

Metabolic Analysis of DFO-Resistant Huh7 Cells and Identification of Targets for Combination Therapy
 , ,
, ,  and
and
Abstract
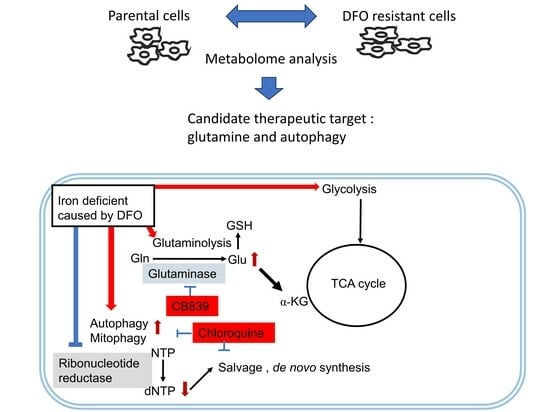
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Metabolome Analysis
2.3. Pathway Analysis with Ingenuity Pathways Analysis (IPA)
2.4. Statistical Analysis
3. Results
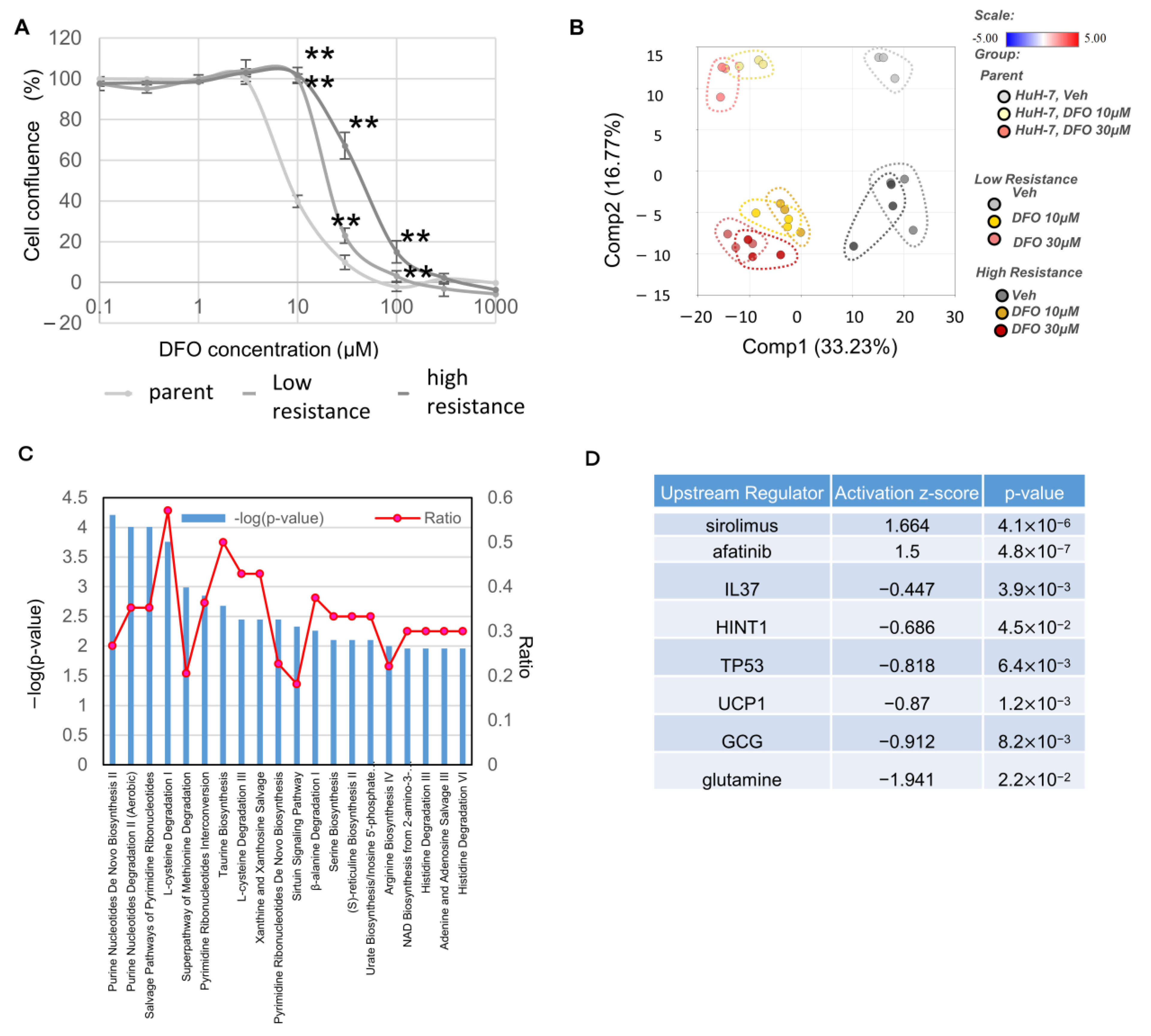
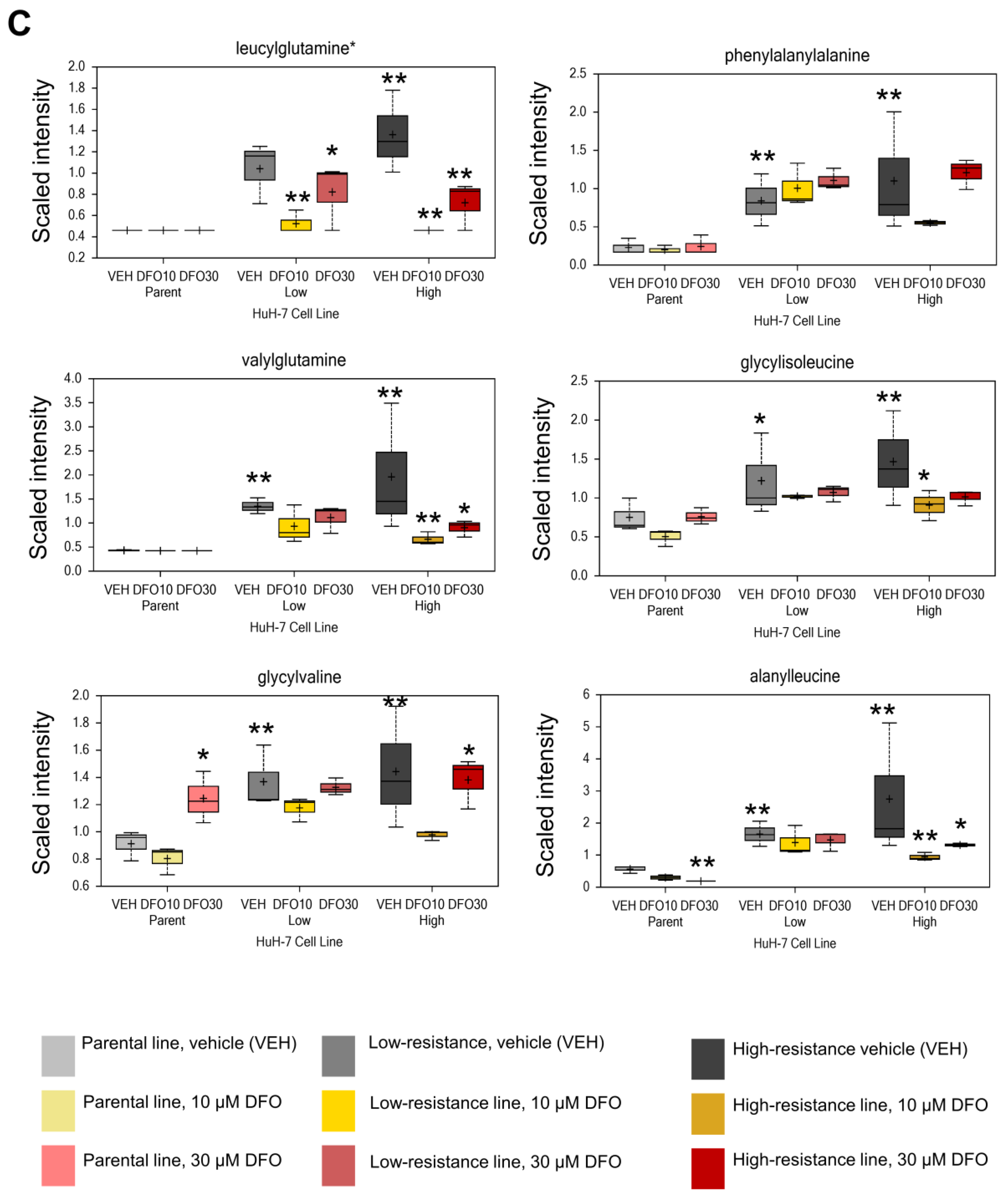
3.1. Enhanced Glycolysis and Salvage Cycle, Altered Glutamine Metabolism, and Accumulation of Dipeptides Occur in DFO-Resistant Huh7 Cells
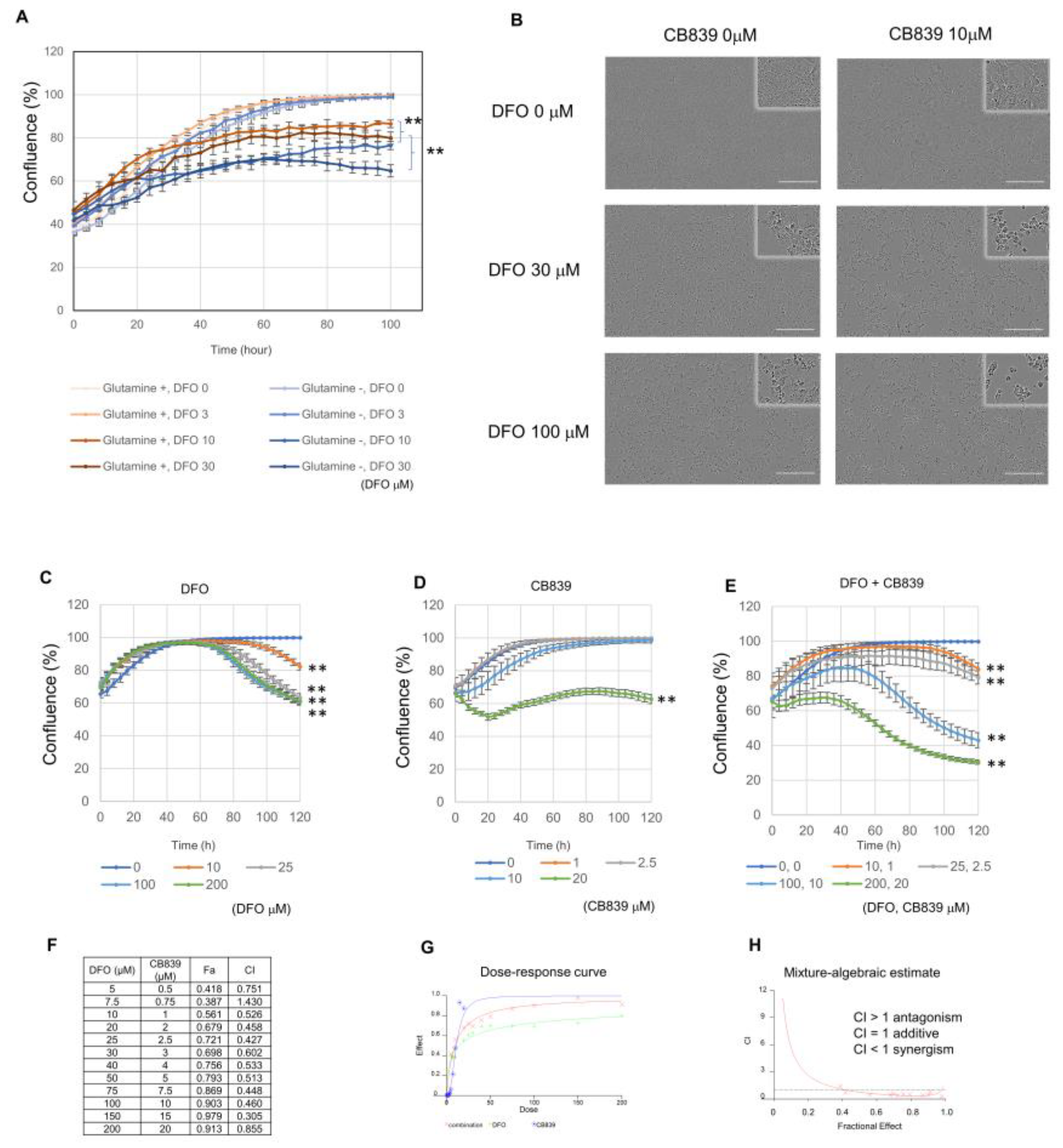
3.2. DFO and GLS Inhibitor Exhibit Synergistic Effects in Huh7 Cells
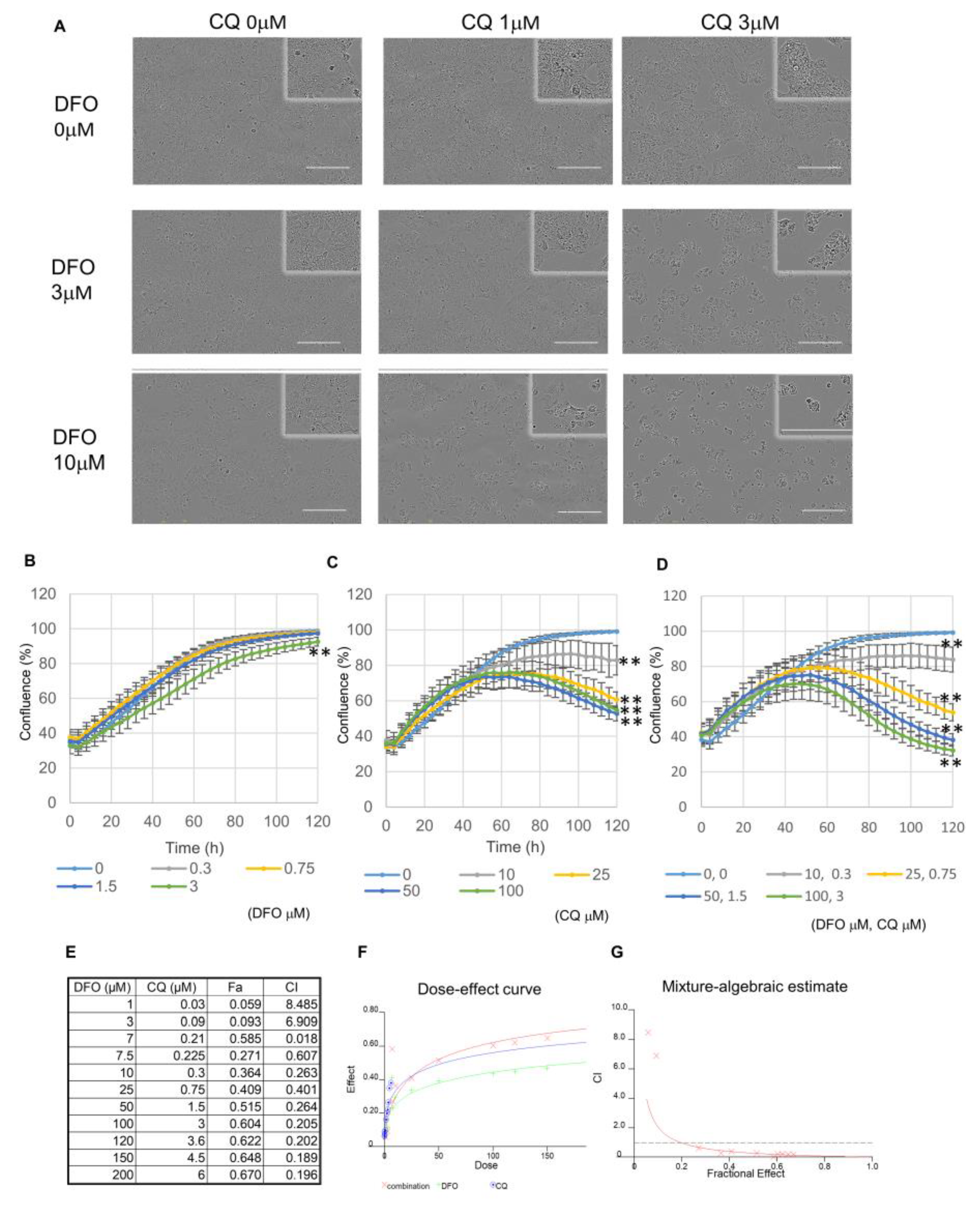
3.3. DFO and Autophagy Inhibitors Exhibit a Synergistic Effect in Huh7 Cells
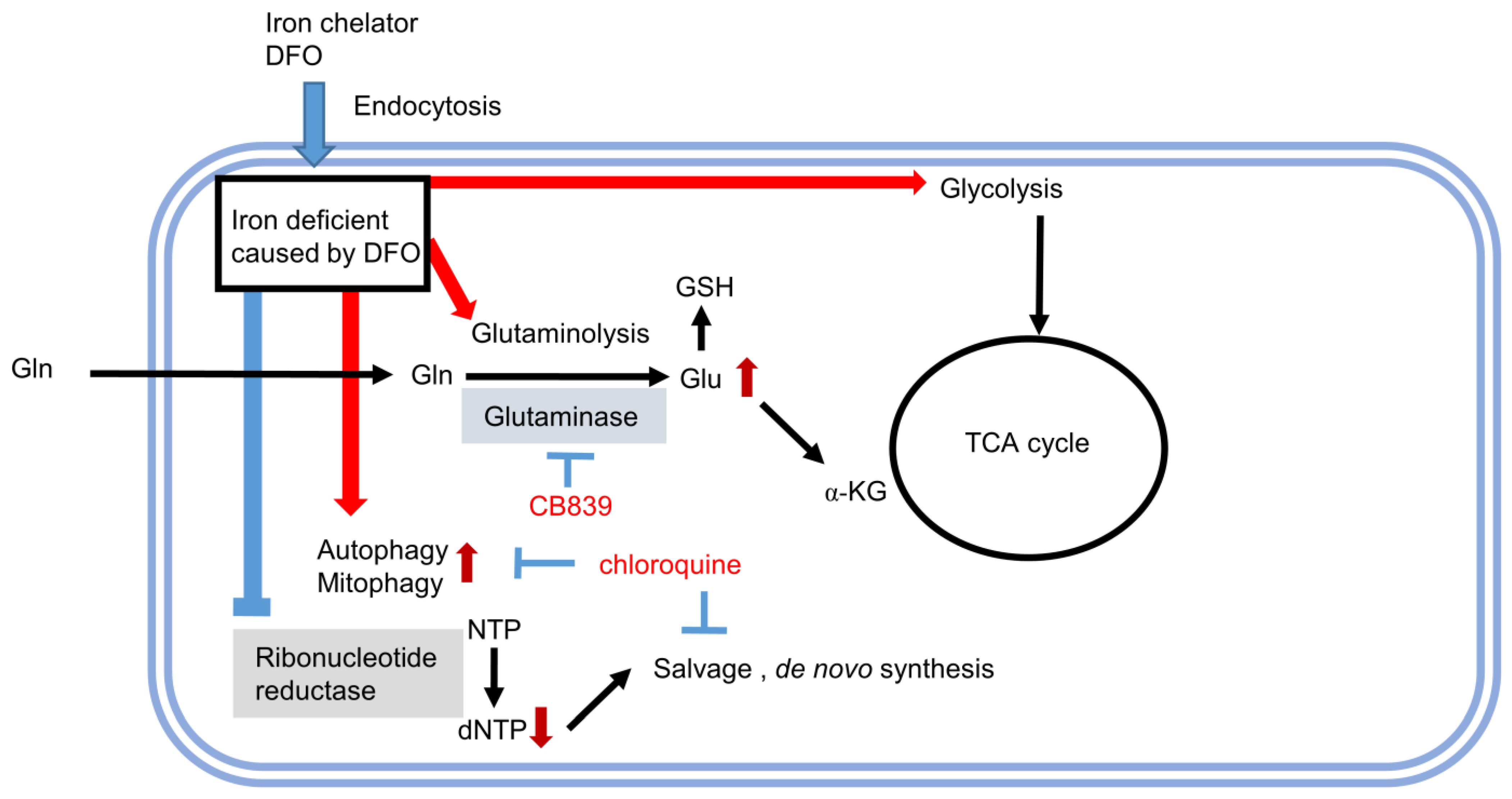
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Serag, H.B.; Marrero, J.A.; Rudolph, L.; Reddy, K.R. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology 2008, 134, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Mauer, K.; O’Kelley, R.; Podda, N.; Flanagan, S.; Gadani, S. New treatment modalities for hepatocellular cancer. Curr. Gastroenterol. Rep. 2015, 17, 442. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: Potential role in the regulation of aging process. Cell. Mol. Life Sci. 2015, 72, 3897–3914. [Google Scholar] [CrossRef] [PubMed]
- Sakaida, I.; Kyle, M.E.; Farber, J.L. Autophagic degradation of protein generates a pool of ferric iron required for the killing of cultured hepatocytes by an oxidative stress. Mol. Pharmacol. 1990, 37, 435–442. [Google Scholar] [PubMed]
- Yu, Y.; Wong, J.; Lovejoy, D.B.; Kalinowski, D.S.; Richardson, D.R. Chelators at the cancer coalface: Desferrioxamine to Triapine and beyond. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6876–6883. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nature reviews. Cancer 2013, 13, 342–355. [Google Scholar] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and Cancer: 2020 Vision. Cancer Res. 2020, 80, 5435–5448. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Kim, J.H.; Aoyama, H.; Hoshino, Y.; Fukumura, H.; Nakakaji, R.; Sato, I.; Ohtake, M.; Akimoto, T.; Narikawa, M.; et al. The iron chelating agent, deferoxamine detoxifies Fe(Salen)-induced cytotoxicity. J. Pharmacol. Sci. 2017, 134, 203–210. [Google Scholar] [CrossRef]
- Lui, G.Y.; Kovacevic, Z.; Richardson, V.; Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget 2015, 6, 18748–18779. [Google Scholar] [CrossRef]
- Sakaida, I.; Hironaka, K.; Uchida, K.; Okita, K. Iron chelator deferoxamine reduces preneoplastic lesions in liver induced by choline-deficient L-amino acid-defined diet in rats. Dig. Dis. Sci. 1999, 44, 560–569. [Google Scholar] [CrossRef]
- Yamasaki, T.; Saeki, I.; Sakaida, I. Efficacy of iron chelator deferoxamine for hepatic arterial infusion chemotherapy in advanced hepatocellular carcinoma patients refractory to current treatments. Hepatol. Int. 2014, 8 (Suppl. 2), 492–498. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Wang, D.; Ren, X.; Zeng, L.; Liu, Y. The growth-inhibitory and apoptosis-inducing effect of deferoxamine combined with arsenic trioxide on HL-60 xenografts in nude mice. Leuk. Res. 2014, 38, 1085–1090. [Google Scholar] [CrossRef]
- Piro, E.; Lentini, M.; Levato, L.; Russo, A.; Molica, S. Sustained Erythroid Response in a Patient with Myelofibrosis Receiving Concomitant Treatment with Ruxolitinib and Deferasirox. Chemotherapy 2018, 63, 107–110. [Google Scholar] [CrossRef]
- Tury, S.; Assayag, F.; Bonin, F.; Chateau-Joubert, S.; Servely, J.L.; Vacher, S.; Becette, V.; Caly, M.; Rapinat, A.; Gentien, D.; et al. The iron chelator deferasirox synergises with chemotherapy to treat triple-negative breast cancers. J. Pathol. 2018, 246, 103–114. [Google Scholar] [CrossRef]
- Shinoda, S.; Kaino, S.; Amano, S.; Harima, H.; Matsumoto, T.; Fujisawa, K.; Takami, T.; Yamamoto, N.; Yamasaki, T.; Sakaida, I. Deferasirox, an oral iron chelator, with gemcitabine synergistically inhibits pancreatic cancer cell growth in vitro and in vivo. Oncotarget 2018, 9, 28434–28444. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Takami, T.; Matsumoto, T.; Yamamoto, N.; Yamasaki, T.; Sakaida, I. An iron chelation-based combinatorial anticancer therapy comprising deferoxamine and a lactate excretion inhibitor inhibits the proliferation of cancer cells. Cancer Metab. 2022, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Dehaven, C.D.; Evans, A.M.; Dai, H.; Lawton, K.A. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Cheminform. 2010, 2, 9. [Google Scholar] [CrossRef]
- Naka, K.; Jomen, Y.; Ishihara, K.; Kim, J.; Ishimoto, T.; Bae, E.J.; Mohney, R.P.; Stirdivant, S.M.; Oshima, H.; Oshima, M.; et al. Dipeptide species regulate p38MAPK-Smad3 signalling to maintain chronic myelogenous leukaemia stem cells. Nat. Commun. 2015, 6, 8039. [Google Scholar] [CrossRef]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta 1999, 1413, 99–107. [Google Scholar] [CrossRef]
- Schalinske, K.L.; Chen, O.S.; Eisenstein, R.S. Iron differentially stimulates translation of mitochondrial aconitase and ferritin mRNAs in mammalian cells. Implications for iron regulatory proteins as regulators of mitochondrial citrate utilization. J. Biol. Chem. 1998, 273, 3740–3746. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 2017, 6, e28083. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Frances, A.; Cordelier, P. The Emerging Role of Cytidine Deaminase in Human Diseases: A New Opportunity for Therapy? Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Zauri, M.; Berridge, G.; Thezenas, M.L.; Pugh, K.M.; Goldin, R.; Kessler, B.M.; Kriaucionis, S. CDA directs metabolism of epigenetic nucleosides revealing a therapeutic window in cancer. Nature 2015, 524, 114–118. [Google Scholar] [CrossRef]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, L.; Zhang, K.; Zhou, B.; Kuo, M.L.; Hu, S.; Chen, L.; Tang, M.; Chen, Y.R.; Yang, L.; et al. Reciprocal regulation of autophagy and dNTP pools in human cancer cells. Autophagy 2014, 10, 1272–1284. [Google Scholar] [CrossRef]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 44, 109–120. [Google Scholar] [CrossRef]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 2012, 30, 671–678. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Hu, T.; Li, P.; Luo, Z.; Chen, X.; Zhang, J.; Wang, C.; Chen, P.; Dong, Z. Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo. Oncol. Rep. 2016, 35, 43–49. [Google Scholar] [CrossRef]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef]
- Yan, X.; Tian, R.; Sun, J.; Zhao, Y.; Liu, B.; Su, J.; Li, M.; Sun, W.; Xu, X. Sorafenib-Induced Autophagy Promotes Glycolysis by Upregulating the p62/HDAC6/HSP90 Axis in Hepatocellular Carcinoma Cells. Front. Pharmacol. 2021, 12, 788667. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujisawa, K.; Matsumoto, T.; Yamamoto, N.; Yamasaki, T.; Takami, T. Metabolic Analysis of DFO-Resistant Huh7 Cells and Identification of Targets for Combination Therapy. Metabolites 2023, 13, 1073. https://doi.org/10.3390/metabo13101073
Fujisawa K, Matsumoto T, Yamamoto N, Yamasaki T, Takami T. Metabolic Analysis of DFO-Resistant Huh7 Cells and Identification of Targets for Combination Therapy. Metabolites. 2023; 13(10):1073. https://doi.org/10.3390/metabo13101073
Chicago/Turabian StyleFujisawa, Koichi, Toshihiko Matsumoto, Naoki Yamamoto, Takahiro Yamasaki, and Taro Takami. 2023. "Metabolic Analysis of DFO-Resistant Huh7 Cells and Identification of Targets for Combination Therapy" Metabolites 13, no. 10: 1073. https://doi.org/10.3390/metabo13101073
APA StyleFujisawa, K., Matsumoto, T., Yamamoto, N., Yamasaki, T., & Takami, T. (2023). Metabolic Analysis of DFO-Resistant Huh7 Cells and Identification of Targets for Combination Therapy. Metabolites, 13(10), 1073. https://doi.org/10.3390/metabo13101073