
Weighted Gene Co-Expression Network Analysis and Support Vector Machine Learning in the Proteomic Profiling of Cerebrospinal Fluid from Extraventricular Drainage in Child Medulloblastoma

 , ,
, ,  and
and
Abstract
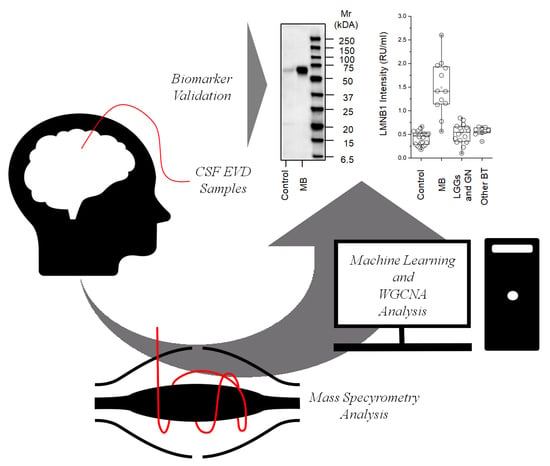
:
1. Introduction
2. Results
2.1. Characterization of Extracellular Vesicles
2.2. Protein Composition
2.3. Western Blot for LMNB1
2.4. ELISA for LMNB1-Validated Proteomic Results
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Patient Information
4.2. Total Fraction
4.3. CPLLs Fraction
4.4. Extracellular Vesicle Fraction
4.5. Dynamic Light Scattering
4.6. Mass Spectrometry (MS) Analysis
4.7. Western Blotting
4.8. ELISA
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.A. Pathology, diagnostics, and classification of medulloblastoma. Brain Pathol. 2020, 30, 664–678. [Google Scholar] [CrossRef]
- Maier, H.; Dalianis, T.; Kostopoulou, O.N. New Approaches in Targeted Therapy for Medulloblastoma in Children. Anticancer Res. 2021, 41, 1715–1726. [Google Scholar] [CrossRef]
- Szalontay, L.; Khakoo, Y. Medulloblastoma: An Old Diagnosis with New Promises. Curr. Oncol. Rep. 2020, 22, 90. [Google Scholar] [CrossRef]
- Zebian, B.; Vergani, F.; Lavrador, J.P.; Mukherjee, S.; Kitchen, W.J.; Stagno, V.; Chamilos, C.; Pettorini, B.; Mallucci, C. Recent technological advances in pediatric brain tumor surgery. CNS Oncol. 2017, 6, 71–82. [Google Scholar] [CrossRef]
- Liu, X.; Ding, C.; Tan, W.; Zhang, A. Medulloblastoma: Molecular understanding, treatment evolution, and new developments. Pharmacol. Ther. 2020, 210, 107516. [Google Scholar] [CrossRef]
- Gottardo, N.G.; Hansford, J.R.; McGlade, J.P.; Alvaro, F.; Ashley, D.M.; Bailey, S.; Baker, D.L.; Bourdeaut, F.; Cho, Y.J.; Clay, M.; et al. Medulloblastoma Down Under 2013: A report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol. 2014, 127, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Laneve, P.; Caffarelli, E. The Non-coding Side of Medulloblastoma. Front. Cell Dev. Biol. 2020, 8, 275. [Google Scholar] [CrossRef]
- Wells, E.M.; Packer, R.J. Pediatric brain tumors. Continuum 2015, 21, 373–396. [Google Scholar] [CrossRef]
- Bookland, M.J.; Kolmakova, A. Peripheral biomarkers for pediatric brain tumors: Current advancements and future challenges. J. Cancer Metastasis Treat. 2019, 5, 33. [Google Scholar] [CrossRef]
- Holtta, M.; Zetterberg, H.; Mirgorodskaya, E.; Mattsson, N.; Blennow, K.; Gobom, J. Peptidome analysis of cerebrospinal fluid by LC-MALDI MS. PLoS ONE 2012, 7, e42555. [Google Scholar] [CrossRef] [Green Version]
- Johanson, C.E.; Duncan, J.A., 3rd; Klinge, P.M.; Brinker, T.; Stopa, E.G.; Silverberg, G.D. Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cereb. Fluid Res. 2008, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Zhang, Y.; Yao, Y.; Hua, W.; Zhang, H.S.; Wu, J.S.; Zhong, P.; Zhou, L.F. Proteomic analysis of cerebrospinal fluid: Toward the identification of biomarkers for gliomas. Neurosurg. Rev. 2014, 37, 367–380. [Google Scholar] [CrossRef]
- Schutzer, S.E.; Liu, T.; Natelson, B.H.; Angel, T.E.; Schepmoes, A.A.; Purvine, S.O.; Hixson, K.K.; Lipton, M.S.; Camp, D.G.; Coyle, P.K.; et al. Establishing the proteome of normal human cerebrospinal fluid. PLoS ONE 2010, 5, e10980. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, M.U.; Hathout, Y.; MacDonald, T.J.; Kieran, M.W.; Gururangan, S.; Blaney, S.M.; Phillips, P.; Packer, R.; Gordish-Dressman, H.; Rood, B.R. Proteomic profiling of cerebrospinal fluid identifies prostaglandin D2 synthase as a putative biomarker for pediatric medulloblastoma: A pediatric brain tumor consortium study. Proteomics 2011, 11, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Spreafico, F.; Bongarzone, I.; Pizzamiglio, S.; Magni, R.; Taverna, E.; De Bortoli, M.; Ciniselli, C.M.; Barzano, E.; Biassoni, V.; Luchini, A.; et al. Proteomic analysis of cerebrospinal fluid from children with central nervous system tumors identifies candidate proteins relating to tumor metastatic spread. Oncotarget 2017, 8, 46177–46190. [Google Scholar] [CrossRef] [Green Version]
- de Bont, J.M.; den Boer, M.L.; Kros, J.M.; Passier, M.M.; Reddingius, R.E.; Smitt, P.A.; Luider, T.M.; Pieters, R. Identification of novel biomarkers in pediatric primitive neuroectodermal tumors and ependymomas by proteome-wide analysis. J. Neuropathol. Exp. Neurol. 2007, 66, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Samuel, N.; Remke, M.; Rutka, J.T.; Raught, B.; Malkin, D. Proteomic analyses of CSF aimed at biomarker development for pediatric brain tumors. J. Neurooncol. 2014, 118, 225–238. [Google Scholar] [CrossRef]
- Saratsis, A.M.; Yadavilli, S.; Magge, S.; Rood, B.R.; Perez, J.; Hill, D.A.; Hwang, E.; Kilburn, L.; Packer, R.J.; Nazarian, J. Insights into pediatric diffuse intrinsic pontine glioma through proteomic analysis of cerebrospinal fluid. Neuro. Oncol. 2012, 14, 547–560. [Google Scholar] [CrossRef]
- Bruschi, M.; Petretto, A.; Cama, A.; Pavanello, M.; Bartolucci, M.; Morana, G.; Ramenghi, L.A.; Garre, M.L.; Ghiggeri, G.M.; Panfoli, I.; et al. Potential biomarkers of childhood brain tumor identified by proteomics of cerebrospinal fluid from extraventricular drainage (EVD). Sci. Rep. 2021, 11, 1818. [Google Scholar] [CrossRef]
- Shnaper, S.; Desbaillets, I.; Brown, D.A.; Murat, A.; Migliavacca, E.; Schluep, M.; Ostermann, S.; Hamou, M.F.; Stupp, R.; Breit, S.N.; et al. Elevated levels of MIC-1/GDF15 in the cerebrospinal fluid of patients are associated with glioblastoma and worse outcome. Int. J. Cancer 2009, 125, 2624–2630. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends. Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Bao, Q.; Huang, Q.; Chen, Y.; Wang, Q.; Sang, R.; Wang, L.; Xie, Y.; Chen, W. Tumor-Derived Extracellular Vesicles Regulate Cancer Progression in the Tumor Microenvironment. Front. Mol. Biosci. 2022, 8, 796385. [Google Scholar] [CrossRef]
- Panfoli, I.; Bruschi, M.; Candiano, G. Exosomes: Key tools for cancer liquid biopsy. Biocell 2022, 46, 2167–2176. [Google Scholar] [CrossRef]
- Nafar, S.; Nouri, N.; Alipour, M.; Fallahi, J.; Zare, F.; Tabei, S.M.B. Exosome as a target for cancer treatment. J. Investig. Med. 2022, 70, 1212–1218. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Achreja, A.; Zhao, H.; Yang, L.; Yun, T.H.; Marini, J.; Nagrath, D. Exo-MFA-A 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab. Eng. 2019, 43, 156–172. [Google Scholar] [CrossRef]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Consortium, U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Candiano, G.; Bruschi, M.; Musante, L.; Santucci, L.; Ghiggeri, G.M.; Carnemolla, B.; Orecchia, P.; Zardi, L.; Righetti, P.G. Blue silver: A very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 2004, 25, 1327–1333. [Google Scholar] [CrossRef]
- Panfoli, I.; Ravera, S.; Podesta, M.; Cossu, C.; Santucci, L.; Bartolucci, M.; Bruschi, M.; Calzia, D.; Sabatini, F.; Bruschettini, M.; et al. Exosomes from human mesenchymal stem cells conduct aerobic metabolism in term and preterm newborn infants. FASEB J. 2016, 30, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, M.; Santucci, L.; Ravera, S.; Bartolucci, M.; Petretto, A.; Calzia, D.; Ghiggeri, G.M.; Ramenghi, L.A.; Candiano, G.; Panfoli, I. Metabolic Signature of Microvesicles from Umbilical Cord Mesenchymal Stem Cells of Preterm and Term Infants. Proteom. Clin. Appl. 2018, 12, e1700082. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Epple, L.M.; Griffiths, S.G.; Dechkovskaia, A.M.; Dusto, N.L.; White, J.; Ouellette, R.J.; Anchordoquy, T.J.; Bemis, L.T.; Graner, M.W. Medulloblastoma exosome proteomics yield functional roles for extracellular vesicles. PLoS ONE 2012, 7, e42064. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.; Montermini, L.; Kim, D.K.; Meehan, B.; Roth, F.P.; Rak, J. The Impact of Oncogenic EGFRvIII on the Proteome of Extracellular Vesicles Released from Glioblastoma Cells. Mol. Cell Proteom. 2018, 17, 1948–1964. [Google Scholar] [CrossRef] [Green Version]
- Hallal, S.; Azimi, A.; Wei, H.; Ho, N.; Lee, M.Y.T.; Sim, H.W.; Sy, J.; Shivalingam, B.; Buckland, M.E.; Alexander-Kaufman, K.L. A Comprehensive Proteomic SWATH-MS Workflow for Profiling Blood Extracellular Vesicles: A New Avenue for Glioma Tumour Surveillance. Int. J. Mol. Sci. 2020, 21, 4754. [Google Scholar] [CrossRef]
- Indira Chandran, V.; Welinder, C.; Mansson, A.S.; Offer, S.; Freyhult, E.; Pernemalm, M.; Lund, S.M.; Pedersen, S.; Lehtio, J.; Marko-Varga, G.; et al. Ultrasensitive Immunoprofiling of Plasma Extracellular Vesicles Identifies Syndecan-1 as a Potential Tool for Minimally Invasive Diagnosis of Glioma. Clin. Cancer Res. 2019, 25, 3115–3127. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, T.A.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef] [Green Version]
- Rivero-Hinojosa, S.; Lau, L.S.; Stampar, M.; Staal, J.; Zhang, H.; Gordish-Dressman, H.; Northcott, P.A.; Pfister, S.M.; Taylor, M.D.; Brown, K.J.; et al. Proteomic analysis of Medulloblastoma reveals functional biology with translational potential. Acta Neuropathol. Commun. 2018, 6, 48. [Google Scholar] [CrossRef]
- Song, Z.Y.; Chao, F.; Zhuo, Z.; Ma, Z.; Li, W.; Chen, G. Identification of hub genes in prostate cancer using robust rank aggregation and weighted gene co-expression network analysis. Aging 2019, 11, 4736–4756. [Google Scholar] [CrossRef]
- Lv, D.; Wu, X.; Wang, M.; Chen, W.; Yang, S.; Liu, Y.; Zeng, G.; Gu, D. Functional Assessment of Four Novel Immune-Related Biomarkers in the Pathogenesis of Clear Cell Renal Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 621618. [Google Scholar] [CrossRef]
- Zhou, D.; Wang, M.; Zhang, Y.; Wang, K.; Zhao, M.; Wang, Y.; Wang, X.; Yu, R.; Zhou, X. Screening and identification of LMNB1 and DLGAP5, two key biomarkers in gliomas. Biosci. Rep. 2021, 41, BSR20210231. [Google Scholar] [CrossRef]
- Wesolowska-Andersen, A.; Borst, L.; Dalgaard, M.D.; Yadav, R.; Rasmussen, K.K.; Wehner, P.S.; Rasmussen, M.; Orntoft, T.F.; Nordentoft, I.; Koehler, R.; et al. Genomic profiling of thousands of candidate polymorphisms predicts risk of relapse in 778 Danish and German childhood acute lymphoblastic leukemia patients. Leukemia 2015, 29, 297–303. [Google Scholar] [CrossRef]
- Li, J.; Sun, Z.; Cui, Y.; Qin, L.; Wu, F.; Li, Y.; Du, N.; Li, X. Knockdown of LMNB1 Inhibits the Proliferation of Lung Adenocarcinoma Cells by Inducing DNA Damage and Cell Senescence. Front. Oncol. 2022, 12, 913740. [Google Scholar] [CrossRef]
- de Angelis, P.M.; Fjell, B.; Kravik, K.L.; Haug, T.; Tunheim, S.H.; Reichelt, W.; Beigi, M.; Clausen, O.P.; Galteland, E.; Stokke, T. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int. J. Oncol. 2004, 24, 1279–1288. [Google Scholar] [CrossRef]
- Michalak, M.; Warnken, U.; Andre, S.; Schnolzer, M.; Gabius, H.J.; Kopitz, J. Detection of Proteome Changes in Human Colon Cancer Induced by Cell Surface Binding of Growth-Inhibitory Human Galectin-4 Using Quantitative SILAC-Based Proteomics. J. Proteome Res. 2016, 15, 4412–4422. [Google Scholar] [CrossRef]
- Wazir, U.; Ahmed, M.H.; Bridger, J.M.; Harvey, A.; Jiang, W.G.; Sharma, A.K.; Mokbel, K. The clinicopathological significance of lamin A/C, lamin B1 and lamin B receptor mRNA expression in human breast cancer. Cell Mol. Biol. Lett. 2013, 18, 595–611. [Google Scholar] [CrossRef]
- Sun, S.; Xu, M.Z.; Poon, R.T.; Day, P.J.; Luk, J.M. Circulating Lamin B1 (LMNB1) biomarker detects early stages of liver cancer in patients. J. Proteome Res. 2010, 9, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Iwayama, Y.; Ohnishi, T.; Toyoshima, M.; Shimamoto, C.; Hisano, Y.; Toyota, T.; Balan, S.; Matsuzaki, H.; Iwata, Y.; et al. Investigation of the fatty acid transporter-encoding genes SLC27A3 and SLC27A4 in autism. Sci. Rep. 2015, 5, 16239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Wang, Q.; Yang, J. LncRNA HOXD-AS1 promotes the metastasis of human hepatocellular carcinoma via modulating miR-326/SLC27A4. Cancer Cell Int. 2020, 20, 161. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Su, J.; Qian, H.; Guo, T. SLC27A4 regulate ATG4B activity and control reactions to chemotherapeutics-induced autophagy in human lung cancer cells. Tumour Biol. 2016, 37, 6943–6952. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.C.; Chou, S.K.; Kan, J.Y.; Kuo, P.L.; Hou, M.F.; Hsu, Y.L. Solute Carrier Family 27 Member 4 (SLC27A4) Enhances Cell Growth, Migration, and Invasion in Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Han, T.; Zhao, Y.; Feng, L. Identification of solute-carrier family 27A molecules (SCL27As) as a potential biomarker of ovarian cancer based on bioinformatics and experiments. Ann. Transl. Med. 2021, 9, 1237. [Google Scholar] [CrossRef]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef]
- Carrillo, C.; Cavia Mdel, M.; Alonso-Torre, S.R. Antitumor effect of oleic acid; mechanisms of action: A review. Nutr. Hosp. 2012, 27, 1860–1865. [Google Scholar]
- Watkins, P.A. Very-long-chain acyl-CoA synthetases. J. Biol. Chem. 2008, 283, 1773–1777. [Google Scholar] [CrossRef] [Green Version]
- Uauy, R.; Dangour, A.D. Nutrition in brain development and aging: Role of essential fatty acids. Nutr. Rev. 2006, 64 Pt 2, S24–S33. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.; Hou, J.; Cui, H. Serine-glycine-one-carbon metabolism: Vulnerabilities in MYCN-amplified neuroblastoma. Oncogenesis 2020, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, M.; Santucci, L.; Ravera, S.; Candiano, G.; Bartolucci, M.; Calzia, D.; Lavarello, C.; Inglese, E.; Ramenghi, L.A.; Petretto, A.; et al. Human urinary exosome proteome unveils its aerobic respiratory ability. J. Proteom. 2016, 136, 25–34. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Albert, T.K.; Interlandi, M.; Sill, M.; Graf, M.; Moreno, N.; Menck, K.; Rohlmann, A.; Melcher, V.; Korbanka, S.; Meyer Zu Horste, G.; et al. An extracellular vesicle-related gene expression signature identifies high-risk patients in medulloblastoma. Neuro-Oncol. 2015, 23, 586–598. [Google Scholar] [CrossRef]
- Yao, M.; Ventura, P.B.; Jiang, Y.; Rodriguez, F.J.; Wang, L.; Perry, J.S.A.; Yang, Y.; Wahl, K.; Crittenden, R.B.; Bennett, M.L.; et al. Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell 2020, 180, 502–520. [Google Scholar] [CrossRef]
- De Braganca, K.C.; Packer, R.J. Treatment Options for Medulloblastoma and CNS Primitive Neuroectodermal Tumor (PNET). Curr. Treat. Options Neurol. 2013, 15, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Dell, R.B.; Holleran, S.; Ramakrishnan, R. Sample size determination. ILAR J. 2002, 43, 207–213. [Google Scholar] [CrossRef]
- Santucci, L.; Candiano, G.; Bruschi, M.; D’Ambrosio, C.; Petretto, A.; Scaloni, A.; Urbani, A.; Righetti, P.G.; Ghiggeri, G.M. Combinatorial peptide ligand libraries for the analysis of low-expression proteins: Validation for normal urine and definition of a first protein MAP. Proteomics 2012, 12, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Granata, S.; Santucci, L.; Candiano, G.; Fabris, A.; Antonucci, N.; Petretto, A.; Bartolucci, M.; Del Zotto, G.; Antonini, F.; et al. Proteomic Analysis of Urinary Microvesicles and Exosomes in Medullary Sponge Kidney Disease and Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2019, 14, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; La Porta, E.; Panfoli, I.; Candiano, G.; Petretto, A.; Vidal, E.; Kajana, X.; Bartolucci, M.; Granata, S.; Ghiggeri, G.M.; et al. Proteomic profile of mesothelial exosomes isolated from peritoneal dialysis effluent of children with focal segmental glomerulosclerosis. Sci. Rep. 2021, 11, 20807. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. 1D and 2D annotation enrichment: A statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinform. 2012, 13 (Suppl. 16), S12. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | MS/ELISA | Sex (F/M) | Age (Year) |
|---|---|---|---|
| Control [24] | |||
| Congenital hydrocephalus [24] | 6/24 | 14/10 | 1(0–22) |
| Low-grade Giomas and Glioneural Tumors [16] | |||
| Pilocytic astrocytoma [8] | 0/12 | 6/6 | 8(3–15) |
| Gangliocytoma/Ganglioglioma [3] | 0/4 | 2/2 | 9(5–11) |
| Medulloblastoma [12] | 6/12 | 4/3 | 5(0–15) |
| Other [9] | |||
| Meningiomas [2], Germ Cell Tumors [2], Epindimomas [2], Plessopapillomas [2], Emangioblastoma [1] | 0/9 | 3/6 | 9(0–18) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruschi, M.; Kajana, X.; Petretto, A.; Bartolucci, M.; Pavanello, M.; Ghiggeri, G.M.; Panfoli, I.; Candiano, G. Weighted Gene Co-Expression Network Analysis and Support Vector Machine Learning in the Proteomic Profiling of Cerebrospinal Fluid from Extraventricular Drainage in Child Medulloblastoma. Metabolites 2022, 12, 724. https://doi.org/10.3390/metabo12080724
Bruschi M, Kajana X, Petretto A, Bartolucci M, Pavanello M, Ghiggeri GM, Panfoli I, Candiano G. Weighted Gene Co-Expression Network Analysis and Support Vector Machine Learning in the Proteomic Profiling of Cerebrospinal Fluid from Extraventricular Drainage in Child Medulloblastoma. Metabolites. 2022; 12(8):724. https://doi.org/10.3390/metabo12080724
Chicago/Turabian StyleBruschi, Maurizio, Xhuliana Kajana, Andrea Petretto, Martina Bartolucci, Marco Pavanello, Gian Marco Ghiggeri, Isabella Panfoli, and Giovanni Candiano. 2022. "Weighted Gene Co-Expression Network Analysis and Support Vector Machine Learning in the Proteomic Profiling of Cerebrospinal Fluid from Extraventricular Drainage in Child Medulloblastoma" Metabolites 12, no. 8: 724. https://doi.org/10.3390/metabo12080724