
Targeted Proteomics for Monitoring One-Carbon Metabolism in Liver Diseases


Abstract
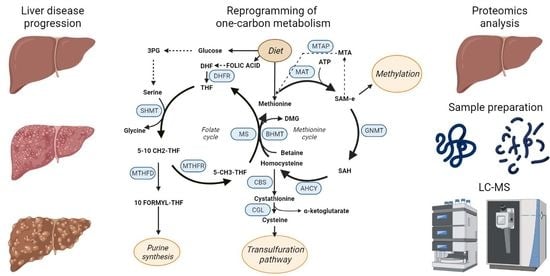
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
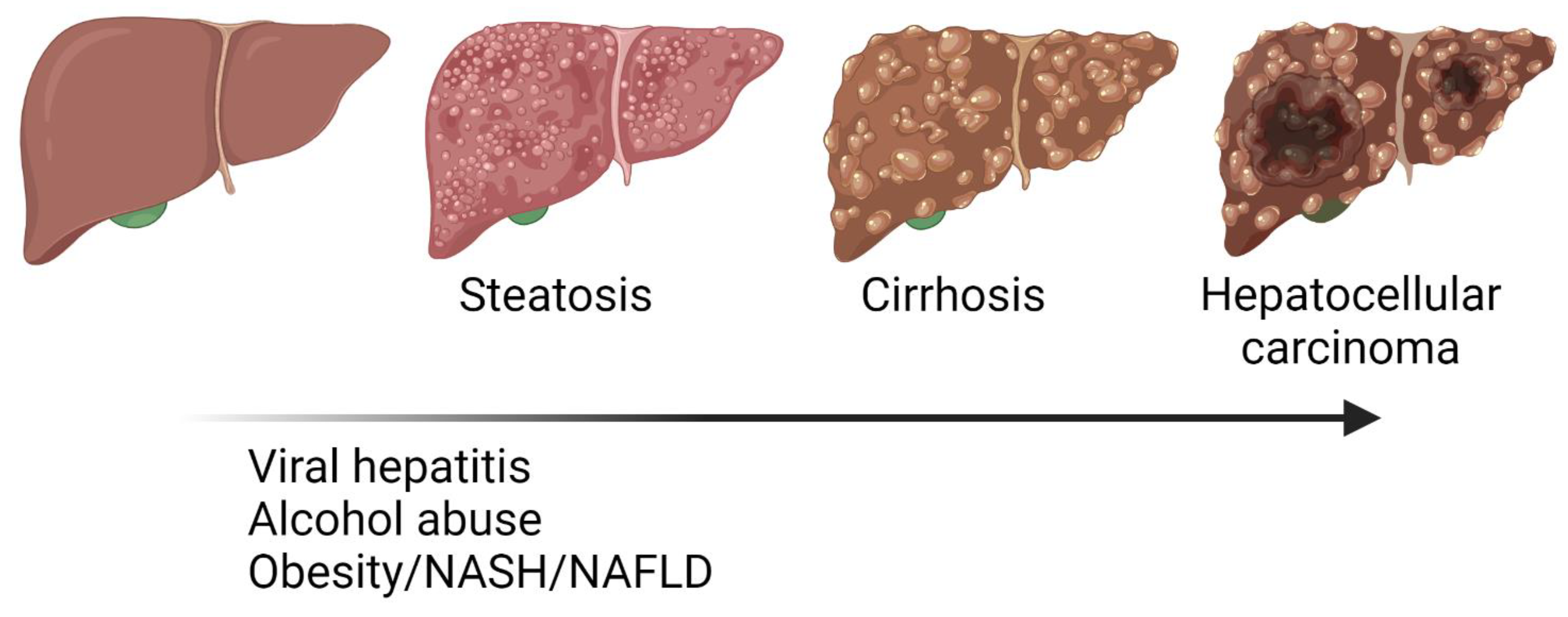
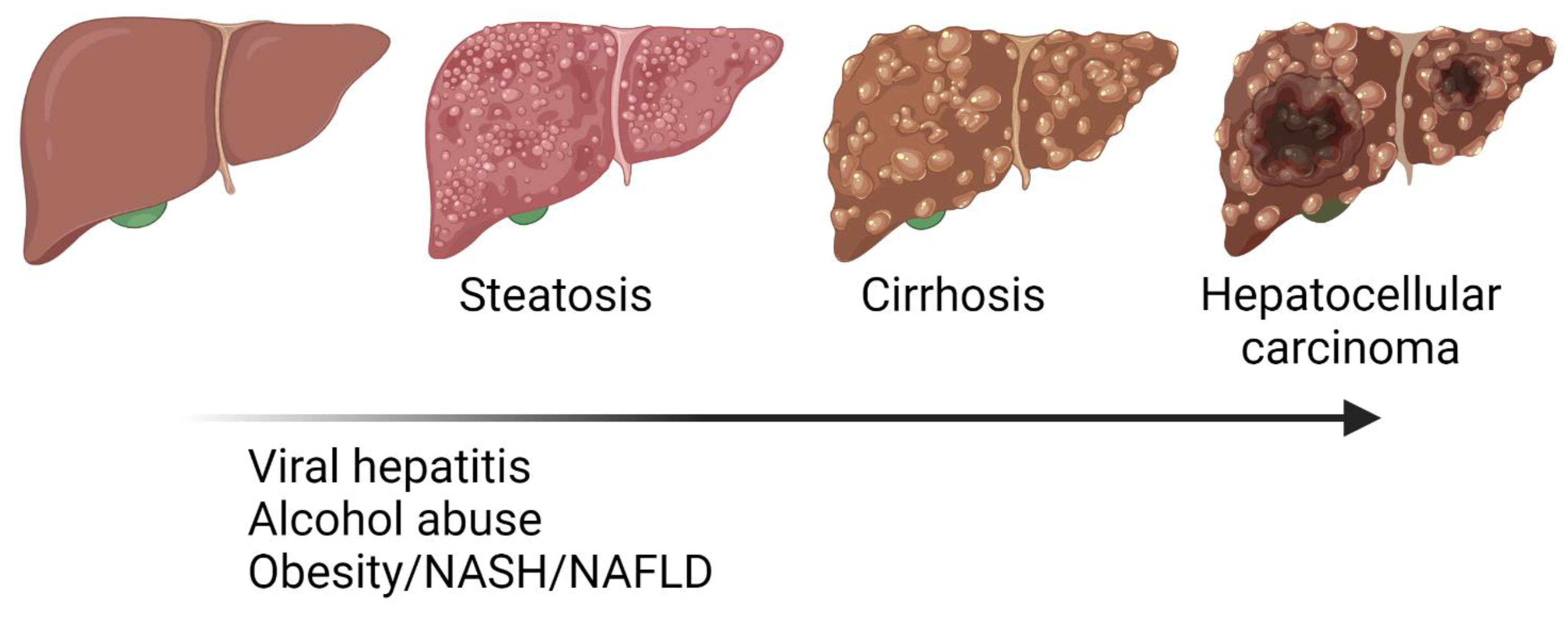
1. Liver Diseases
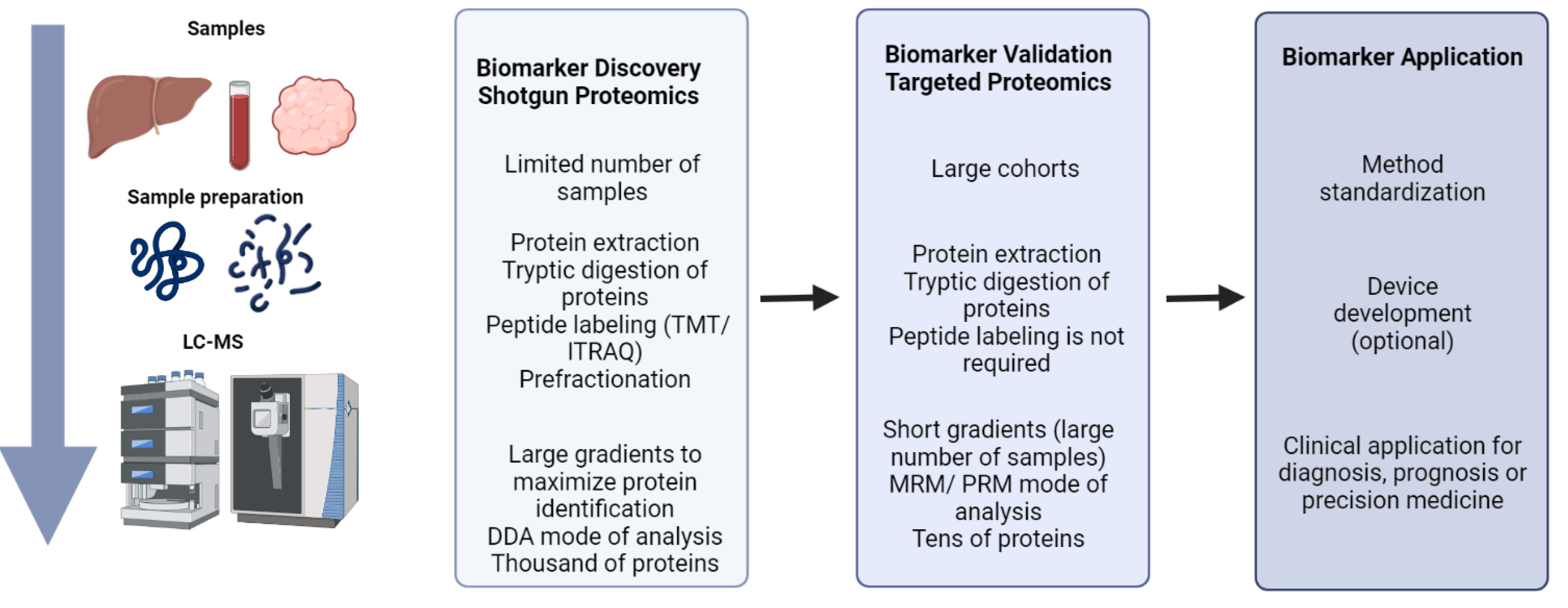
2. The Role of Proteomics in Biomarker Discovery and Validation
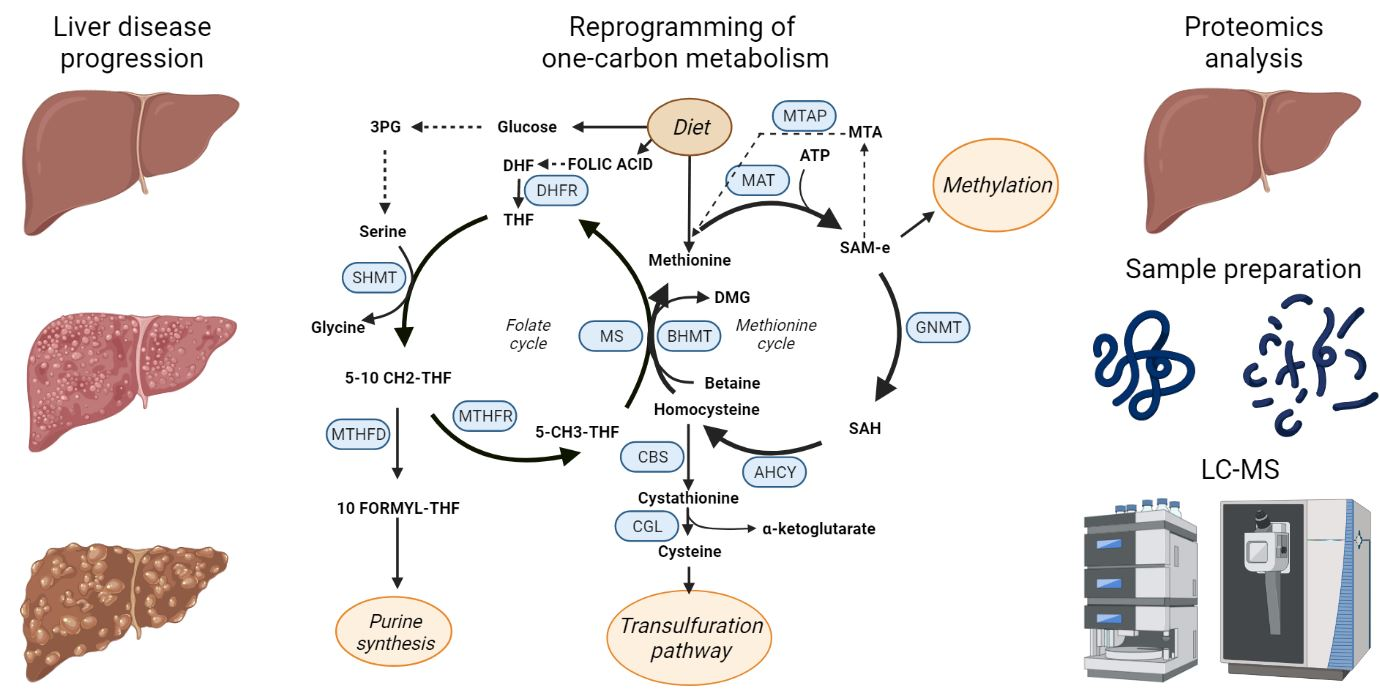
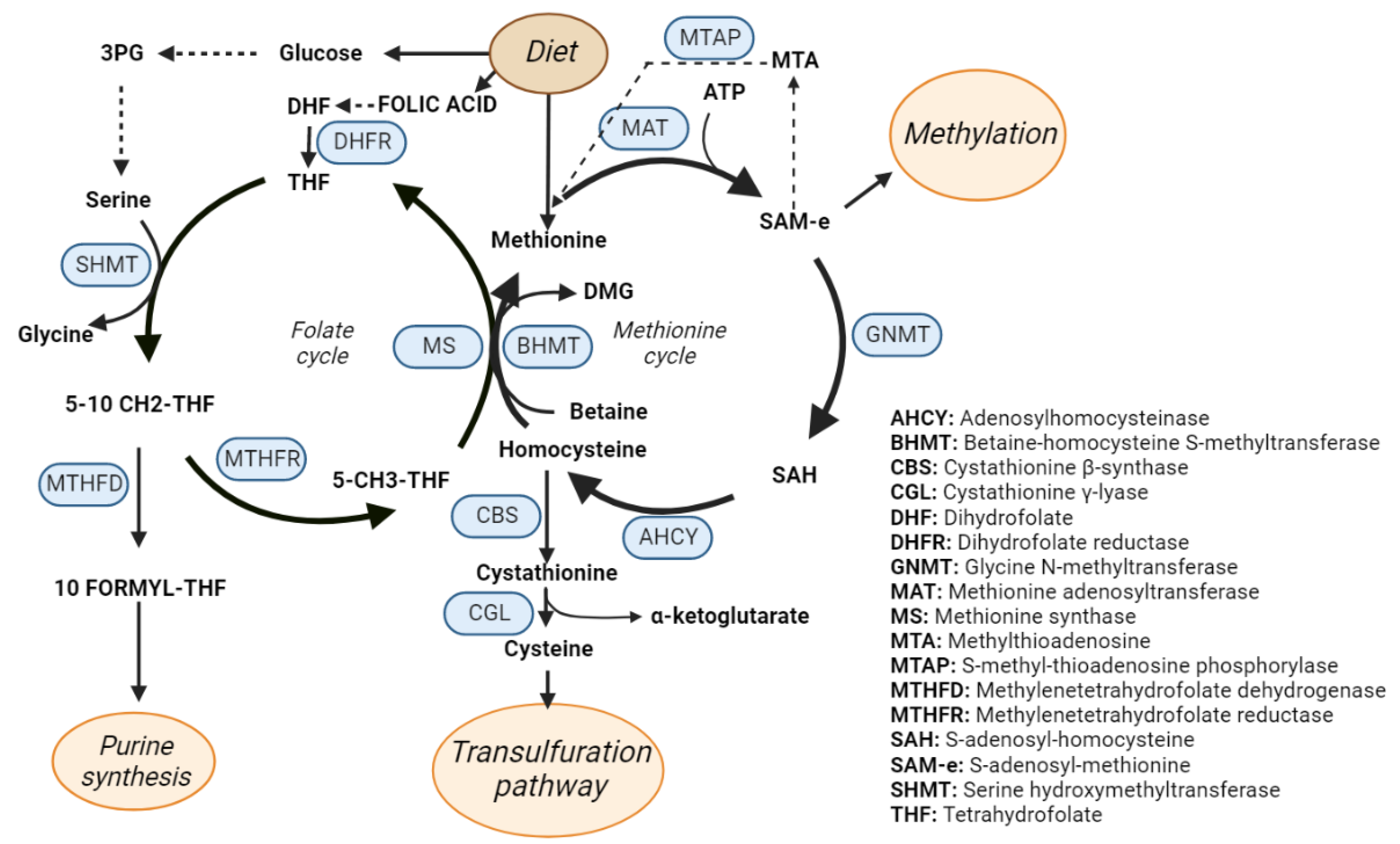
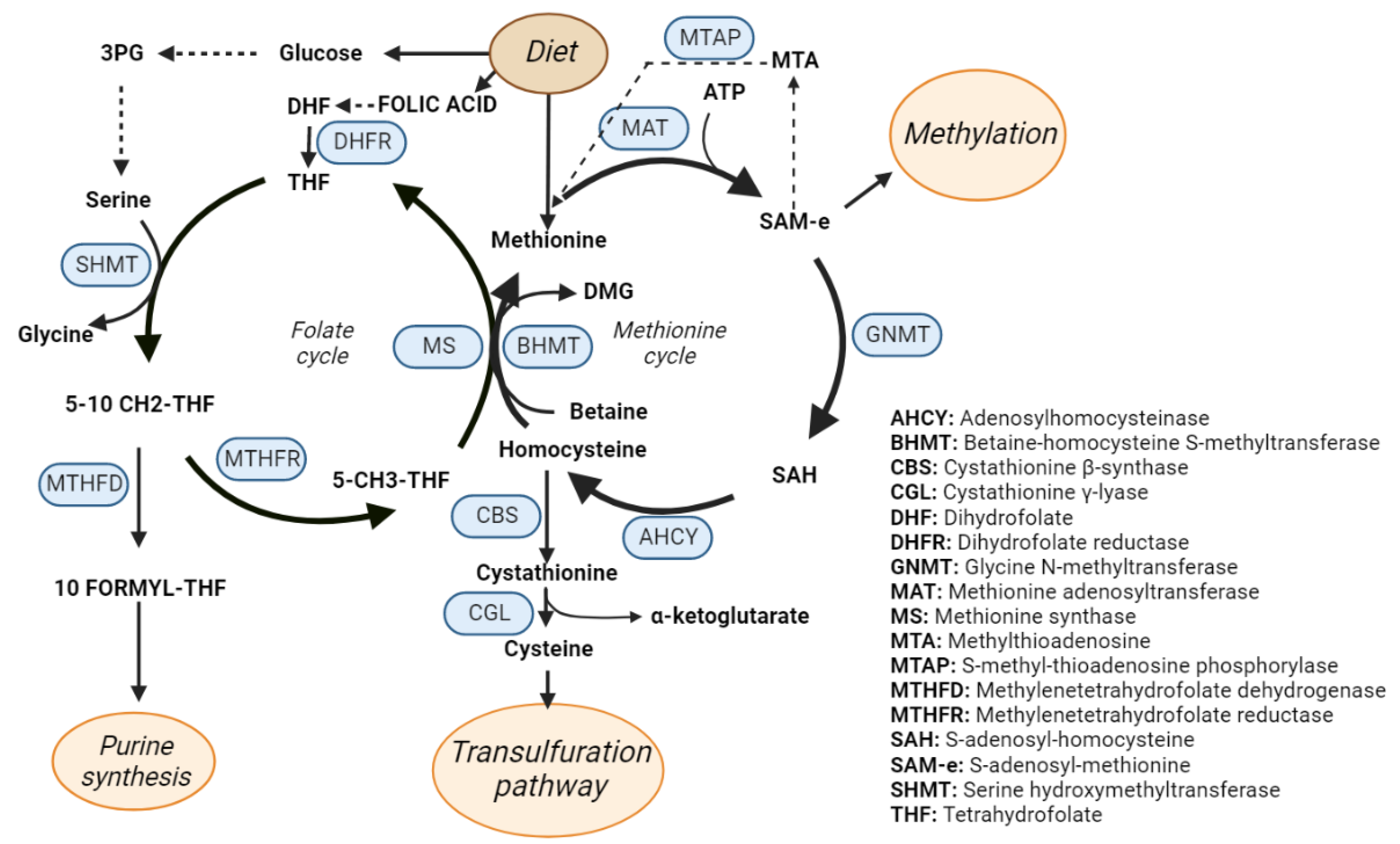
3. One-Carbon Metabolism
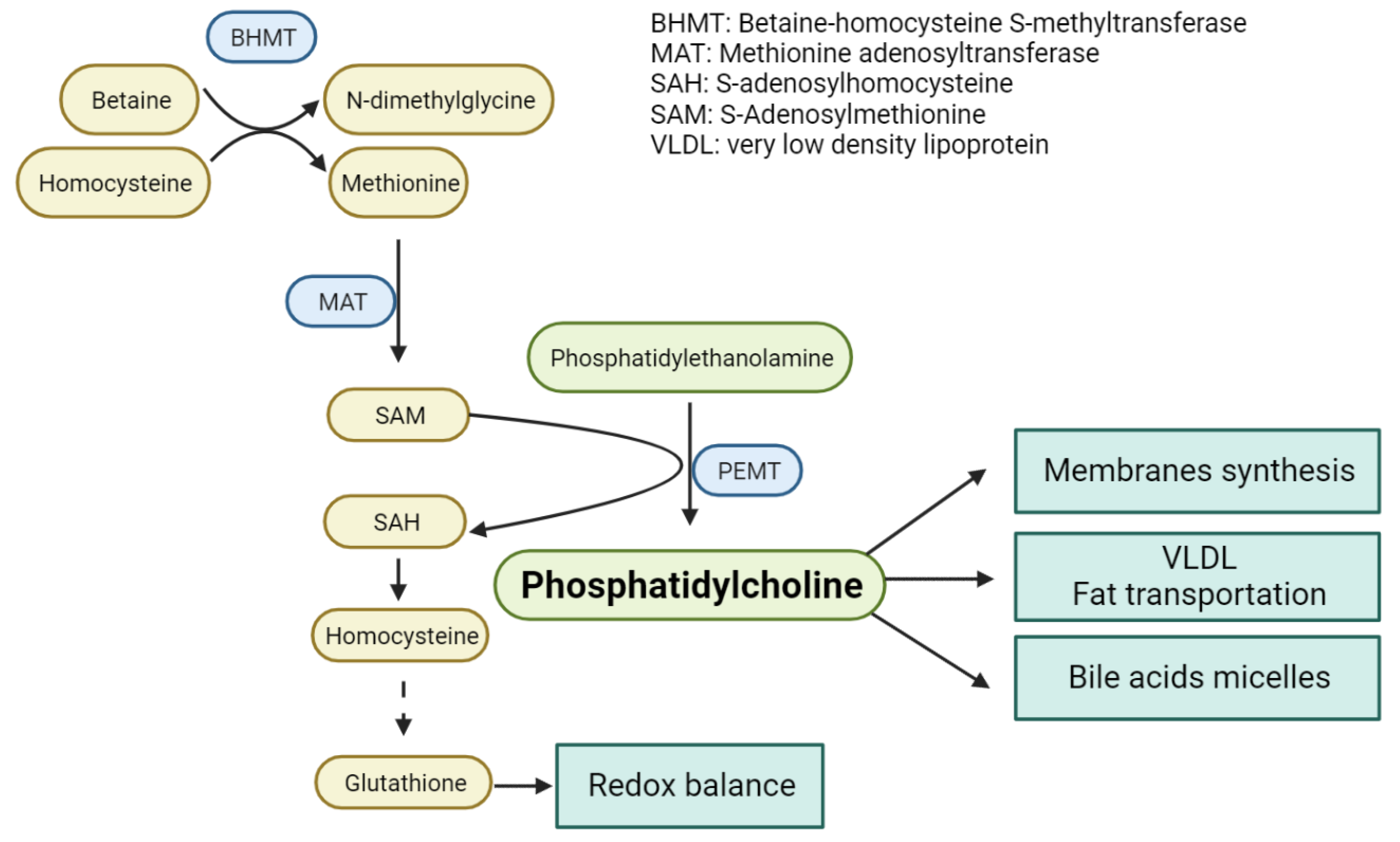
4. One-Carbon Metabolism in NAFLD, NASH and Fibrosis
5. One-Carbon Metabolism in Cirrhosis and Hepatocellular Carcinoma
6. Proteomics for Biomarker Discovery. Monitoring of OCM in Liver Diseases
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AHCY | Adenosylhomocysteinase |
| BHMT | Betaine-homocysteine S-methyltransferase |
| CBS | Cystathionine β-synthase |
| CGL | Cystathionine γ-lyase |
| DHF | Dihydrofolate |
| DHFR | Dihydrofolate reductase |
| GNMT | Glycine N-methyltransferase |
| HCC | Hepatocellular carcinoma |
| HBV | Hepatitis virus B |
| HCV | Hepatitis C virus |
| HPLC | high-permormance liquid chromatography |
| MAT1A/MAT2A | Methionine adenosyltransferase 1A/2A |
| MS | Methionine synthase |
| MTA | Methylthioadenosine |
| MTAP | S-methyl-thioadenosine phosphorylase |
| MTHFD | Methylenetetrahydrofolate dehydrogenase |
| MTHFR | Methylenetetrahydrofolate reductase |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| PC | Phosphatidylcholine |
| PEMT | Phosphatidylethanolamine N-ethyltransferase |
| PFIC | Progressive familiar intrahepatic cholestasis |
| SAH | S-adenosyl-homocysteine |
| SAM | S-adenosyl-methionine |
| SHMT | Serine hydroxymethyltransferase |
| THF | Tetrahydrofolate |
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Williams, R. Global challenges in liver disease. Hepatology 2006, 44, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef]
- Chuang, S.-C.; Vecchia, C.; la Boffetta, P. Liver cancer: Descriptive epidemiology and risk factors other than HBV and HCV infection. Cancer Lett. 2009, 286, 9–14. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Status Report on Alcohol and Health 2018. In Global Status Report on Alcohol; World Health Organization: Geneva, Switzerland, 2019; Volume 65. [Google Scholar]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Panchal, S.K.; Poudyal, H.; Iyer, A.; Nazer, R.; Alam, A.; Diwan, V.; Kauter, K.; Sernia, C.; Campbell, F.; Ward, L.; et al. High-carbohydrate high-fat diet-induced metabolic syndrome and cardiovascular remodeling in rats. J. Cardiovasc. Pharmacol. 2011, 57, 51–64. [Google Scholar] [CrossRef]
- Siegel, A.B.; Zhu, A.X. Metabolic syndrome and hepatocellular carcinoma. Cancer 2009, 115, 5651–5661. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73, 4–13. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Iwarson, S. The main five types of viral hepatitis: An alphabetical update. Scand. J. Infect. Dis. 1992, 24, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef]
- Asselah, T. Sofosbuvir for the treatment of hepatitis C virus. Expert Opin. Pharm. 2014, 15, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Li, H.C.; Lo, S.Y. Hepatitis C virus: Virology, diagnosis and treatment. World J. Hepatol. 2015, 7, 1377–1389. [Google Scholar] [CrossRef]
- Bull, L.N.; Thompson, R.J. Progressive Familial Intrahepatic Cholestasis. Clin. Liver Dis. 2018, 22, 657–669. [Google Scholar] [CrossRef]
- Sticova, E.; Jirsa, M.; Pawłowska, J. New Insights in Genetic Cholestasis: From Molecular Mechanisms to Clinical Implications. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2313675. [Google Scholar] [CrossRef]
- Vitale, G.; Gitto, S.; Raimondi, F.; Mattiaccio, A.; Mantovani, V.; Vukotic, R.; D’Errico, A.; Seri, M.; Russell, R.B.; Andreone, P. Cryptogenic cholestasis in young and adults: ATP8B1, ABCB11, ABCB4, and TJP2 gene variants analysis by high-throughput sequencing. J. Gastroenterol. 2018, 53, 945–958. [Google Scholar] [CrossRef]
- Paulusma, C.C.; Groen, A.; Kunne, C.; Ho-Mok, K.S.; Spijkerboer, A.L.; De Waart, D.R.; Hoek, F.J.; Vreeling, H.; Hoeben, K.A.; Van Marle, J.; et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology 2006, 44, 195–204. [Google Scholar] [CrossRef]
- Cai, S.; Gautam, S.; Nguyen, T.; Soroka, C.J.; Rahner, C.; Boyer, J.L. ATP8B1 Deficiency Disrupts the Bile Canalicular Membrane Bilayer Structure in Hepatocytes, But FXR Expression and Activity Are Maintained. Gastroenterology 2009, 136, 1060–1069. [Google Scholar] [CrossRef] [Green Version]
- Klomp, L.W.J.; Vargas, J.C.; van Mil, S.W.C.; Pawlikowska, L.; Strautnieks, S.S.; van Eijk, M.J.T.; Juijn, J.A.; Pabón-Peña, C.; Smith, L.B.; DeYoung, J.A.; et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 2004, 40, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Strautnieks, S.S.; A Byrne, J.; Pawlikowska, L.; Cebecauerová, D.; Rayner, A.; Dutton, L.; Meier, Y.; Antoniou, A.; Stieger, B.; Arnell, H.; et al. Severe Bile Salt Export Pump Deficiency: 82 Different ABCB11 Mutations in 109 Families. Gastroenterology 2008, 134, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Sohn, M.J.; Woo, M.H.; Seong, M.-W.; Park, S.S.; Kang, G.H.; Moon, J.S.; Ko, J.S. Benign Recurrent Intrahepatic Cholestasis Type 2 in Siblings with Novel ABCB11 Mutations. Pediatric Gastroenterol. Hepatol. Nutr. 2019, 22, 201–206. [Google Scholar] [CrossRef] [PubMed]
- de Vree, J.M.L.; Jacquemin, E.; Sturm, E.; Cresteil, D.; Bosma, P.J.; Aten, J.; Deleuze, J.-F.; Desrochers, M.; Burdelski, M.; Bernard, O.; et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc. Natl. Acad. Sci. 1998, 95, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P.; Weerasekera, N.; Linton, K.; Donaldson, O.; Chambers, J.; Egginton, E.; Weaver, J.; Nelson-Piercy, C.; De Swiet, M.; Warnes, G.; et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: Evidence for a defect in protein trafficking. Hum. Mol. Genet. 2000, 9, 1209–1217. [Google Scholar] [CrossRef]
- Gonzales, E.; Davit-Spraul, A.; Baussan, C.; Buffet, C.; Maurice, M.; Jacquemin, E.; et al. Liver diseases related to MDR3 (ABCB4) gene deficiency. Front. Biosci. (Landmark Ed.) 2009, 14, 4242–4256. [Google Scholar] [CrossRef]
- Gautherot, J.; Delautier, D.; Maubert, M.-A.; Aït-Slimane, T.; Bolbach, G.; Delaunay, J.-L.; et al. Phosphorylation of ABCB4 impacts its function: Insights from disease-causing mutations. Hepatology 2014, 60, 610–621. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Debakey, M.E. Epidemiology of HCC: Consider the Population. J. Clin. Gastroenterol. 2014, 47, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Vogel, A.; Martinelli, E.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; et al. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yu, S.J.; Yeo, I.; Cho, Y.Y.; Lee, D.H.; Cho, Y.; Cho, E.J.; Lee, J.-H.; Kim, Y.J.; Lee, S.; et al. Prediction of response to sorafenib in hepatocellular carcinoma: A putative marker panel by multiple reaction monitoring-mass spectrometry (MRM-MS). Mol. Cell. Proteom. 2017, 16, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Santoro, A. Sorafenib A Review of its Use in Advanced Hepatocellular Carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef]
- Bialecki, E.S.; di Bisceglie, A.M. Diagnosis of hepatocellular carcinoma. HPB Off. J. Int. Hepato Pancreato Biliary Assoc. 2005, 7, 26. [Google Scholar] [CrossRef]
- Bertuccio, P.; Turati, F.; Carioli, G.; Rodriguez, T.; La Vecchia, C.; Malvezzi, M.; Negri, E.; et al. Global trends and predictions in hepatocellular carcinoma mortality. J. Hepatol. 2017, 67, 302–309. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Sawada, Y.; Endo, I.; Saito, K.; Uemura, Y.; Nakatsura, T. Biomarkers for the early diagnosis of hepatocellular carcinoma 2015 Advances in Hepatocellular Carcinoma. World J. Gastroenterol 2015, 21, 10573–10583. [Google Scholar] [CrossRef]
- Xu, G.-P.; Zhao, Q.; Wang, D.; Xie, W.-Y.; Zhang, L.-J.; Zhou, H.; Chen, S.-Z.; Wu, L.-F.; et al. The association between BRCA1 gene polymorphism and cancer risk: A meta-analysis. Oncotarget 2018, 9, 8681. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Nelson Hayes, C.; Chayama, K. MicroRNAs as Biomarkers for Liver Disease and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2016, 17, 280. [Google Scholar] [CrossRef]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Giannini, E.; Botta, F.; Fasoli, A.; Ceppa, P.; Risso, D.; Lantieri, P.B.; Celle, G.; Testa, R. Progressive Liver Functional Impairment Is Associated with an Increase in AST/ALT Ratio. Dig. Dis. Sci. 1999, 44, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Adams, L.; Prins, A.; Bulsara, M.; De Boer, B.; Garas, G.; MacQuillan, G.; Speers, D.; Jeffrey, G. Validation of the FibroTest Biochemical Markers Score in Assessing Liver Fibrosis in Hepatitis C Patients. Clin. Chem. 2003, 49, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Coco, B.; Oliveri, F.; Maina, A.M.; Ciccorossi, P.; Sacco, R.; Colombatto, P.; Bonino, F.; Brunetto, M.R. Transient elastography: A new surrogate marker of liver fibrosis influenced by major changes of transaminases. J. Viral. Hepat. 2007, 14, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, A.A.M.; Wan, A.F.; Myers, R.P. FibroTest and FibroScan for the prediction of hepatitis C-related fibrosis: A systematic review of diagnostic test accuracy. Am. J. Gastroenterol. 2007, 102, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Allmaier, G.; Apweiler, R.; Attwood, T.; Baumann, M.; Benigni, A.; Bennett, S.E.; Bischoff, R.; Bongcam-Rudloff, E.; Capasso, G.; et al. Recommendations for Biomarker Identification and Qualification in Clinical Proteomics. Sci. Transl. Med. 2010, 2, 46ps42. [Google Scholar] [CrossRef]
- Masuda, T.; Mori, A.; Ito, S.; Ohtsuki, S. Quantitative and targeted proteomics-based identification and validation of drug efficacy biomarkers. Drug Metab. Pharmacokinet. 2021, 36, 100361. [Google Scholar] [CrossRef]
- Cong, Y.; Liang, Y.; Motamedchaboki, K.; Huguet, R.; Truong, T.; Zhao, R.; Shen, Y.; Lopez-Ferrer, D.; Zhu, Y.; Kelly, R.T. Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal. Chem. 2020, 92, 2665–2671. [Google Scholar] [CrossRef]
- Kelly, R.T. Single-cell Proteomics: Progress and Prospects. Mol. Cell. Proteom. 2020, 19, 1739–1748. [Google Scholar] [CrossRef]
- Pribluda, A.; de La Cruz, C.C.; Jackson, E.L. Intratumoral heterogeneity: From diversity comes resistance. Clin. Cancer Res. 2015, 21, 2916–2923. [Google Scholar] [CrossRef] [Green Version]
- McDonald, W.H.; Yates, J.R., III. Shotgun proteomics and biomarker discovery. Dis. Markers 2002, 18, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N.; Yates, J.R. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef] [PubMed]
- Doerr, A. DIA mass spectrometry. Nat. Methods 2015, 12, 35. [Google Scholar] [CrossRef]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef]
- Mermelekas, G.; Vlahou, A.; Zoidakis, J. SRM/MRM targeted proteomics as a tool for biomarker validation and absolute quantification in human urine. Expert Rev. Mol. Diagn. 2015, 15, 1441–1454. [Google Scholar] [CrossRef]
- Santamaría, E.; MuñozMu, J.; FernándezFern, J.; Prıèto, J.; Corrales, F.J. Toward the discovery of new biomarkers of hepatocellular carcinoma by proteomics. Liver Int. 2007, 27, 163–173. [Google Scholar] [CrossRef]
- Steel, L.F.; Shumpert, D.; Trotter, M.; Seeholzer, S.H.; Evans, A.A.; London, W.T.; Dwek, R.; Block, T.M. A strategy for the comparative analysis of serum proteomes for the discovery of biomarkers for hepatocellular carcinoma. Proteomics 2003, 3, 601–609. [Google Scholar] [CrossRef]
- Li, Y.; Tang, Z.Y.; Tian, B.; Ye, S.L.; Qin, L.X.; Xue, Q.; Sun, R. Serum CYFRA 21-1 level reflects hepatocellular carcinoma metastasis: Study in nude mice model and clinical patients. J. Cancer Res. Clin. Oncol. 2006, 132, 515–520. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, A.; Blenis, J.; Gomes, A.P. Beyond the Warburg Effect: How Do Cancer Cells Regulate One-Carbon Metabolism? Front. Cell Dev. Biol. 2018, 90. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Udali, S.; de Santis, D.; Choi, S.W. One-carbon metabolism and epigenetics. Mol. Asp. Med. 2017, 54, 28–36. [Google Scholar] [CrossRef]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian Folate-Mediated One-Carbon Metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef]
- Hebbring, S.J.; Chai, Y.; Ji, Y.; Abo, R.P.; Jenkins, G.D.; Fridley, B.; Zhang, J.B.; Eckloff, B.W.; Wieben, E.D.; Weinshilboum, R.M. Serine hydroxymethyltransferase 1 and 2: Gene sequence variation and functional genomic characterization. J. Neurochem. 2012, 120, 881–890. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- McGuire, J.J. Anticancer Antifolates: Current Status and Future Directions. Curr. Pharm. Des. 2005, 9, 2593–2613. [Google Scholar] [CrossRef]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Gibson, J.B.; Carson, N.A.J.; Neill, D.W. Pathological findings in homocystinuria. J. Clin. Pathol. 1964, 17, 427. [Google Scholar] [CrossRef] [PubMed]
- Nygård, O.; Vollset, S.E.; Refsum, H.; Stensvold, I.; Tverdal, A.; Nordrehaug, J.E.; Ueland, M.; Kvåle, G. Total Plasma Homocysteine and Cardiovascular Risk Profile: The Hordaland Homocysteine Study. JAMA 1995, 274, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Platt, R.; Wu, Q.; Leclerc, D.; Christensen, B.; Yang, H.; Gravel, R.A.; Rozen, R. A Common Variant in Methionine Synthase Reductase Combined with Low Cobalamin (Vitamin B12) Increases Risk for Spina Bifida. Mol. Genet. Metab. 1999, 67, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Zatz, M.; Dudley, P.; Kloog, Y.; Markey, S. Nonpolar lipid methylation. Biosynthesis of fatty acid methyl esters by rat lung membranes using S-adenosylmethionine. J. Biol. Chem. 1981, 256, 10028–10032. [Google Scholar] [CrossRef]
- Spector, A.A.; Yorek, M.A. Membrane lipid composition and cellular function. J. Lipid Res. 1985, 26, 1015–1035. [Google Scholar] [CrossRef]
- Hickman, M.J.; Petti, A.A.; Ho-Shing, O.; Silverman, S.J.; McIsaac, R.S.; Lee, T.A.; Botstein, D. Coordinated regulation of sulfur and phospholipid metabolism refl ects the importance of methylation in the growth of yeast. Mol. Biol. Cell 2011, 22, 4192–4204. [Google Scholar] [CrossRef]
- Struck, A.-W.; Thompson, M.L.; Wong, L.S.; Micklefield, J. S-Adenosyl-Methionine-Dependent Methyltransferases: Highly Versatile Enzymes in Biocatalysis, Biosynthesis and Other Biotechnological Applications. Chem. Bio. Chem. 2012, 13, 2642–2655. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Pérez, S.; Torres-Cuevas, I.; Martí-Andrés, P.; Taléns-Visconti, R.; Paradela, A.; Guerrero, L.; Franco, L.; López-Rodas, G.; Torres, L.; et al. Blockade of the trans-sulfuration pathway in acute pancreatitis due to nitration of cystathionine β-synthase. Redox Biol. 2020, 28, 101324. [Google Scholar] [CrossRef]
- Mato, J.M.; Corrales, F.J.; Lu, S.C.; Avila, M. S-Adenosylmethionine: A control switch that regulates liver function. FASEB J. 2002, 16, 15–26. [Google Scholar] [CrossRef]
- Sanchez del Pino, M.M.; Corrales, F.J.; Mato, J.M. Hysteretic Behavior of Methionine Adenosyltransferase III: Methionine switches between two conformations of the enzyme with different specific activity. J. Biol. Chem. 2000, 275, 23476–23482. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, F.; Corrales, F.J.; Miqueo, C.; Mato, J.M. Nitric oxide inactivates rat hepatic methionine adenosyltransferase in vivo by S-nitrosylation. Hepatology 1998, 28, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- García-Trevijano, E.R.; Latasa, M.U.; Carretero, M.V.; Berasain, C.; Mato, J.M.; Avila, M.A. S-Adenosylmethionine regulates MAT1A and MAT2A gene expression in cultured rat hepatocytes: A new role for S-adenosylmethionine in the maintenance of the differentiated status of the liver. FASEB J. 2000, 14, 2511–2518. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.; Avila, M.A.; Carretero, M.V.; Latasa, M.U.; Caballería, J.; López-Rodas, G.; Boukaba, A.; Lu, S.C.; Franco, L.; Mato, J.M. Liver-specific methionine adenosyltransferase MAT1A gene expression is associated with a specific pattern of promoter methylation and histone acetylation: Implications for MAT1A silencing during transformation. FASEB J. 2000, 14, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Chantar, M.L.; Vázquez-Chantada, M.; Ariz, U.; Martínez, N.; Varela, M.; Luka, Z.; Capdevila, A.; Rodríguez, J.; Aransay, A.M.; Matthiesen, R.; et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 2008, 47, 1191–1199. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Morales, A.; Colell, A.; Ballesta, A.; Rodés, J.; Kaplowitz, N.; Fernández-Checa, J.C. Feeding S-Adenosyl-L-Methionine Attenuates Both Ethanol-Induced Depletion of Mitochondrial Glutathione and Mitochondrial Dysfunction in Periportal and Perivenous Rat Hepatocytes. Hepatology 1995, 21, 207–214. [Google Scholar] [CrossRef]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar] [CrossRef]
- Friedel, H.A.; Goa, K.L.; Benfield, P. S-Adenosyl-L-Methionine: A Review of its Pharmacological Properties and Therapeutic Potential in Liver Dysfunction and Affective Disorders in Relation to its Physiological Role in Cell Metabolism. Drugs 1989, 38, 389–416. [Google Scholar] [CrossRef]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Mato, J.M.; Cámara, J.; de Paz, J.F.; Caballeria, L.; Coll, S.; Caballero, A.; García-Buey, L.; Beltrán, J.; Benita, V.; Caballería, J.; et al. S-Adenosylmethionine in alcoholic liver cirrhosis: A randomized, placebo-controlled, double-blind, multicenter clinical trial. J. Hepatol. 1999, 30, 1081–1089. [Google Scholar] [CrossRef]
- Simile, M.M.; Banni, S.; Angioni, E.; Carta, G.; De Miglio, M.R.; Muroni, M.R.; Calvisi, D.F.; Carru, A.; Pascale, R.M.; Feo, F. 5′-Methylthioadenosine administration prevents lipid peroxidation and fibrogenesis induced in rat liver by carbon-tetrachloride intoxication. J. Hepatol. 2001, 34, 386–394. [Google Scholar] [CrossRef]
- Fickert, P.; Fuchsbichler, A.; Wagner, M.; Zollner, G.; Kaser, A.; Tilg, H.; Krause, R.; Lammert, F.; Langner, C.; Zatloukal, K.; et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004, 127, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Latasa, M.U.; Gil-Puig, C.; Fernández-Barrena, M.G.; Rodríguez-Ortigosa, C.M.; Banales, J.M.; Urtasun, R.; Goñi, S.; Méndez, M.; Arcelus, S.; Juanarena, N.; et al. Oral Methylthioadenosine Administration Attenuates Fibrosis and Chronic Liver Disease Progression in Mdr2 −/− Mice. PLoS ONE 2010, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.J. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 521–533. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.P.; Eudy, B.J.; Deminice, R. One-Carbon Metabolism in Fatty Liver Disease and Fibrosis: One-Carbon to Rule Them All. J. Nutr. 2020, 150, 994–1003. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Et Biophys. Acta (BBA) Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Walker, A.K. 1-Carbon Cycle Metabolites Methylate Their Way to Fatty Liver. Trends Endocrinol. Metab. 2017, 28, 63–72. [Google Scholar] [CrossRef]
- Mato, J.M.; Lu, S.C. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 2007, 45, 1306–1312. [Google Scholar] [CrossRef]
- Lyman, R.L.; Thenen, S.; Cook, C.R. Methionine Deficiency and Fatty Liver in Male and Female Rats. Proc. Soc. Exp. Biol. Med. 1964, 117, 696–699. [Google Scholar] [CrossRef]
- Lu, S.C.; Alvarez, L.; Huang, Z.-Z.; Chen, L.; An, W.; Corrales, F.J.; Avila, M.A.; Kanel, G.; Mato, J.M. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved. Pnas 2011, 98, 5560–5565. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.; Zhao, Y.; Koonen, D.P.Y.; Sletten, T.; Su, B.; Lingrell, S.; Cao, G.; Peake, D.A.; Kuo, M.-S.; Proctor, S.; et al. Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem. 2010, 285, 22403–22413. [Google Scholar] [CrossRef] [PubMed]
- Radziejewska, A.; Muzsik, A.; Milagro, F.I.; Martínez, J.A.; Chmurzynska, A. One-Carbon Metabolism and Nonalcoholic Fatty Liver Disease: The Crosstalk between Nutrients, Microbiota, and Genetics. Lifestyle Genom. 2020, 13, 53–63. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.P.; Leonard, K.A.; Jacobs, R.L. Dietary creatine supplementation lowers hepatic triacylglycerol by increasing lipoprotein secretion in rats fed high-fat diet. J. Nutr. Biochem. 2017, 50, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Alam, S.F.; Ganguly, P.K. Obesity-Induced Non-alcoholic Fatty Liver Disease (NAFLD): Role of Hyperhomocysteinemia. In Pathophysiology of Obesity-Induced Health Complications; Springer International Publishing: New York, NY, USA, 2020; pp. 181–192. [Google Scholar] [CrossRef]
- Watanabe, M.; Osada, J.; Aratani, Y.; Kluckman, K.; Reddick, R.; Malinow, M.R.; Maeda, N. Mice deficient in cystathionine β-synthase: Animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. USA 1995, 92, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Deminice, R.; Da Silva, R.P.; Lamarre, S.G.; Kelly, K.B.; Jacobs, R.L.; Brosnan, M.E.; Brosnan, J.T. Betaine supplementation prevents fatty liver induced by a high-fat diet: Effects on one-carbon metabolism. Amino Acids 2015, 47, 839–846. [Google Scholar] [CrossRef]
- Augoustides-Savvopoulou, P.; Luka, Z.; Karyda, S.; Stabler, S.P.; Allen, R.H.; Patsiaoura, K.; Wagner, C.; Mudd, S.H. Glycine N-methyltransferase deficiency: A new patient with a novel mutation. J. Inherit. Metab. Dis. 2003, 26, 745–759. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Ali, R.; Cusi, K. New diagnostic and treatment approaches in non-alcoholic fatty liver disease (NAFLD). Ann. Med. 2009, 41, 265–278. [Google Scholar] [CrossRef]
- Cheemerla, S.; Balakrishnan, M. Global epidemiology of Chronic liver Disease. Clin. Liver Dis. 2021, 17, 365–370. [Google Scholar] [CrossRef]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in hepatocellular carcinoma incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Pillai, A.; Tiro, J. Early Detection, Curative Treatment, and Survival Rates for Hepatocellular Carcinoma Surveillance in Patients with Cirrhosis: A Meta-analysis. PLoS Med. 2014, 11, e1001624. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.; Capocaccia, R.; Hackl, M.; Lemmens, V.; Molina, E.; Pierannunzio, D.; Sant, M.; Trama, A.; Faivre, J. Survival in patients with primary liver cancer, gallbladder and extrahepatic biliary tract cancer and pancreatic cancer in Europe 1999–2007: Results of EUROCARE-5. Eur. J. Cancer 2015, 51, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.; Berasain, C.; Torres, L.; Martín-Duce, A.; Corrales, F.J.; Yang, H.; Prieto, J.; Lu, S.C.; Caballería, J.; Rodés, J.; et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J. Hepatol. 2000, 33, 907–914. [Google Scholar] [CrossRef]
- Kharbanda, K.K. Alcoholic liver disease and methionine metabolism. Semin. Liver Dis. 2009, 29, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.I.; Molina, M.; Odriozola, L.; Elortza, F.; Mato, J.M.; Sitek, B.; Zhang, P.; He, F.; Latasa, M.U.; Ávila, M.A.; et al. Prioritizing Popular Proteins in Liver Cancer: Remodelling One-Carbon Metabolism. J. Proteome Res. 2017, 16, 4506–4514. [Google Scholar] [CrossRef] [PubMed]
- Frau, M.; Tomasi, M.L.; Simile, M.M.; Demartis, M.I.; Salis, F.; Latte, G.; Calvisi, D.F.; Seddaiu, M.A.; Daino, L.M.E.; Feo, C.F.; et al. Role of transcriptional and posttranscriptional regulation of methionine adenosyltransferases in liver cancer progression. Hepatology 2012, 56, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jin, Y.; Yao, X.-H.; Fan, W.; Zhang, J.; Cao, Y.; Li, J. A novel mechanism of the M1–M2 methionine adenosyltransferase switch-mediated hepatocellular carcinoma metastasis. Mol. Carcinog. 2018, 57, 1201–1212. [Google Scholar] [CrossRef]
- Mato, J.M.; Luz Martínez-Chantar, M.; Lu, S.C. S-adenosylmethionine metabolism and liver disease. Ann. Hepatol. 2013, 12, 183–189. [Google Scholar] [CrossRef]
- Avila, M.A.; García-Trevijano, E.R.; Lu, S.C.; Corrales, F.J.; Mato, J.M. Methylthioadenosine. Int. J. Biochem. Cell Biol. 2004, 36, 2125–2130. [Google Scholar] [CrossRef]
- Bigaud, E.; Corrales, F.J. Methylthioadenosine (MTA) regulates liver cells proteome and methylproteome: Implications in liver biology and disease. Mol. Cell. Proteom. 2016, 15, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; E Geyer, P.; Albrechtsen, N.J.W.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsbøll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef] [PubMed]
- Kockmann, T.; Trachsel, C.; Panse, C.; Wahlander, A. Targeted proteomics coming of age–SRM, PRM and DIA performance evaluated from a core facility perspective. Proteomics 2016, 16, 2183–2192. [Google Scholar] [CrossRef]
- Sabidó, E.; Wu, Y.; Bautista, L.; Porstmann, T.; Chang, C.; Vitek, O.; Stoffel, M.; Aebersold, R. Targeted proteomics reveals strain-specific changes in the mouse insulin and central metabolic pathways after a sustained high-fat diet. Mol. Syst. Biol. 2013, 9, 681. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, L.; Paradela, A.; Corrales, F.J. Development of a Standardized MRM Method for the Quantification of One Carbon Metabolism Enzymes. In Methods in Molecular Biology; Humana: New York, NY, USA, 2022; Volume 2420, pp. 159–175. [Google Scholar]
- Whiteaker, J.R.; the Clinical Proteomic Tumor Analysis Consortium (CPTAC); Halusa, G.N.; Hoofnagle, A.N.; Sharma, V.; MacLean, B.; Yan, P.; A Wrobel, J.; Kennedy, J.J.; Mani, D.R.; et al. CPTAC Assay Portal: A repository of targeted proteomic assays. Nat. Methods 2014, 11, 703–704. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, L.; Sangro, B.; Ambao, V.; Granero, J.I.; Ramos-Fernández, A.; Paradela, A.; Corrales, F.J. Monitoring one-carbon metabolism by mass spectrometry to assess liver function and disease. J. Physiol. Biochem. 2022, 78, 229–243. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero, L.; Paradela, A.; Corrales, F.J. Targeted Proteomics for Monitoring One-Carbon Metabolism in Liver Diseases. Metabolites 2022, 12, 779. https://doi.org/10.3390/metabo12090779
Guerrero L, Paradela A, Corrales FJ. Targeted Proteomics for Monitoring One-Carbon Metabolism in Liver Diseases. Metabolites. 2022; 12(9):779. https://doi.org/10.3390/metabo12090779
Chicago/Turabian StyleGuerrero, Laura, Alberto Paradela, and Fernando J. Corrales. 2022. "Targeted Proteomics for Monitoring One-Carbon Metabolism in Liver Diseases" Metabolites 12, no. 9: 779. https://doi.org/10.3390/metabo12090779
APA StyleGuerrero, L., Paradela, A., & Corrales, F. J. (2022). Targeted Proteomics for Monitoring One-Carbon Metabolism in Liver Diseases. Metabolites, 12(9), 779. https://doi.org/10.3390/metabo12090779