


Genetic Predisposition to Hepatocellular Carcinoma



Abstract
:
1. Introduction
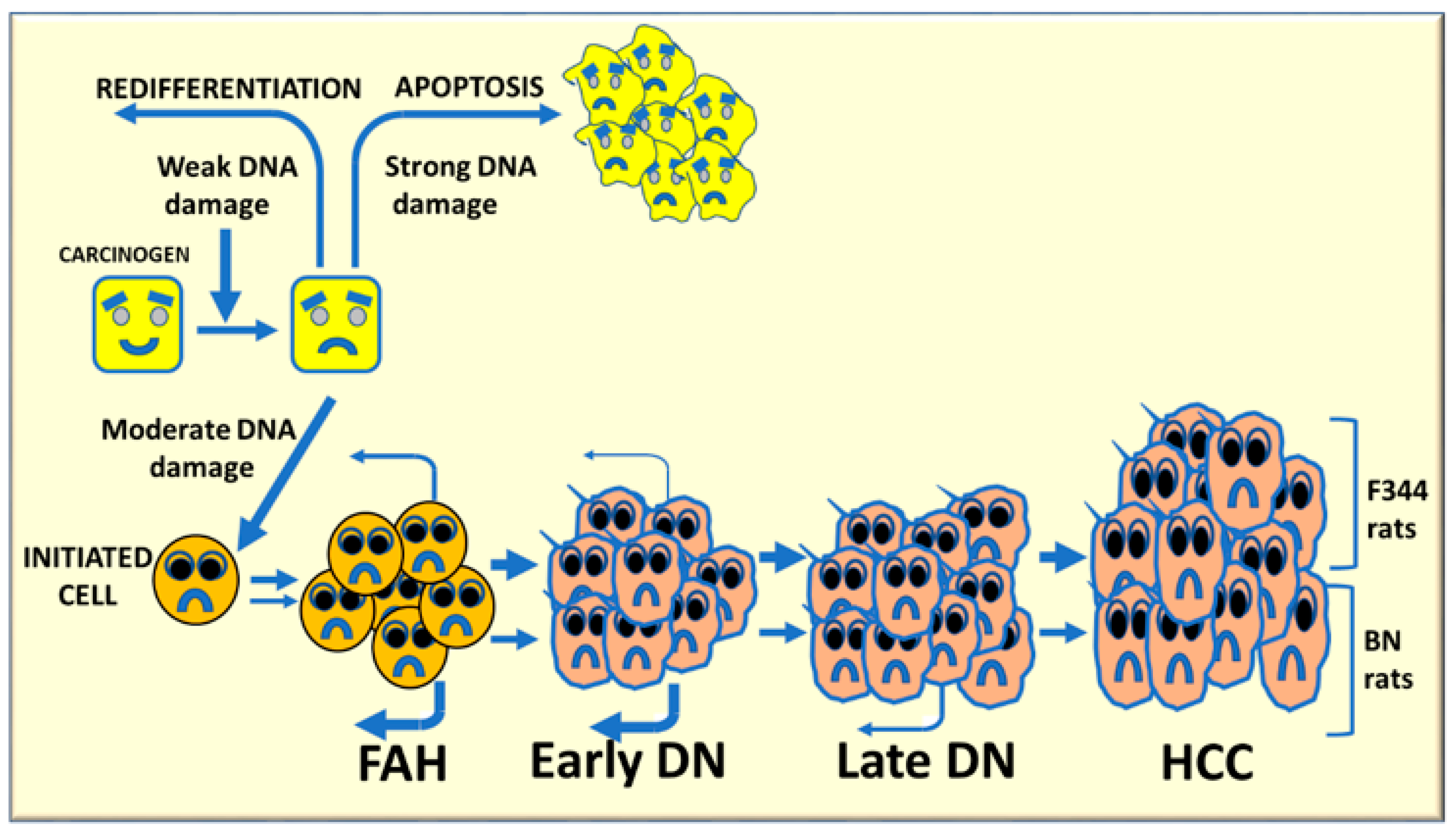
2. Molecular Mechanisms Determining the Susceptibility to Hepatocarcinogenesis
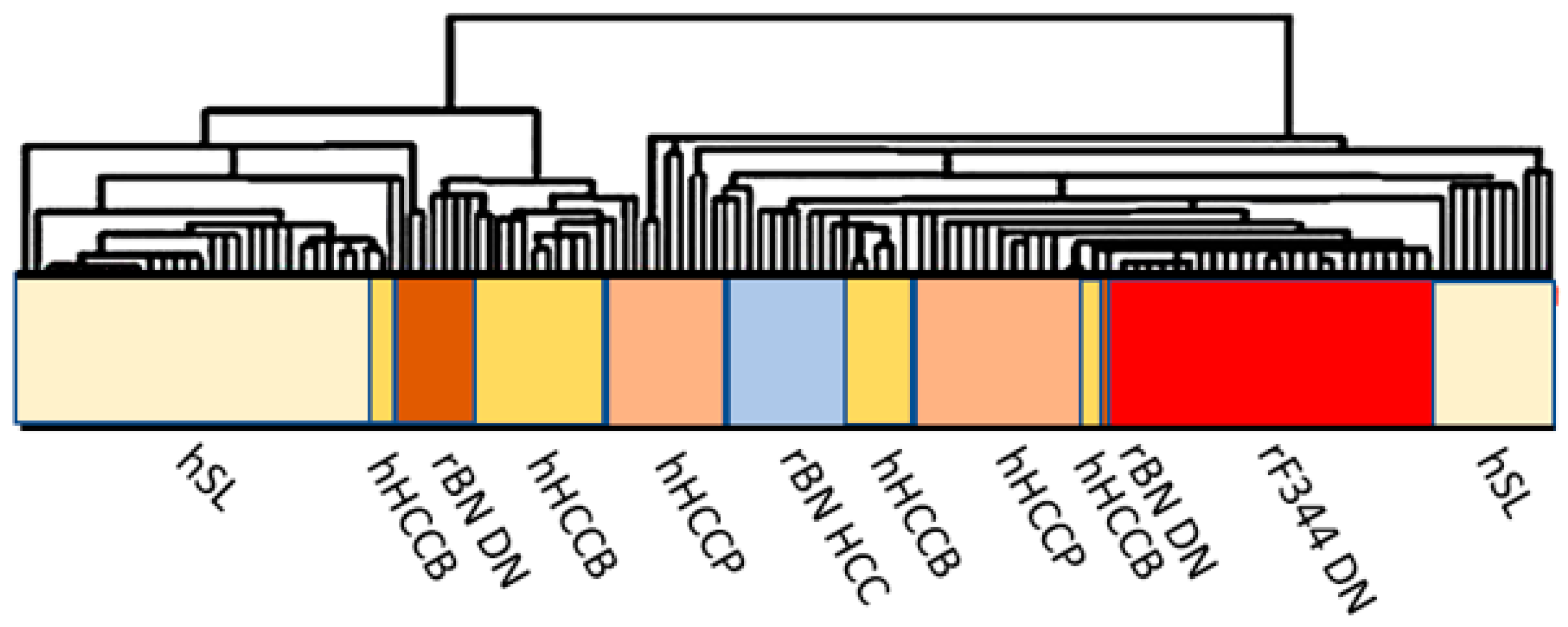
3. The Role of Genetic Mechanisms Regulating the Susceptibility to HCC
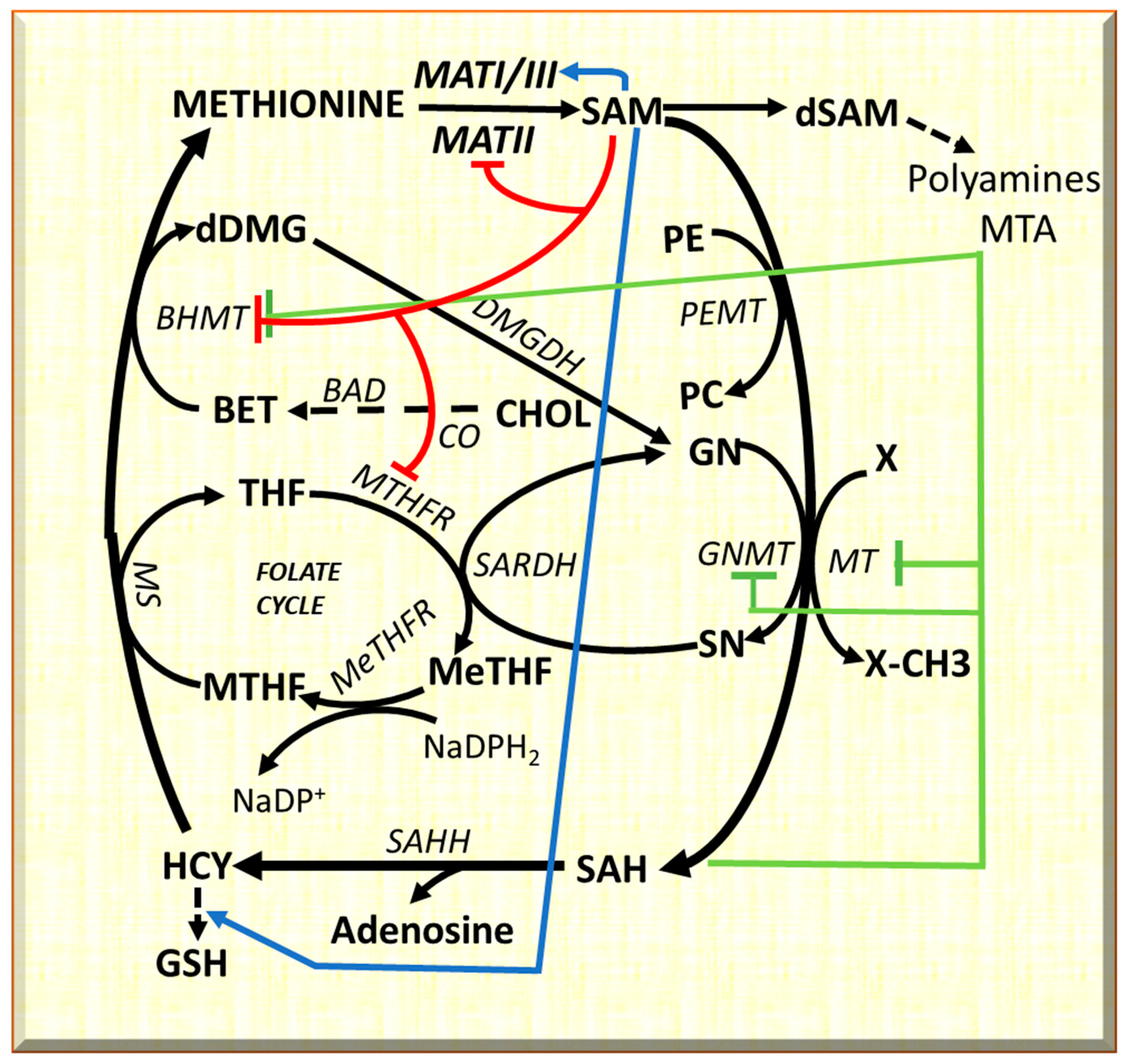
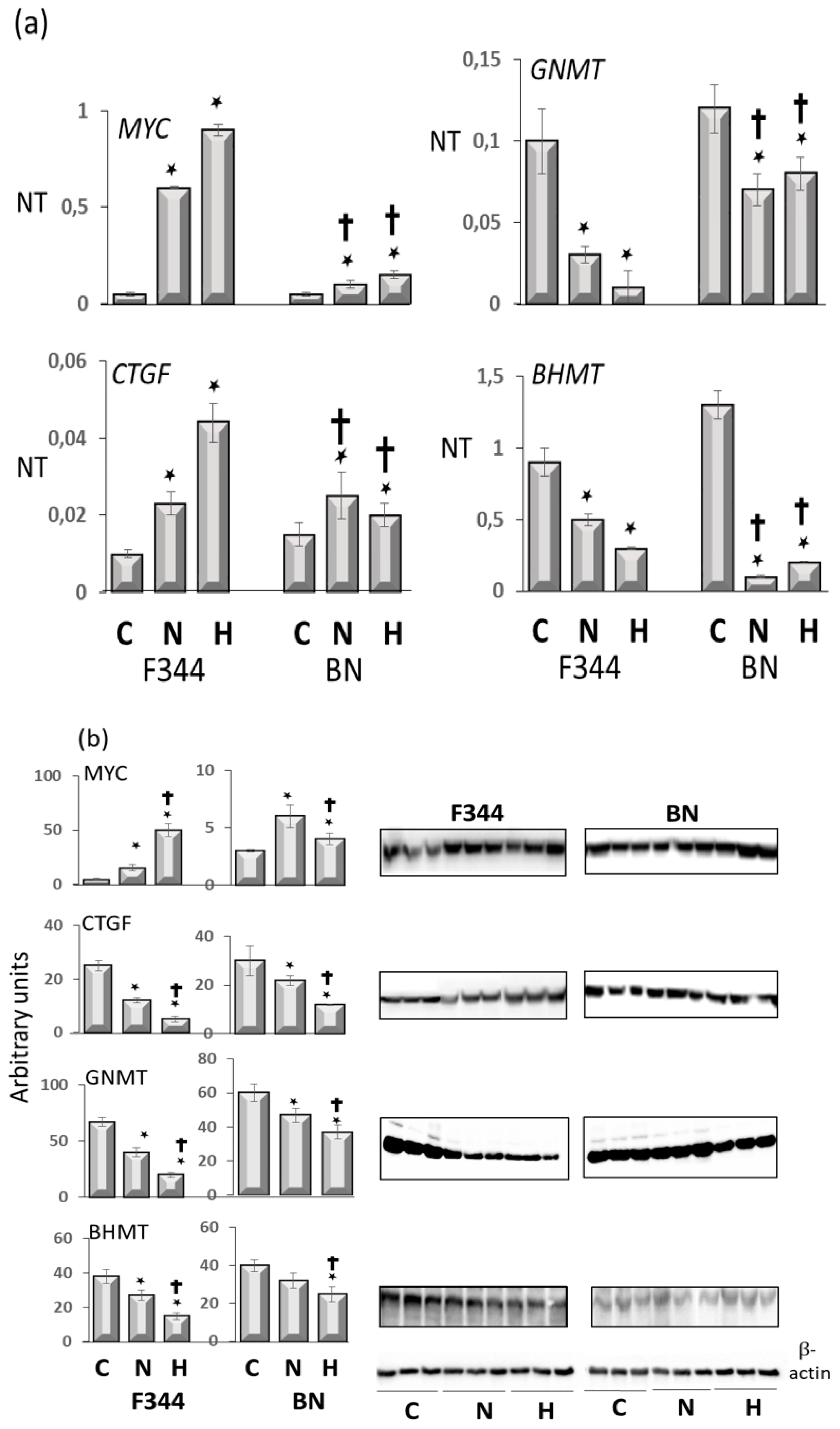
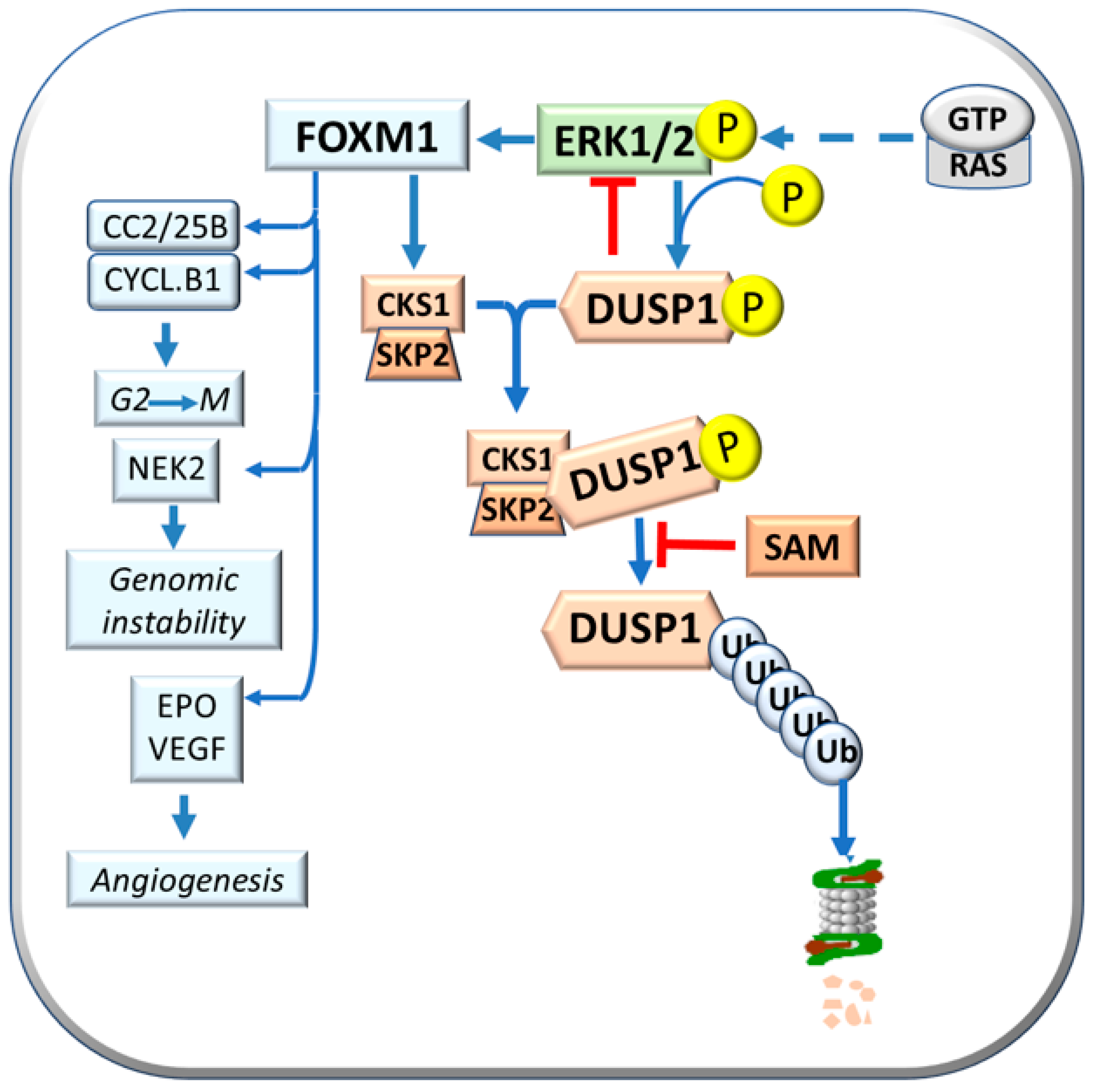
4. GNMT and the Genetic Predisposition to HCC
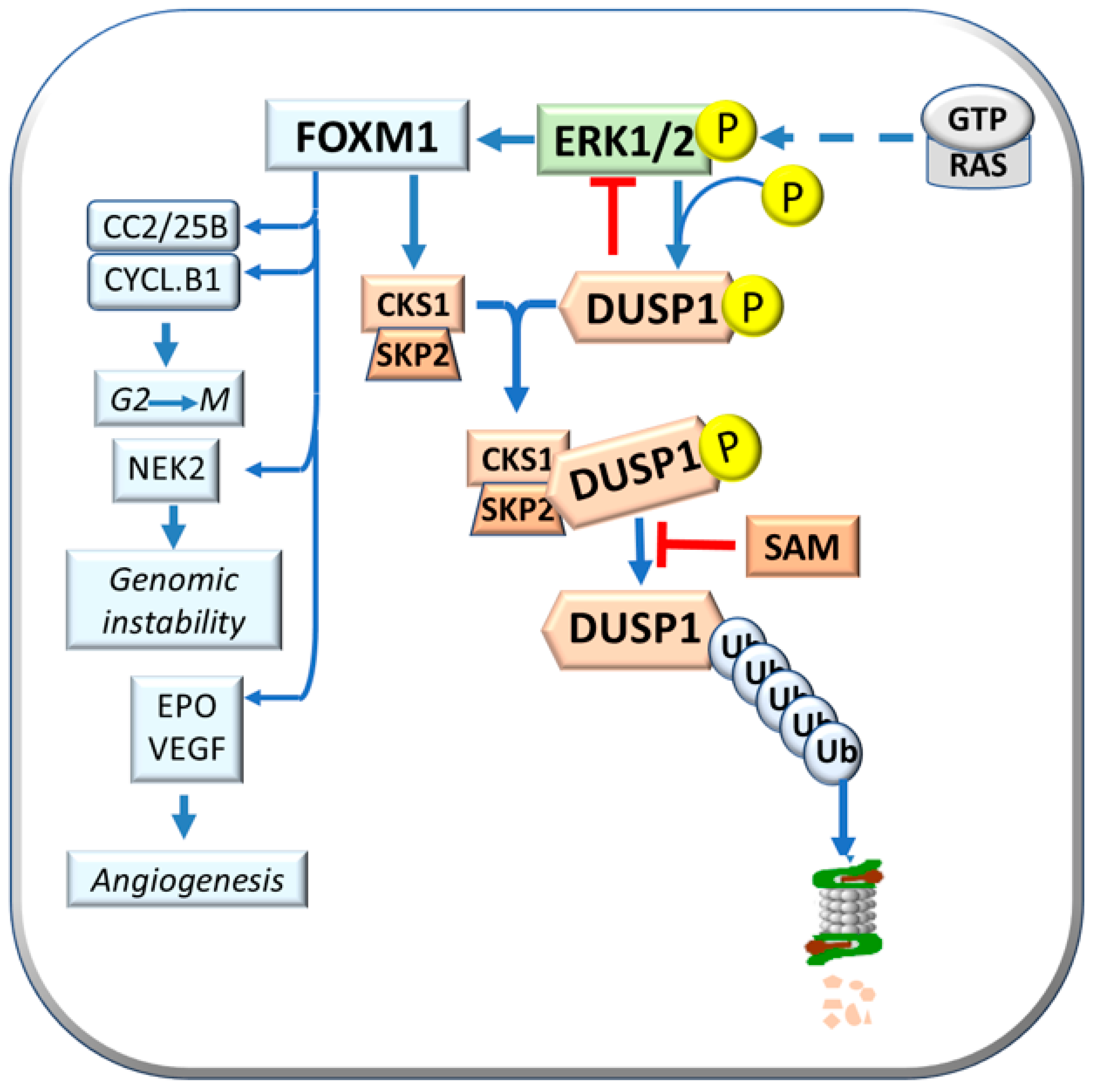
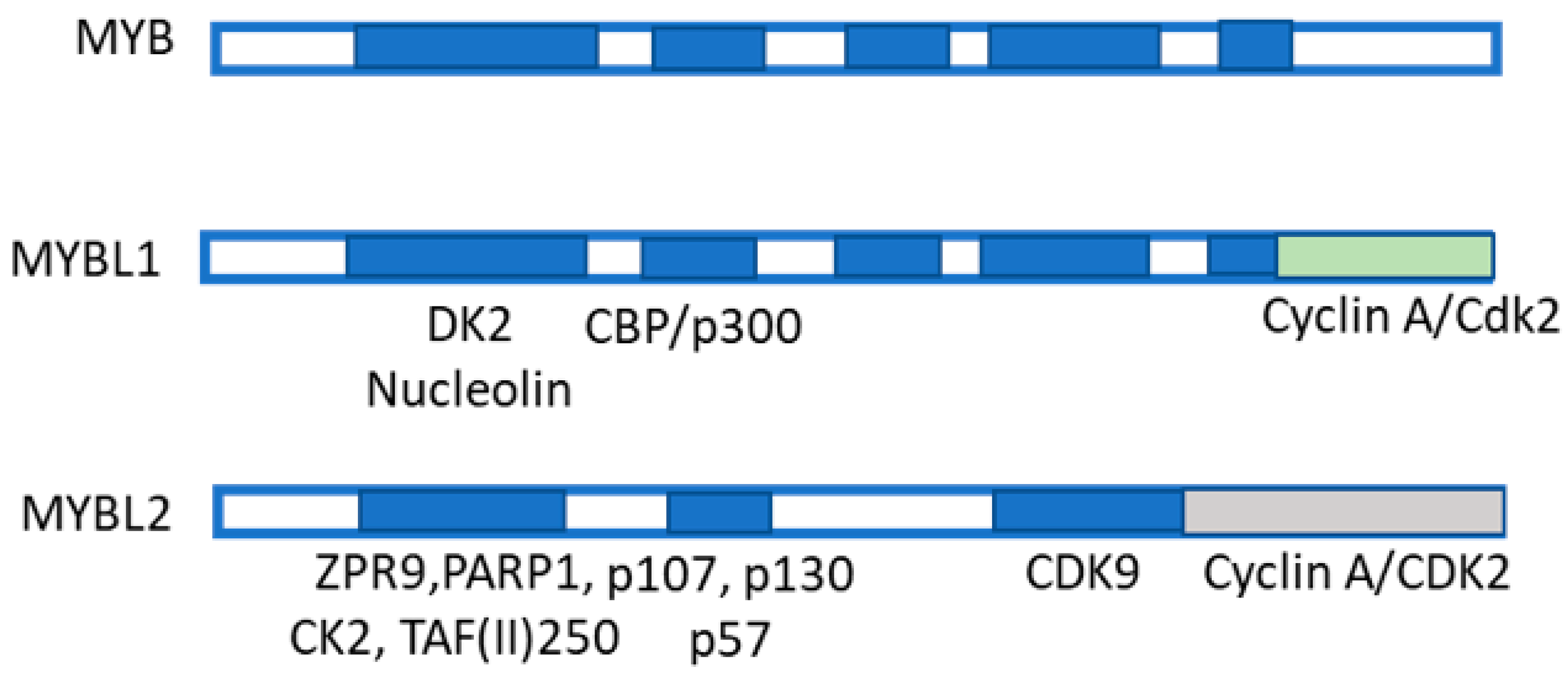
5. MYBL2 Gene and Liver Cancer
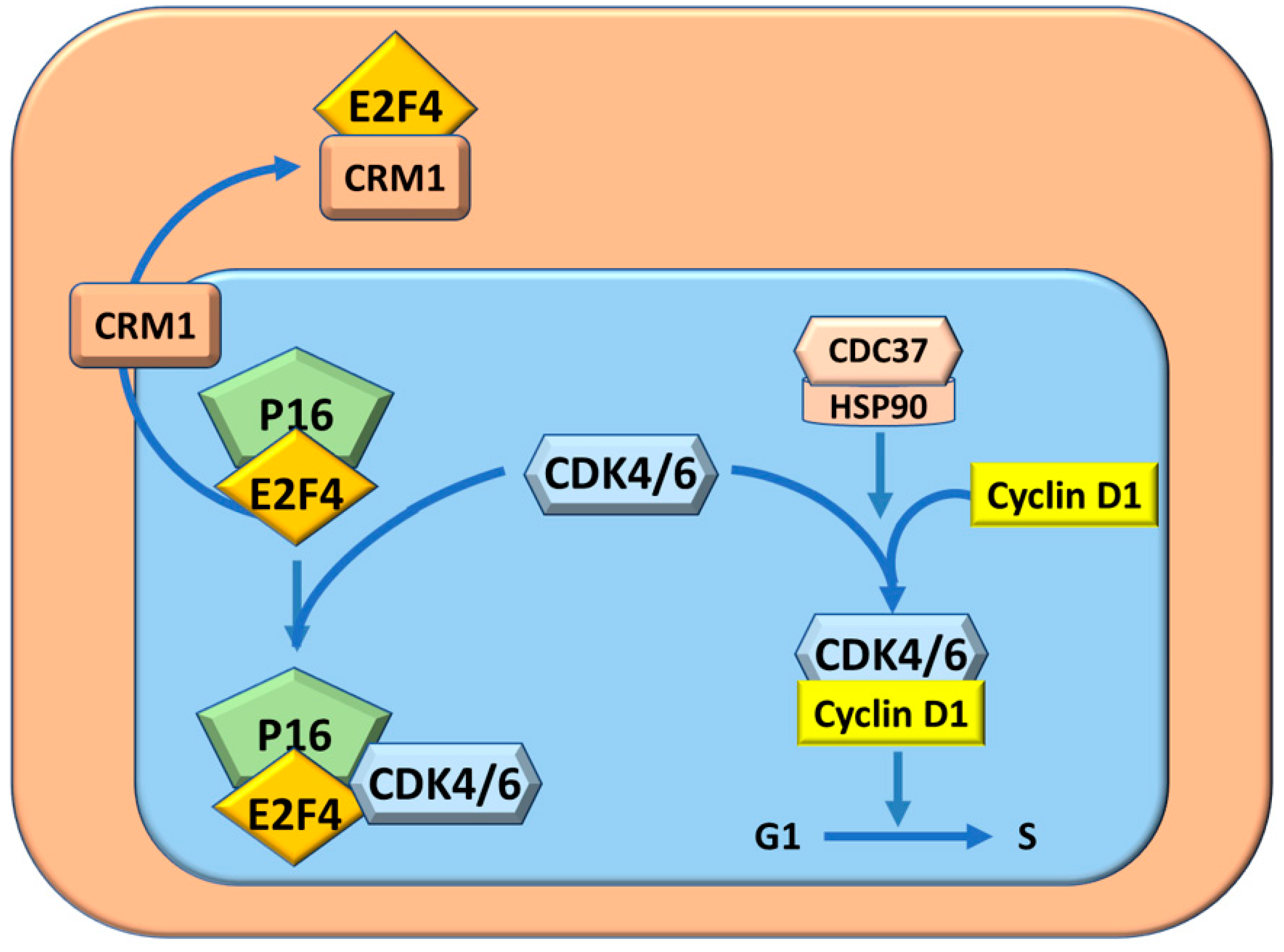
6. HCC Modifiers and Genetic Predisposition
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, L.; Zhou, T.; Zhang, Z.; Zeng, C. p53 Mutation at Serine 249 and Its Gain of Function Are Highly Related to Hepatocellular Carcinoma after Smoking Exposure. Public Health Genomics 2021, 24, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Yoshida, E.M.; Rathi, S.; Marquez, V.; Kim, P.; Erb, S.R.; Salh, B.S. Biomarkers for hepatocellular cancer. World J. Hepatol. 2020, 12, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Reboui, S.; Sou, S.; Bioulac-Sage, P.; Zucman-Rossi, J. Molecular pathogenesis of focal nodular hyperplasia and hepatocellular adenoma. J. Hepatol. 2008, 48, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Farber, E.; Sarma, D.S. Hepatocarcinogenesis: A dynamic cellular perspective. Lab. Invest. 1987, 56, 4–22. [Google Scholar]
- Feo, F.; Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Calvisi, D. Genetic alterations in liver carcinogenesis: Implications for new preventive and therapeutic strategies. Crit. Rev. Oncog. 2000, 11, 19–62. [Google Scholar] [CrossRef]
- Lee, J.S.; Chu, I.S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef]
- Dragani, T.A. Risk of HCC: Genetic heterogeneity and complex genetics. J. Hepatol. 2010, 52, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Leone, P.; Solimando, A.G.; Fasano, R.; Argentiero, A.; Malerba, E.; Buonavoglia, A.; Lupo, L.G.; De Valli, R.; Silvestris, N.; Racanelli, V. The Evolving Role of Immune Checkpoint Inhibitors in Hepatocellular Carcinoma Treatment. Vaccines 2021, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.; Ro, S.W.; Seo, S.H.; Jeon, Y.; Moon, H.; Kim, D.Y.; Kim, S.U. Genetically Engineered Mouse Models for Liver Cancer. Cancers 2019, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragani, T.A.; Manenti, G.; Della Porta, G. Genet susceptibility to murine hepatocarcinogenesis is associated with high growth rate of NDEA-initiated hepatocytes. J. Cancer Res. Clin. Oncol. 1987, 113, 223–229. [Google Scholar] [CrossRef]
- Manenti, G.; Binelli, G.; Gariboldi, M.; Canzian, F.; De Gregorio, L.; Falvella, F.S.; Dragani, T.A.; Pierotti, M.A. Multiple loci affect genetic predisposition to hepatocarcinogenesis in mice. Genomics 1994, 23, 118–124. [Google Scholar] [CrossRef]
- Kaposi-Novak, P.; Libbrecht, L.; Woo, H.G.; Lee, Y.H.; Sears, N.C.; Coulouarn, C.; Conner, E.A.; Factor, V.M.; Roskams, M.T.; Thorgeirsson, S.S. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009, 69, 2775–2782. [Google Scholar] [CrossRef] [Green Version]
- Pascale, R.M.; Simile, M.M.; Peitta, G.; Seddaiu, M.A.; Feo, F.; Calvisi, D.F. Experimental Models to Define the Genetic Predisposition to Liver Cancer. Cancers 2019, 11, 1450. [Google Scholar] [CrossRef] [Green Version]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Garcea, R.; Daino, L.; Pascale, R.; Simile, M.M.; Puddu, M.; Frassetto, S.; Cozzolino, P.; Seddaiu, M.A.; Gaspa, L.; Feo, F. Inhibition of promotion and persistent nodule growth by S-adenosyl-L-methionine in rat liver carcinogenesis: Role of remodeling and apoptosis. Cancer Res. 1989, 49, 1850–1856. [Google Scholar]
- Frau, M.; Biasi, F.; Feo, F.; Pascale, R.M. Prognostic markers and putative therapeutic targets for hepatocellular carcinoma. Mol. Aspects Med. 2010, 31, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.C.; Gamble, M.V.; Hall, M.N.; Nijhout, H.F. Mathematical analysis of the regulation of competing methyltransferases. BMC Syst. Biol. 2015, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.J.; Wagner, C. Glycine N-methyltransferase is a folate binding protein of rat liver cytosol. Proc Natl. Acad. Sci. USA 1984, 81, 3631–3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.C.; Wu, M.T.; Lin, Y.J.; Tang, F.Y.; Ko, H.A.; Chiang, E.P. Regulation of Folate-Mediated One-Carbon metabolism by Glycine-N-Methyltransferase (GNMT) and Methylenetetrahydrofolate Reductase (MTHFR). J. Nutr. Sci. Vitaminol. 2015, 61, S148–S150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.T.; Schalinske, K.L. New insights into the regulation of methyl group and homocysteine metabolism. J. Nutr. 2007, 137, 311–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutzbach, C.; Stokstad, E.L.R. Feedback inhibition in methylenetetrahydrofolate reductase in rat liver by S-adenosylmethionine. Biochim. Biophys. Acta 1967, 139, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Jencks, D.A.; Matthews, R.G. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine. Effects of adenosylmethionine and NADPH on the equilibrium between active and inactive forms of the enzyme and on the kinetics of approach to equilibrium. J. Biol. Chem. 1987, 262, 2493. [Google Scholar] [CrossRef]
- Wagner, C.; Briggs, W.T.; Cook, R.J. Inhibition of glycine N-methyltransferase activity by folate derivatives: Implications for regulation of methyl group metabolism. Biochem. Biophys. Res. Commun. 1985, 127, 746–752. [Google Scholar] [CrossRef]
- Ou, X.; Yang, H.; Ramani, K.; Ara, A.I.; Chen, H.; Mato, J.M.; Lu, S.C. Inhibition of human betaine-homocysteine methyltransferase expression by S-adenosylmethionine and methylthioadenosine. Biochem. J. 2007, 401, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Calvisi, D.F.; Pinna, F.; Pellegrino, R.; Sanna, V.; Sini, M.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Frau, M.; Tomasi, M.L.; et al. Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int. J. Cancer 2008, 123, 2057–2064. [Google Scholar] [CrossRef]
- Frau, M.; Simile, M.M.; Tomasi, M.L.; Demartis, M.I.; Daino, L.; Brozzetti, S.; Feo, C.F.; Massarelli, G.; Solinas, G.; Feo, F.; et al. An expression signature of phenotypic resistance to hepatocellular carcinoma identified by cross-species gene expression analysis. Cell. Oncol. 2012, 35, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasi, M.L.; Ramani, K.; Lopitz-Otsoa, F.; Rodríguez, M.S.; Li, T.W.; Ko, K.; Yang, H.; Bardag-Gorce, F.; Iglesias-Ara, A.; Feo, F.; et al. S-adenosylmethionine regulates dual-specificity mitogen-activated protein kinase phosphatase expression in mouse and human hepatocytes. Hepatology 2010, 51, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Pinna, F.; Meloni, F.; Ladu, S.; Pellegrino, R.; Sini, L.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Virdis, P.; et al. Dual specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res. 2008, 68, 4192–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvisi, D.F.; Pinna, F.; Ladu, S.; Pellegrino, R.; Simile, M.M.; Frau, M.; De Miglio, M.R.; Tomasi, M.L.; Sanna, V.; Muroni, M.R.; et al. Forkhead box M1B is a determinant of rat susceptibility to hepatocarcinogenesis and sustains ERK activity in human HCC. Gut 2009, 58, 679–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feo, F.; Frau, M.; Pascale, R.M. Interaction of major genes predisposing to hepatocellular carcinoma with genes encoding signal transduction pathways influences tumor phenotype and prognosis. World. J. Gastroenterol. 2008, 14, 6601–6615. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Krupczak-Hollis, K.; Tan, Y.; Dennewitz, M.B.; Adami, G.R.; Costa, R.H. Increased hepatic Forkhead Box M1B (FoxM1B) levels in old-aged mice stimulated liver regeneration through diminished p27Kip1 protein levels and increased Cdc25B expression. J. Biol. Chem. 2002, 277, 44310–44316. [Google Scholar] [CrossRef] [Green Version]
- Pascale, R.M.; Simile, M.M.; Calvisi, D.F.; Frau, M.; Muroni, M.R.; Seddaiu, M.A.; Daino, M.D.; Muntoni, M.R.; De Miglio, S.S.; Thorgeirsson, F.; et al. Role of HSP90, CDC37, and CRM1 as modulators of P16(INK4A) activity in rat liver carcinogenesis and human liver cancer. Hepatology 2005, 42, 1310–1319. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Calvisi, D.F.; Asara, G.; Casabona, D.; Frau, M.; Seddaiu, M.A.; Feo, F. Cell cycle deregulation in liver lesions of rats with and without genetic predisposition to hepatocarcinogenesis. Hepatology 2002, 35, 1341–1350. [Google Scholar] [CrossRef]
- Hunter, T.; Poon, R.Y. Cdc37: A protein kinase chaperone? Trends Cell Biol. 1997, 7, 157–161. [Google Scholar] [CrossRef]
- Stepanova, L.; Leng, X.; Parker, S.B.; Harper, J.W. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996, 10, 1491–1502. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Liu, T.; Wang, K.; Wang, X.; Xu, S.; He, D.; Zeng, J. Transcriptional regulation of FoxM1 by HIF-1α mediates hypoxia-induced EMT in prostate cancer. Oncol. Rep. 2019, 42, 1307–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Mo, P.; Huang, W.; Zhang, L.; Wang, Y.; Zhu, H.; Tian, D.; Liu, J.; Chen, Z.; Zhang, Y.; et al. The TNF-a/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis 2012, 33, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
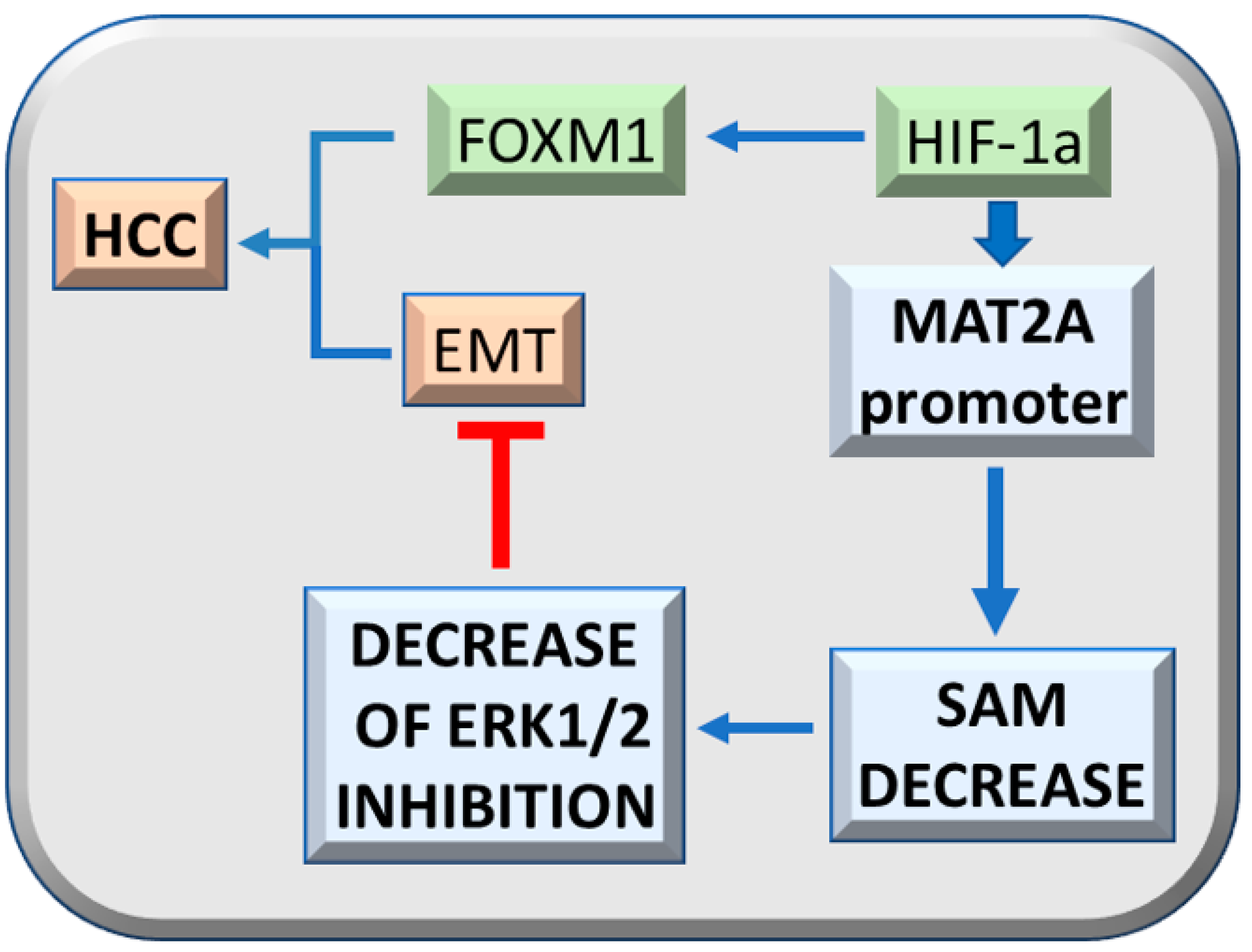
- Liu, Q.; Liu, L.; Zhao, Y.; Zhang, J.; Wang, D.; Chen, J.; He, Y.; Wu, J.; Zhang, Z.; Liu, Z. Hypoxia induces genomic DNA demethylation through the activation of HIF-1a and transcriptional upregulation of MAT2A in hepatoma cells. Mol. Cancer Ther. 2011, 10, 1113–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, R.; Bresnick, E. Glycine N-methyltransferase is an example of functional diversity. Role as a polycyclic aromatic hydrocarbon-binding receptor. J. Biol. Chem. 1997, 272, 21221–21226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simile, M.M.; Cigliano, A.; Paliogiannis, P.; Daino, L.; Manetti, R.; Feo, C.F.; Calvisi, D.F.; Feo, F.; Pascale, R.M. Nuclear localization dictates hepatocarcinogenesis suppression by glycine N-methyltransferase. Transl. Oncol. 2022, 15, 101239. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, C.; Wang, B.; Yu, B.; Ge, I.; Shi, X. Quercetin suppresses the chymotrypsin-like activity of proteasome via inhibition of MEK1/ERK1/2 signaling pathway in hepatocellular carcinoma HepG2 cells. Can. J. Physiol. Pharmacol. 2018, 96, 521–526. [Google Scholar] [CrossRef]
- El-Hamoly, T.; Hajnády, Z.; Nagy-Pénzes, M.; Bakondi, E.; Regdon, Z.; Demény, M.A.; Kovács, K.; Hegedűs, C.; Abd El-Rahman, S.S.; Szabó, É.; et al. Poly(ADP-Ribose) Polymerase 1 Promotes Inflammation and Fibrosis in a Mouse Model of Chronic Pancreatitis. Int. J. Mol. Sci. 2021, 22, 3593. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Cao, Z.; Horvth, B.; Park, O.; Wang, H.; Erdelyi, K.; Holovac, E.; Wang, Y.; Liaudet, L.; et al. Poly (ADP-ribose) polymerase-1 is a key mediator of liver inflammation and fibrosis. Hepatology 2014, 59, 1998–2009. [Google Scholar] [CrossRef] [Green Version]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2020, 8, 287–297. [Google Scholar] [CrossRef]
- Li, C.H.; Yen, C.H.; Chen, Y.F.; Lee, K.J.; Fang, C.C.; Zhang, X.; Lai, C.C.; Huang, S.F.; Lin, H.K.; Arthur Chen, Y.M. Characterization of the GNMT-HectH9-PREX2 tripartite relationship in the pathogenesis of hepatocellular carcinoma. Int. J. Cancer. 2017, 140, 2284–2297. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dong, W. Oxidative stress and bronchopulmonary dysplasia. Gene 2018, 678, 177–183. [Google Scholar] [CrossRef]
- Perrone, S.; Tataranno, M.L.; Buonocore, G. Oxidative stress and bronchopulmonary dysplasia. J. Clin. Neonatol. 2012, 1, 109–114. [Google Scholar] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mense, S.M.; Barrows, D.; Hodakoski, C.; Steinbach, N.; Schoenfeld, D.; Su, W.; Hopkins, B.D.; Su, T.; Fine, B.; Hibshoosh, H.; et al. PTEN inhibits PREX2-catalyzed activation of RAC1 to restrain tumor cell invasion. Sci. Signal. 2015, 8, ra32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lissanu Deribe, Y. Interplay between PREX2 mutations and the PI3K pathway and its effect on epigenetic regulation of gene expression in NRAS-mutant melanoma. Small GTPases 2016, 7, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Kaur, S.; Hu, Y. Lab review: Molecular dissection of the signal transduction pathways associated with PTEN deletion-induced optic nerve regeneration. Restor. Neurol. Neurosci. 2019, 37, 545–552. [Google Scholar] [CrossRef]
- Liao, Y.J.; Lee, T.S.; Twu, Y.C.; Hsu, S.M.; Yang, C.P.; Wang, C.K.; Liang, Y.C.; Chen, Y.A. Glycine N-methyltransferase deficiency in female mice impairs insulin signaling and promotes gluconeogenesis by modulating the PI3K/Akt pathway in the liver. J. Biomed. Sci. 2016, 23, 69. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.H.; Hung, J.H.; Ueng, Y.F.; Liu, S.P.; Chen, S.Y.; Liu, H.H.; Chou, T.Y.; Tsai, T.F.; Darbha, R.; Hsieh, L.L.; et al. Glycine N-methyltransferase affects the metabolism of aflatoxin B1 and blocks its carcinogenic effect. Toxicol. Appl. Pharmacol. 2009, 235, 296–304. [Google Scholar] [CrossRef]
- Yan, H.; Huang, Z.Z.; Wang, J.; Lu, S.C. The role of c-Myb and Sp1 in the up-regulation of methionine adenosyltransferase 2A gene expression in human hepatocellular carcinoma. FASEB J. 2001, 15, 1507–1516. [Google Scholar]
- Liu, M.; Du, Q.; Mao, G.; Dai, N.; Zhang, F. MYB proto-oncogene like 2 promotes hepatocellular carcinoma growth and glycolysis via binding to the Optic atrophy 3 promoter and activating its expression. Bioengineered 2022, 13, 5344–5356. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Zhou, S.; Wang, S.; Zheng, K.; Xu, D.; Liu, Y.; Wang, X.; Wang, X.; Yan, H.; et al. Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Mol. Cancer 2015, 7, 14–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
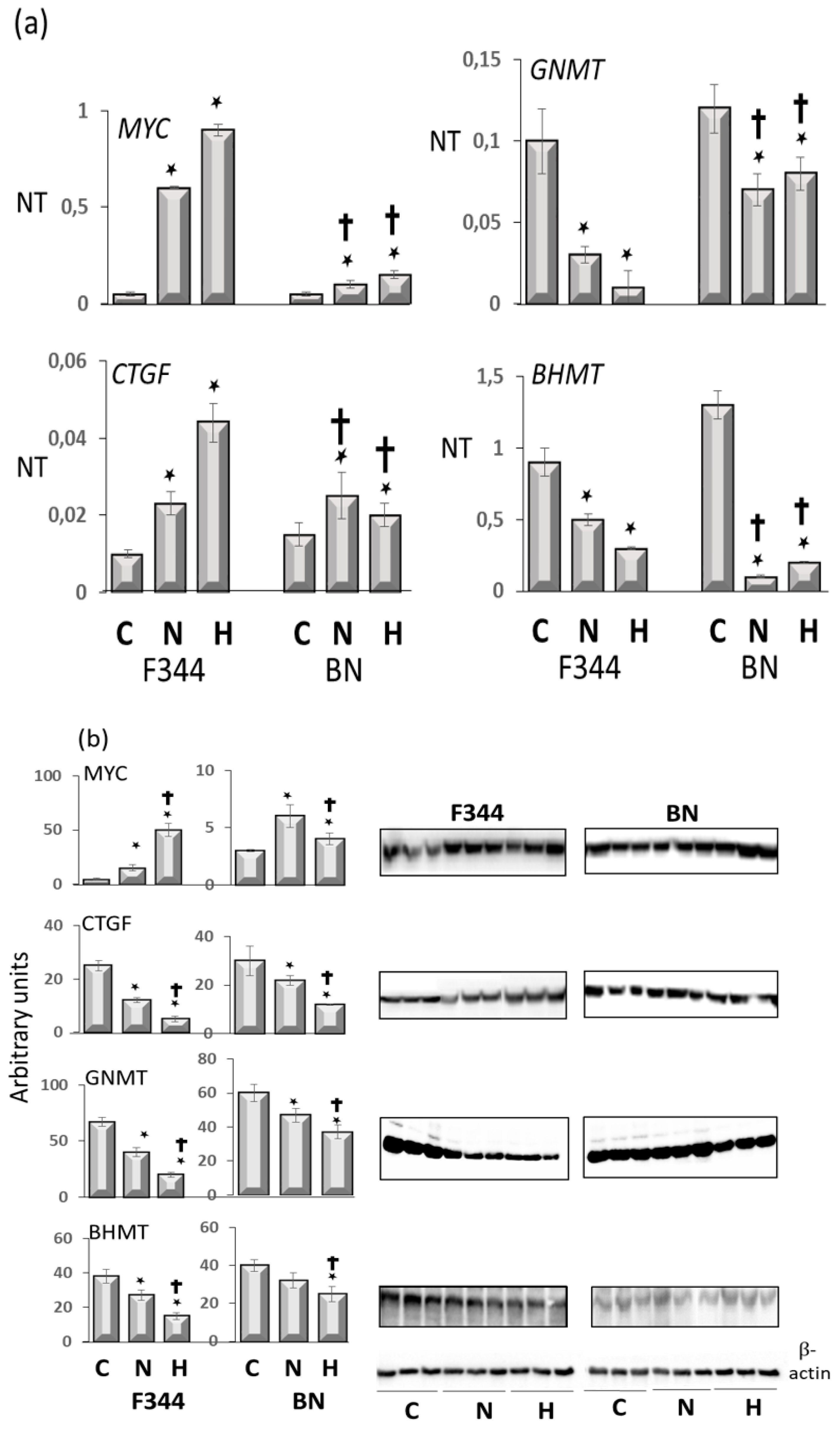
- Frau, M.; Ladu, S.; Calvisi, D.F.; Simile, M.M.; Bonelli, P.; Daino, L.; Tomasi, M.L.; Seddaiu, M.A.; Feo, F.; Pascale, R.M. Mybl2 expression is under genetic control and contributes to determine a hepatocellular carcinoma susceptible phenotype. J. Hepatol. 2011, 55, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Cheng, W.; Huang, D.; Wei, A. High MYBL2 expression and transcription regulatory activity is associated with poor overall survival in patients with hepatocellular carcinoma. Curr. Res. Transl. Med. 2018, 66, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Solt, D.B.; Medline, A.; Farber, E. Rapid emergence of carcinogen induced hyperplastic lesions in a new model of sequential analysis of liver carcinogenesis. Am. J. Pathol. 1997, 88, 595–618. [Google Scholar]
- Enomoto, K.; Farber, E. Kinetics of phenotypic maturation of remodeling of hyperplastic nodules during liver carcinogenesis. Cancer Res. 1982, 42, 2330–2335. [Google Scholar]
- Wood, G.A.; Sarma, D.S.R.; Archer, M.C. Resistance to the promotion of glutathione S-transferase 7-7-positive liver lesions in Copenhagen rats. Carcinogenesis 1999, 20, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Wood, G.A.; Korkola, J.E.; Lee, V.M.; Sarma, D.S.R.; Archer, M.C. Resistance of Copenhagen rats to chemical induction of glutathione S-transferase 7-7-positive liver foci. Carcinogenesis 1997, 18, 1745–1750. [Google Scholar] [CrossRef] [Green Version]
- Wood, G.A.; Sarma, D.S.R.; Archer, M.C. Inheritance of resistance to promotion of preneoplastic liver lesions in Copenhagen rats. Exp. Biol. Med. 2001, 226, 831–835. [Google Scholar] [CrossRef]
- Mazzantini, R.P.; de Conti, A.; Moreno, F.S. Persistent and remodeling hepatic preneoplastic lesions present differences in cell proliferation and apoptosis, as well as in p53, Bcl-2 and NF-kappaB pathways. J. Cell Biochem. 2008, 103, 538–546. [Google Scholar] [CrossRef]
- Frau, M.; Tomasi, M.L.; Simile, M.M.; Demartis, M.I.; Salis, F.; Latte, G.; Calvisi, D.F.; Seddaiu, M.A.; Daino, L.; Feo, C.F.; et al. Role of transcriptional and posttranscriptional regulation of methionine adenosyltransferases in liver cancer progression. Hepatology 2012, 56, 165–175. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Muroni, M.R.; Gaspa, L.; Dragani, T.A.; Feo, F. The BN rat strain carries dominant hepatocarcinogen resistance loci. Carcinogenesis 1996, 17, 1765–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feo, F.; De Miglio, M.R.; Simile, M.M.; Muroni, M.R.; Calvisi, D.F.; Frau, M.; Pascale, R.M. Hepatocellular carcinoma as a complex polygenic disease. Interpretative analysis of recent developments on genetic predisposition. Biochim. Biopjhys. Acta. 2006, 1765, 126–147. [Google Scholar]
- Feo, F.; Frau, M.; Tomasi, M.L.; Brozzetti, S.; Pascale, R.M. Genetic and epigenetic control of molecular alterations in hepatocellular carcinoma. Exp. Biol. Med. 2009, 234, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Rego de Carvalho, L.; Borrego, A.; Jensen, J.R.; Koury Cabrera, W.A.; Marques Santos, A.; Ribeiro, O.R.; Starobinas, S.; De Franco, M.; Dragani, T.A.; Manenti, G.; et al. Genetic Predisposition to Hepatocarcinogenesis in Inbred and Outbred Mouse Lines Selected for High or Low Inflammatory Response. Immunol. Res. 2019, 5298792. [Google Scholar] [CrossRef]
- Liu, H.; Chen, S.; Liu, M.; Nie, H.; Lu, H. Comorbid Chronic Diseases are Strongly Correlated with Disease Severity among COVID-19 Patients: A Systematic Review and Meta-Analysis. Aging Dis. 2020, 11, 668–678. [Google Scholar] [CrossRef]
- Chan, S.L.; Kudo, M. Impacts of COVID-19 on Liver Cancers: During and after the Pandemic. Liver Cancer 2020, 9, 491–502. [Google Scholar] [CrossRef]
- Li, Y.; Lu, L.; Tu, J.; Zhang, J.; Xiong, T.; Fan, W.; Wang, J.; Li, M.; Chen, Y.; Steggerda, J.; et al. Reciprocal Regulation Between Forkhead Box M1/NF-kB and Methionine Adenosyltransferase 1A Drives Liver Cancer. Hepatology 2020, 72, 1682–1700. [Google Scholar] [CrossRef]
- De Matteis, S.; Ragusa, A.; Marisi, G.; De Domenico, S.; Casadei Gardini, A.; Bonafè, M.; Giudetti, A.M. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxid. Med. Cell Longev. 2018, 2018, 7512159. [Google Scholar] [CrossRef] [Green Version]
- Martínez-López, N.; Varela-Rey, M.; Fernández-Ramos, D.; Woodhoo, A.; Vázquez-Chantada, M.; Embade, N.; Espinosa-Hevia, L.; Bustamante, F.J.; Parada, L.A.; Rodriguez, M.S.; et al. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology 2010, 52, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.; Feo, F. The Warburg Effect 97 Years after Its Discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENES | ROLE IN HCC |
|---|---|
| MAT I/III (Adult liver) | Synthesis of SAM |
| MAT II (widely distributed) | Synthesis of SAM |
| PEMT/GNMT | Synthesis of SAH |
| SAHH | Synthesis of homocysteine |
| FOXM1 | Contribution to DUSP1 proteasomal degradation |
| HIF-1 alpha | Activation of FOXM1 |
| MYC, CTGF | Overexpressed in F-344 rat strain |
| PCNA | Overexpressed in F-344 rat strain |
| GNMT | Overexpressed in BN rat strain |
| BHMT | Overexpressed in BN rat strain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascale, R.M.; Calvisi, D.F.; Feo, F.; Simile, M.M. Genetic Predisposition to Hepatocellular Carcinoma. Metabolites 2023, 13, 35. https://doi.org/10.3390/metabo13010035
Pascale RM, Calvisi DF, Feo F, Simile MM. Genetic Predisposition to Hepatocellular Carcinoma. Metabolites. 2023; 13(1):35. https://doi.org/10.3390/metabo13010035
Chicago/Turabian StylePascale, Rosa M., Diego F. Calvisi, Francesco Feo, and Maria M. Simile. 2023. "Genetic Predisposition to Hepatocellular Carcinoma" Metabolites 13, no. 1: 35. https://doi.org/10.3390/metabo13010035
APA StylePascale, R. M., Calvisi, D. F., Feo, F., & Simile, M. M. (2023). Genetic Predisposition to Hepatocellular Carcinoma. Metabolites, 13(1), 35. https://doi.org/10.3390/metabo13010035