
Paternal Obesity Induced by High-Fat Diet Impairs the Metabolic and Reproductive Health of Progeny in Rats

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Diet
2.3. Intraperitoneal Glucose Tolerance Test (IPGTT) and Insulin Tolerance Test (ITT)
2.4. Estrus Cycle Analysis
2.5. Sperm Analysis
2.6. Tissue Extraction
2.7. Histological Analysis
2.8. Immunofluorescence Staining
2.9. Testosterone Determination
2.10. Statistical Analysis
3. Results
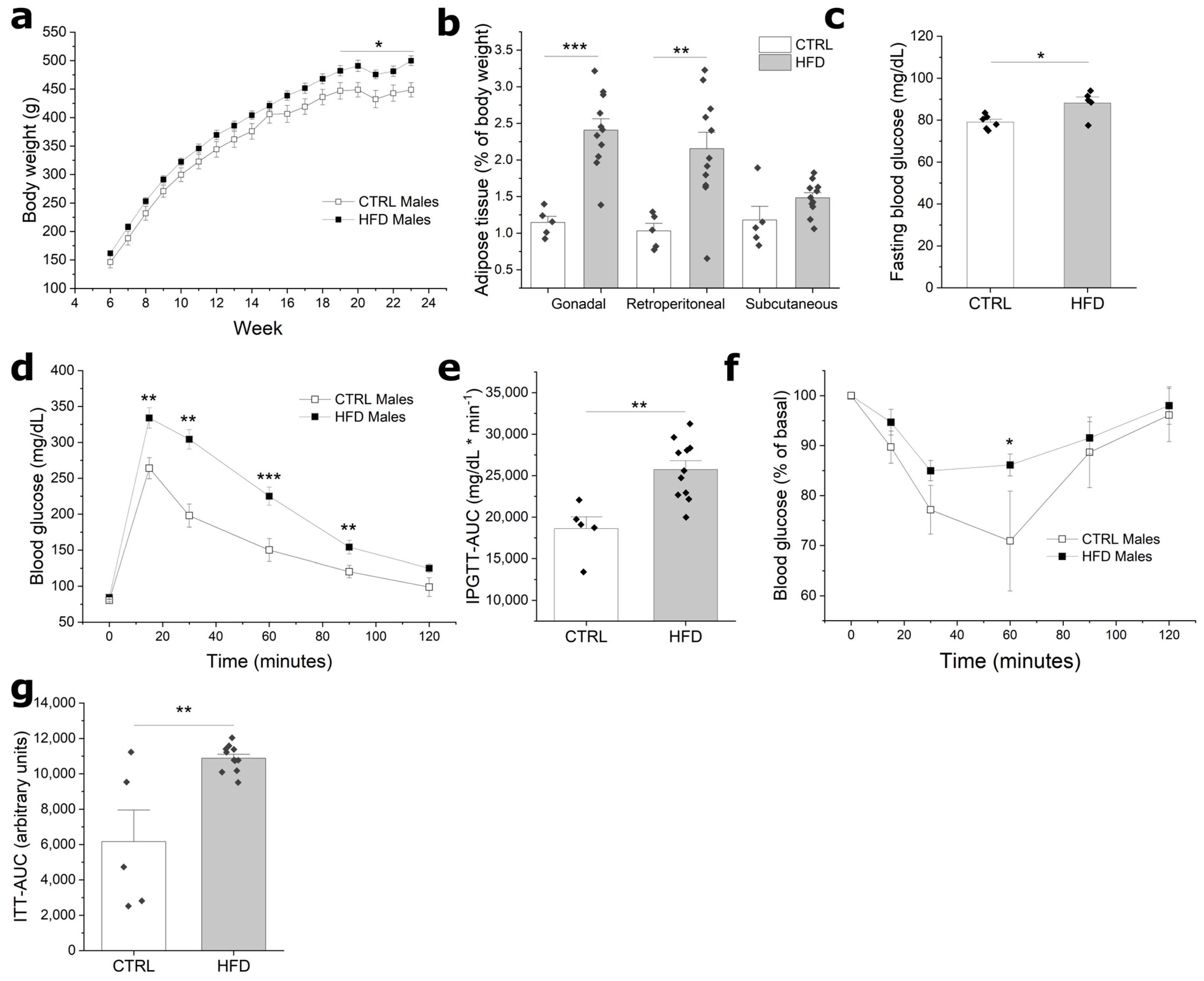
3.1. HFD Induces Obesity and Impaired Glucose Homeostasis in F0 Males
3.2. Paternal Obesity Reduces Reproductive Potential
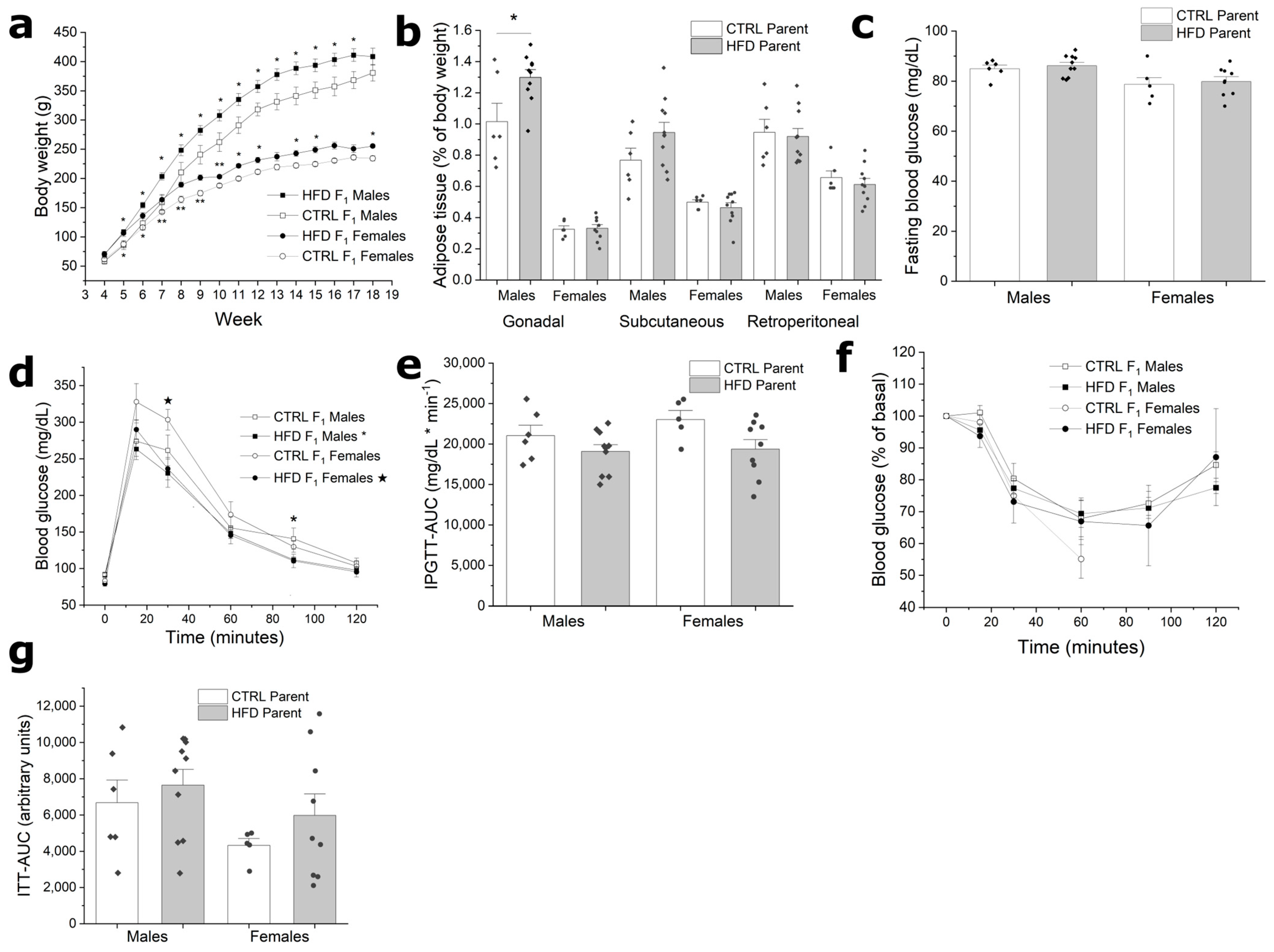
3.3. F1 from HFD Fathers Have Increased Body Weight without Metabolic Alterations
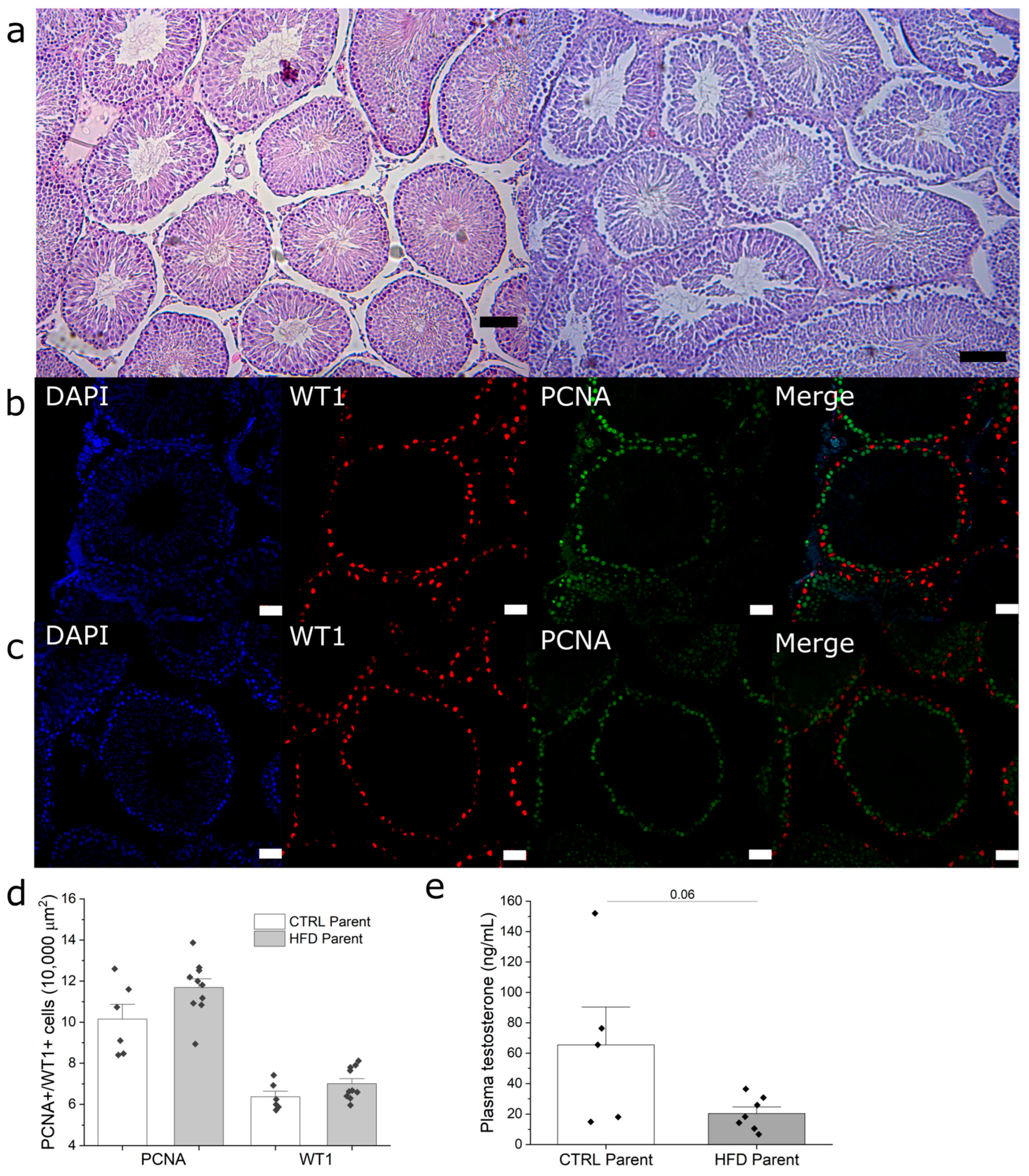
3.4. Paternal Obesity Impairs Sperm Production in F1 Males
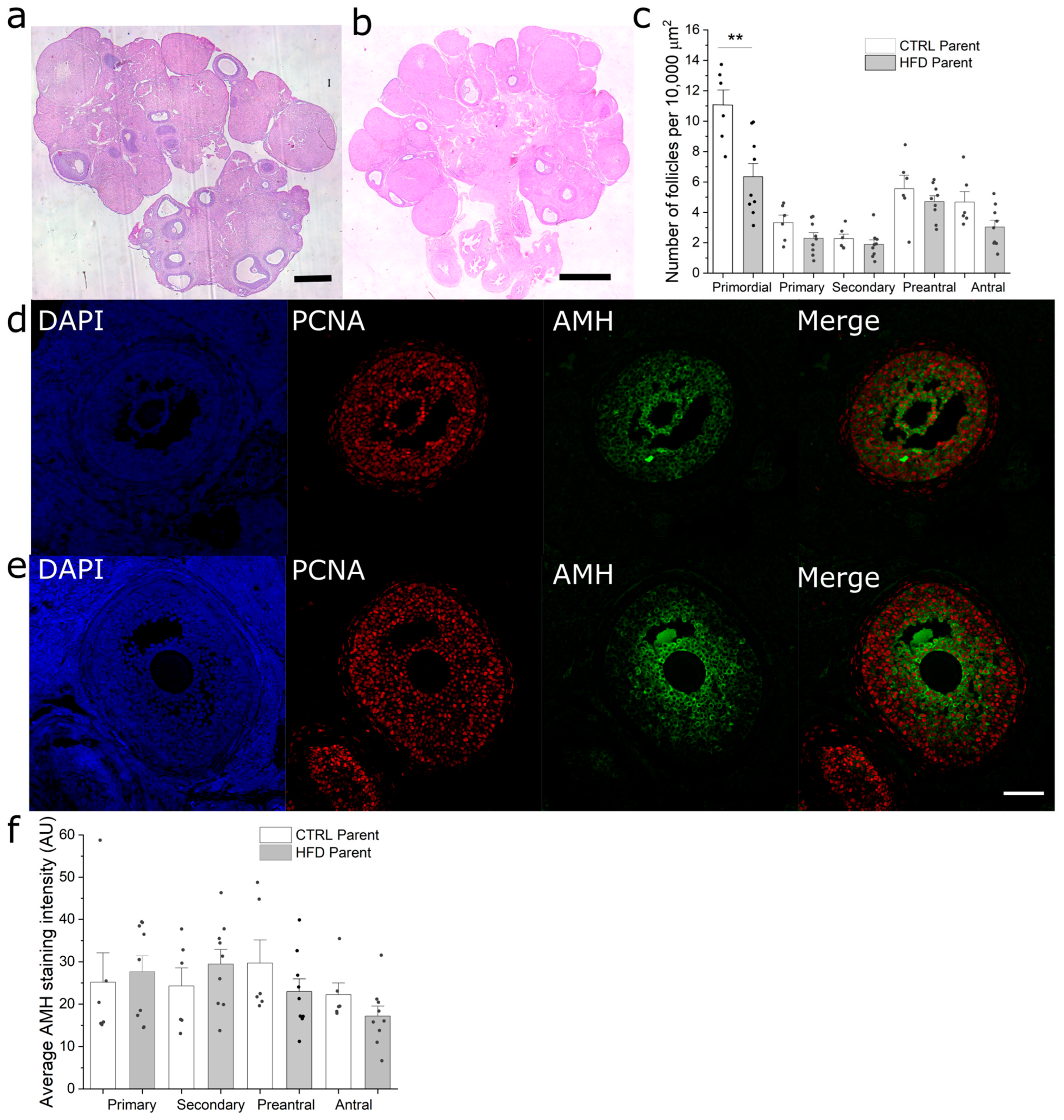
3.5. Paternal Obesity Reduces Follicular Reserve in F1 Females
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metab. Clin. Exp. 2019, 92, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, M.N.; Flaxman, S.R.; Boerma, T.; Vanderpoel, S.; Stevens, G.A. National, regional, and global trends in infertility prevalence since 1990: A systematic analysis of 277 health surveys. PLoS Med. 2012, 9, e1001356. [Google Scholar] [CrossRef] [PubMed]
- Law, D.C.G.; Maclehose, R.F.; Longnecker, M.P. Obesity and time to pregnancy. Hum. Reprod. 2007, 22, 414–420. [Google Scholar] [CrossRef]
- Knight, M.; Kurinczuk, J.J.; Spark, P.; Brocklehurst, P. UK Obstetric Surveillance System Extreme obesity in pregnancy in the United Kingdom. Obstet. Gynecol. 2010, 115, 989–997. [Google Scholar] [CrossRef]
- Kasturi, S.S.; Tannir, J.; Brannigan, R.E. The metabolic syndrome and male infertility. J. Androl. 2008, 29, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.K.; Andersson, A.M.; Jørgensen, N.; Andersen, A.G.; Carlsen, E.; Petersen, J.H.; Skakkebaek, N.E. Body mass index in relation to semen quality and reproductive hormones among 1558 Danish men. Fertil. Steril. 2004, 82, 863–870. [Google Scholar] [CrossRef]
- Hoffman, D.J.; Powell, T.L.; Barrett, E.S.; Hardy, D.B. Developmental origins of metabolic diseases. Physiol. Rev. 2021, 101, 739–795. [Google Scholar] [CrossRef]
- Nomura, Y.; Lambertini, L.; Rialdi, A.; Lee, M.; Mystal, E.Y.; Grabie, M.; Manaster, I.; Huynh, N.; Finik, J.; Davey, M.; et al. Global methylation in the placenta and umbilical cord blood from pregnancies with maternal gestational diabetes, preeclampsia, and obesity. Reprod. Sci. 2014, 21, 131–137. [Google Scholar] [CrossRef]
- Masuyama, H.; Mitsui, T.; Nobumoto, E.; Hiramatsu, Y. The Effects of High-Fat Diet Exposure In Utero on the Obesogenic and Diabetogenic Traits Through Epigenetic Changes in Adiponectin and Leptin Gene Expression for Multiple Generations in Female Mice. Endocrinology 2015, 156, 2482–2491. [Google Scholar] [CrossRef]
- Campbell, J.M.; McPherson, N.O. Influence of increased paternal BMI on pregnancy and child health outcomes independent of maternal effects: A systematic review and meta-analysis. Obes. Res. Clin. Pract. 2019, 13, 511–521. [Google Scholar] [CrossRef]
- Ng, S.F.; Lin, R.C.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Fullston, T.; Ohlsson Teague, E.M.; Palmer, N.O.; DeBlasio, M.J.; Mitchell, M.; Corbett, M.; Print, C.G.; Owens, J.A.; Lane, M. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 4226–4243. [Google Scholar] [CrossRef]
- Galarza, R.A.; Rhon Calderón, E.A.; Cortez, A.E.; Faletti, A.G. Maternal Overweight Disrupts the Sexual Maturation of the Offspring. Reprod. Sci. 2017, 24, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Reame, V.; Pytlowanciv, E.Z.; Ribeiro, D.L.; Pissolato, T.F.; Taboga, S.R.; Góes, R.M.; Pinto-Fochi, M.E. Obesogenic environment by excess of dietary fats in different phases of development reduces spermatic efficiency of wistar rats at adulthood: Correlations with metabolic status. Biol. Reprod. 2014, 91, 151. [Google Scholar] [CrossRef]
- Fullston, T.; McPherson, N.O.; Owens, J.A.; Kang, W.X.; Sandeman, L.Y.; Lane, M. Paternal obesity induces metabolic and sperm disturbances in male offspring that are exacerbated by their exposure to an “obesogenic” diet. Physiol. Rep. 2015, 3, e12336. [Google Scholar] [CrossRef]
- Fullston, T.; Shehadeh, H.; Sandeman, L.Y.; Kang, W.X.; Wu, L.L.; Robker, R.L.; McPherson, N.O.; Lane, M. Female offspring sired by diet induced obese male mice display impaired blastocyst development with molecular alterations to their ovaries, oocytes and cumulus cells. J. Assist. Reprod. Genet. 2015, 32, 725–735. [Google Scholar] [CrossRef]
- Fan, Y.; Liu, Y.; Xue, K.; Gu, G.; Fan, W.; Xu, Y.; Ding, Z. Diet-induced obesity in male C57BL/6 mice decreases fertility as a consequence of disrupted blood-testis barrier. PLoS ONE 2015, 10, e0120775. [Google Scholar] [CrossRef]
- Hu, X.; Ge, X.; Liang, W.; Shao, Y.; Jing, J.; Wang, C.; Zeng, R.; Yao, B. Effects of saturated palmitic acid and omega-3 polyunsaturated fatty acids on Sertoli cell apoptosis. Syst. Biol. Reprod. Med. 2018, 64, 368–380. [Google Scholar] [CrossRef]
- Verderame, M.; Migliaccio, V.; Scudiero, R. Role of estrogen receptors, P450 aromatase, PCNA and p53 in high-fat-induced impairment of spermatogenesis in rats. Comptes Rendus Biol. 2018, 341, 371–379. [Google Scholar] [CrossRef]
- Li, X.J.; Wang, H.; Lu, D.Y.; Yu, T.T.; Ullah, K.; Shi, X.Y.; Shen, Y.H.; Fei, X.Y.; Lin, Z.Y.; Huang, H.F.; et al. Anti-Müllerian Hormone Accelerates Pathological Process of Insulin Resistance in Polycystic Ovary Syndrome Patients. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Metab. 2021, 53, 504–511. [Google Scholar] [CrossRef]
- Hubscher, C.H.; Brooks, D.L.; Johnson, J.R. A quantitative method for assessing stages of the rat estrous cycle. Biotech. Histochem. Off. Publ. Biol. Stain Comm. 2005, 80, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Elgawish, R.A.R.; Abdelrazek, H.M.A. Effects of lead acetate on testicular function and caspase-3 expression with respect to the protective effect of cinnamon in albino rats. Toxicol. Rep. 2014, 1, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Björndahl, L.; Söderlund, I.; Kvist, U. Evaluation of the one-step eosin-nigrosin staining technique for human sperm vitality assessment. Hum. Reprod. 2003, 18, 813–816. [Google Scholar] [CrossRef]
- Myers, M.; Britt, K.L.; Wreford, N.G.; Ebling, F.J.; Kerr, J.B. Methods for quantifying follicular numbers within the mouse ovary. Reproduction 2004, 127, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Bakos, H.W.; Henshaw, R.C.; Mitchell, M.; Lane, M. Paternal body mass index is associated with decreased blastocyst development and reduced live birth rates following assisted reproductive technology. Fertil. Steril. 2011, 95, 1700–1704. [Google Scholar] [CrossRef]
- Freeman, E.; Fletcher, R.; Collins, C.E.; Morgan, P.J.; Burrows, T.; Callister, R. Preventing and treating childhood obesity: Time to target fathers. Int. J. Obes. 2012, 36, 12–15. [Google Scholar] [CrossRef]
- Hillman, S.; Peebles, D.M.; Williams, D.J. Paternal metabolic and cardiovascular risk factors for fetal growth restriction: A case-control study. Diabetes Care 2013, 36, 1675–1680. [Google Scholar] [CrossRef]
- Chen, Y.P.; Xiao, X.M.; Li, J.; Reichetzeder, C.; Wang, Z.N.; Hocher, B. Paternal body mass index (BMI) is associated with offspring intrauterine growth in a gender dependent manner. PLoS ONE 2012, 7, e36329. [Google Scholar] [CrossRef]
- Kirk, S.L.; Samuelsson, A.M.; Argenton, M.; Dhonye, H.; Kalamatianos, T.; Poston, L.; Taylor, P.D.; Coen, C.W. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE 2009, 4, e5870. [Google Scholar] [CrossRef]
- Oshio, L.T.; Andreazzi, A.E.; Lopes, J.F.; Sá, J.P.; Bolotari, M.; Costa, V.M.G.; Guerra, M.O.; Peters, V.M. A paternal hypercaloric diet affects the metabolism and fertility of F1 and F2 Wistar rat generations. J. Dev. Orig. Health Dis. 2020, 11, 653–663. [Google Scholar] [CrossRef]
- César, H.; Sertorio, M.N.; de Souza, E.A.; Jamar, G.; Santamarina, A.; Jucá, A.; Casagrande, B.P.; Pisani, L.P. Parental high-fat high-sugar diet programming and hypothalamus adipose tissue axis in male Wistar rats. Eur. J. Nutr. 2022, 61, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garrido, M.A.; Ruiz-Pino, F.; Velasco, I.; Barroso, A.; Fernandois, D.; Heras, V.; Manfredi-Lozano, M.; Vazquez, M.J.; Castellano, J.M.; Roa, J.; et al. Intergenerational Influence of Paternal Obesity on Metabolic and Reproductive Health Parameters of the Offspring: Male-Preferential Impact and Involvement of Kiss1-Mediated Pathways. Endocrinology 2018, 159, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.G.; Morgan, M.D.; Heger, A.H.; Sharpe, R.M.; Drake, A.J. High-fat diet disrupts metabolism in two generations of rats in a parent-of-origin specific manner. Sci. Rep. 2016, 6, 31857. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fadeil, M.R.; Abd Allah, E.S.H.; Iraqy, H.M.; Elgamal, D.A.; Abdel-Ghani, M.A. Experimental obesity and diabetes reduce male fertility: Potential involvement of hypothalamic Kiss-1, pituitary nitric oxide, serum vaspin and visfatin. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2019, 26, 181–189. [Google Scholar] [CrossRef]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar] [CrossRef]
- Fullston, T.; Palmer, N.O.; Owens, J.A.; Mitchell, M.; Bakos, H.W.; Lane, M. Diet-induced paternal obesity in the absence of diabetes diminishes the reproductive health of two subsequent generations of mice. Hum. Reprod. 2012, 27, 1391–1400. [Google Scholar] [CrossRef]
- McPherson, N.O.; Fullston, T.; Bakos, H.W.; Setchell, B.P.; Lane, M. Obese father’s metabolic state, adiposity, and reproductive capacity indicate son’s reproductive health. Fertil. Steril. 2014, 101, 865–873. [Google Scholar] [CrossRef]
- Sadler-Riggleman, I.; Klukovich, R.; Nilsson, E.; Beck, D.; Xie, Y.; Yan, W.; Skinner, M.K. Epigenetic transgenerational inheritance of testis pathology and Sertoli cell epimutations: Generational origins of male infertility. Environ. Epigenetics 2019, 5, dvz013. [Google Scholar] [CrossRef]
- Suleiman, J.B.; Nna, V.U.; Othman, Z.A.; Zakaria, Z.; Bakar, A.B.A.; Mohamed, M. Orlistat attenuates obesity-induced decline in steroidogenesis and spermatogenesis by up-regulating steroidogenic genes. Andrology 2020, 8, 1471–1485. [Google Scholar] [CrossRef]
- Crisóstomo, L.; Jarak, I.; Rato, L.P.; Raposo, J.F.; Batterham, R.L.; Oliveira, P.F.; Alves, M.G. Inheritable testicular metabolic memory of high-fat diet causes transgenerational sperm defects in mice. Sci. Rep. 2021, 11, 9444. [Google Scholar] [CrossRef]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS ONE 2013, 8, e55387. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-González, G.L.; Vega, C.C.; Boeck, L.; Vázquez, M.; Bautista, C.J.; Reyes-Castro, L.A.; Saldaña, O.; Lovera, D.; Nathanielsz, P.W.; Zambrano, E. Maternal obesity and overnutrition increase oxidative stress in male rat offspring reproductive system and decrease fertility. Int. J. Obes. 2015, 39, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Ambrosetti, V.; Guerra, M.; Ramírez, L.A.; Reyes, A.; Álvarez, D.; Olguín, S.; González-Mañan, D.; Fernandois, D.; Sotomayor-Zárate, R.; Cruz, G. Increase in endogenous estradiol in the progeny of obese rats is associated with precocious puberty and altered follicular development in adulthood. Endocrine 2016, 53, 258–270. [Google Scholar] [CrossRef]
- Xia, D.; Parvizi, N.; Zhou, Y.; Xu, K.; Jiang, H.; Li, R.; Hang, Y.; Lu, Y. Paternal fenvalerate exposure influences reproductive functions in the offspring. Reprod. Sci. 2013, 20, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Zhang, X.; Tan, X.; Ji, M.; Chen, Y.; Wan, Z.; Yu, Z. Multigenerational and transgenerational effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on ovarian reserve and follicular development through AMH/AMHR2 pathway in adult female rats. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2020, 140, 111309. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Live OffSpring Per Litter | Testis Weight (% of Body Mass) | Seminiferous Tubule Area (μm2) | Germinal Epithelium Thickness (μm) | Sperm Per mL (Millions) | % of Live Sperm | Plasma Testosterone (ng/mL) |
|---|---|---|---|---|---|---|---|
| Control | 13.0 ± 1.3 | 0.416 ± 0.015 | 63,567 ± 1563 | 76.66 ± 1.16 | 30.64 ± 1.81 | 87 ± 3.59 | 37.21 ± 13.79 |
| HFD | 11.4 ± 1.2 | 0.378 ± 0.009 * | 65,264 ± 1027 | 77.74 ± 0.86 | 20.64 ± 1.10 ** | 72 ± 3.55 ** | 17.25 ± 4.65 |
| Condition | Testis Weight (% of Body Mass) | Seminiferous Tubule Area (μm2) | Sperm Per mL (millions) | % Of Live Sperm |
|---|---|---|---|---|
| F1 CTR | 0.48 ± 0.02 | 6187 ± 3427 | 24.18 ± 2.63 | 92.8 ± 2.0 |
| F1 HFD | 0.47 ± 0.02 | 52,675 ± 2352 | 24.21 ± 1.34 | 75.2 ± 4.1 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larqué, C.; Lugo-Martínez, H.; Mendoza, X.; Nochebuena, M.; Novo, L.; Vilchis, R.; Sánchez-Bringas, G.; Ubaldo, L.; Velasco, M.; Escalona, R. Paternal Obesity Induced by High-Fat Diet Impairs the Metabolic and Reproductive Health of Progeny in Rats. Metabolites 2023, 13, 1098. https://doi.org/10.3390/metabo13101098
Larqué C, Lugo-Martínez H, Mendoza X, Nochebuena M, Novo L, Vilchis R, Sánchez-Bringas G, Ubaldo L, Velasco M, Escalona R. Paternal Obesity Induced by High-Fat Diet Impairs the Metabolic and Reproductive Health of Progeny in Rats. Metabolites. 2023; 13(10):1098. https://doi.org/10.3390/metabo13101098
Chicago/Turabian StyleLarqué, Carlos, Haydée Lugo-Martínez, Xiadany Mendoza, Monserrat Nochebuena, Luis Novo, Ricardo Vilchis, Guadalupe Sánchez-Bringas, Laura Ubaldo, Myrian Velasco, and Rene Escalona. 2023. "Paternal Obesity Induced by High-Fat Diet Impairs the Metabolic and Reproductive Health of Progeny in Rats" Metabolites 13, no. 10: 1098. https://doi.org/10.3390/metabo13101098
APA StyleLarqué, C., Lugo-Martínez, H., Mendoza, X., Nochebuena, M., Novo, L., Vilchis, R., Sánchez-Bringas, G., Ubaldo, L., Velasco, M., & Escalona, R. (2023). Paternal Obesity Induced by High-Fat Diet Impairs the Metabolic and Reproductive Health of Progeny in Rats. Metabolites, 13(10), 1098. https://doi.org/10.3390/metabo13101098