS) Metabolomics Data. Metabolites 2022, 12, 429. [Google Scholar] [CrossRef]
- Fahrmann, J.F.; Kim, K.; DeFelice, B.C.; Taylor, S.L.; Gandara, D.R.; Yoneda, K.Y.; Cooke, D.T.; Fiehn, O.; Kelly, K.; Miyamoto, S. Investigation of metabolomic blood biomarkers for detection of adenocarcinoma lung cancer. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1716–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrmann, J.F.; Grapov, D.; DeFelice, B.C.; Taylor, S.L.; Kim, K.; Kelly, K.; Wikoff, W.R.; Pass, H.I.; Rom, W.N.; Fiehn, O.; et al. Serum phosphatidylethanolamine levels distinguish benign from malignant solitary pulmonary nodules and represent a potential diagnostic biomarker for lung cancer. Cancer Biomark. 2016, 16, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.L.; Ponzini, M.; Wilson, M.; Kim, K. Comparison of imputation and imputation-free methods for statistical analysis of mass spectrometry data with missing data. Brief. Bioinform. 2021, 23, bbab353. [Google Scholar] [CrossRef] [PubMed]
- Scheel, I.; Aldrin, M.; Glad, I.K.; Sorum, R.; Lyng, H.; Frigessi, A. The influence of missing value imputation on detection of differentially expressed genes from microarray data. Bioinformatics 2005, 21, 4272–4279. [Google Scholar] [CrossRef] [PubMed]
- Kokla, M.; Virtanen, J.; Kolehmainen, M.; Paananen, J.; Hanhineva, K. Random forest-based imputation outperforms other methods for imputing LC-MS metabolomics data: A comparative study. BMC Bioinform. 2019, 20, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honaker, J.; King, G.; Blackwell, M. Amelia II: A Program for Missing Data. J. Stat. Softw. 2011, 45, 1–47. [Google Scholar] [CrossRef]
- Hastie, T.T.R.; Narasimhan, B.; Chu, G. Impute: Impute: Imputation for Microarray Data; R package: Madison, WI, USA, 2022. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, M.D.; Ponzini, M.D.; Taylor, S.L.; Kim, K. Imputation of Missing Values for Multi-Biospecimen Metabolomics Studies: Bias and Effects on Statistical Validity. Metabolites 2022, 12, 671. https://doi.org/10.3390/metabo12070671
Wilson MD, Ponzini MD, Taylor SL, Kim K. Imputation of Missing Values for Multi-Biospecimen Metabolomics Studies: Bias and Effects on Statistical Validity. Metabolites. 2022; 12(7):671. https://doi.org/10.3390/metabo12070671
Chicago/Turabian StyleWilson, Machelle D., Matthew D. Ponzini, Sandra L. Taylor, and Kyoungmi Kim. 2022. "Imputation of Missing Values for Multi-Biospecimen Metabolomics Studies: Bias and Effects on Statistical Validity" Metabolites 12, no. 7: 671. https://doi.org/10.3390/metabo12070671
APA StyleWilson, M. D., Ponzini, M. D., Taylor, S. L., & Kim, K. (2022). Imputation of Missing Values for Multi-Biospecimen Metabolomics Studies: Bias and Effects on Statistical Validity. Metabolites, 12(7), 671. https://doi.org/10.3390/metabo12070671