

Key Therapeutic Targets to Treat Hyperglycemia-Induced Atherosclerosis Analyzed Using a Petri Net-Based Model



Abstract
:1. Introduction
1.1. Research Context
1.2. Biological Background
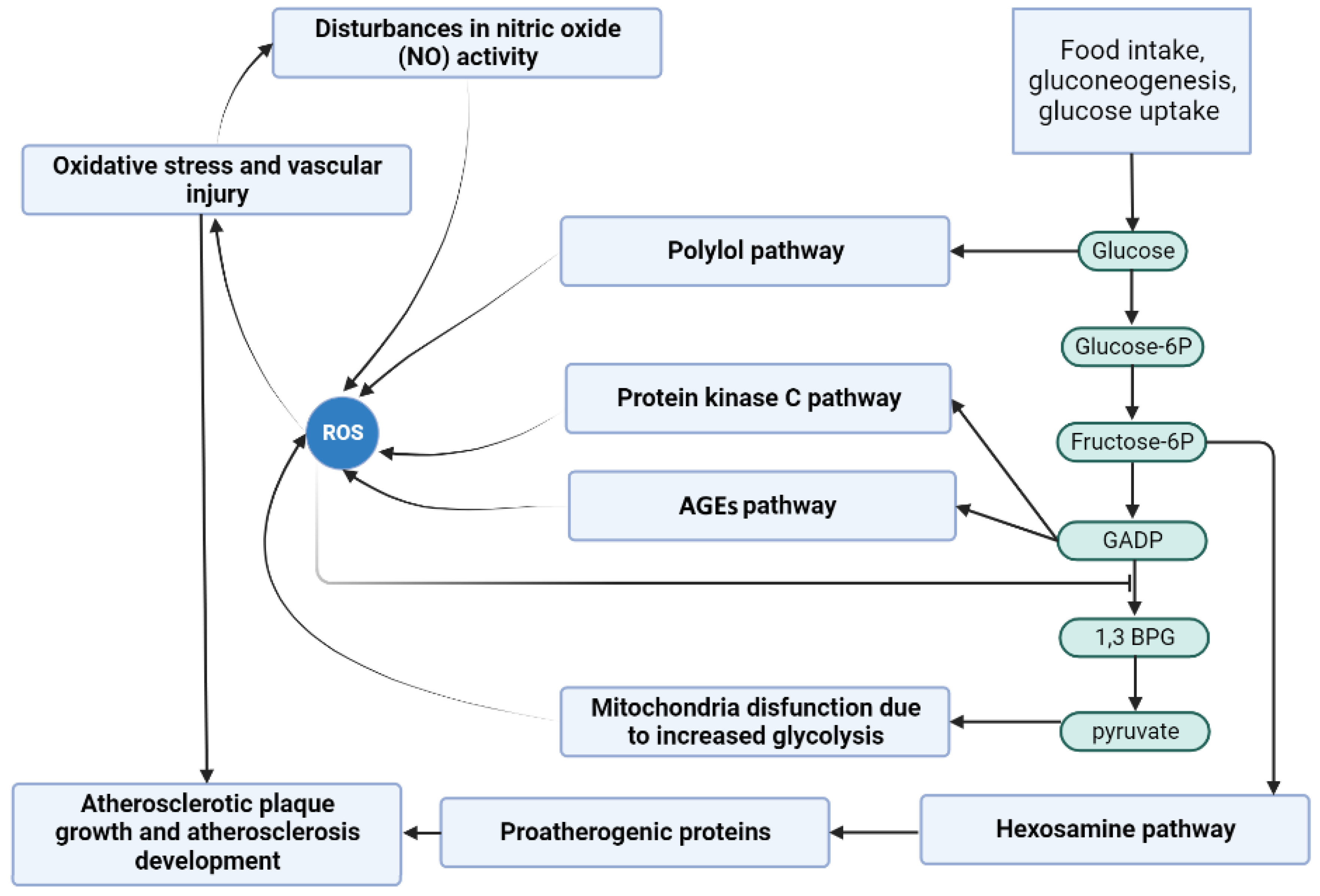
1.2.1. Glycolytic Pathway
1.2.2. Reactive Oxygen Species
1.2.3. Protein Kinase C (PKC) Pathway
1.2.4. Hexosamine Pathway
1.2.5. Polyol Pathway
1.2.6. Advanced Glycation End Products (AGEs) Pathway
1.2.7. Oxidative Stress
1.2.8. Diabetes Mellitus and Atherosclerosis
2. Materials and Methods
3. Results and Discussion
3.1. Petri Net-Based Model Presentation and the Results of Its Formal Analysis
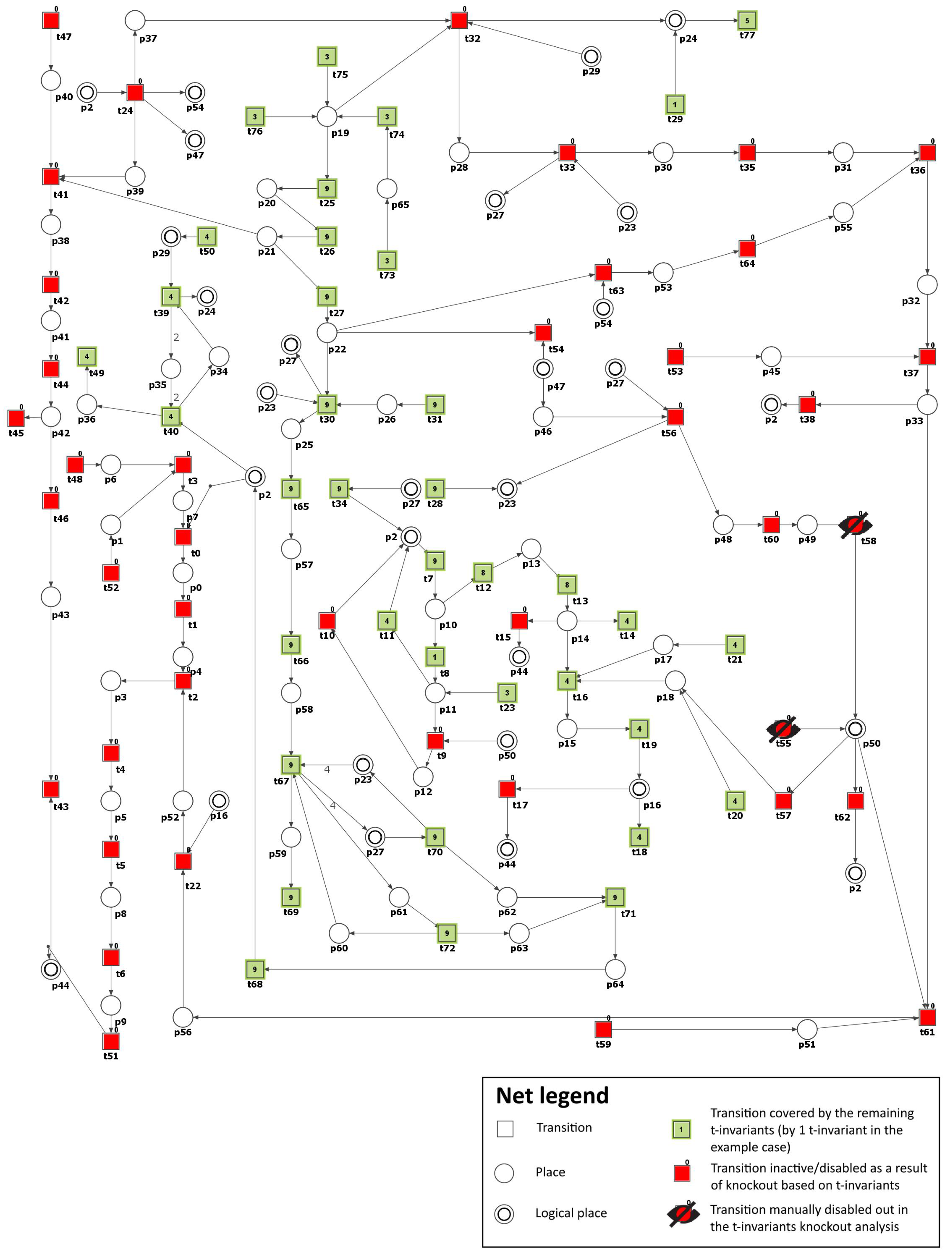
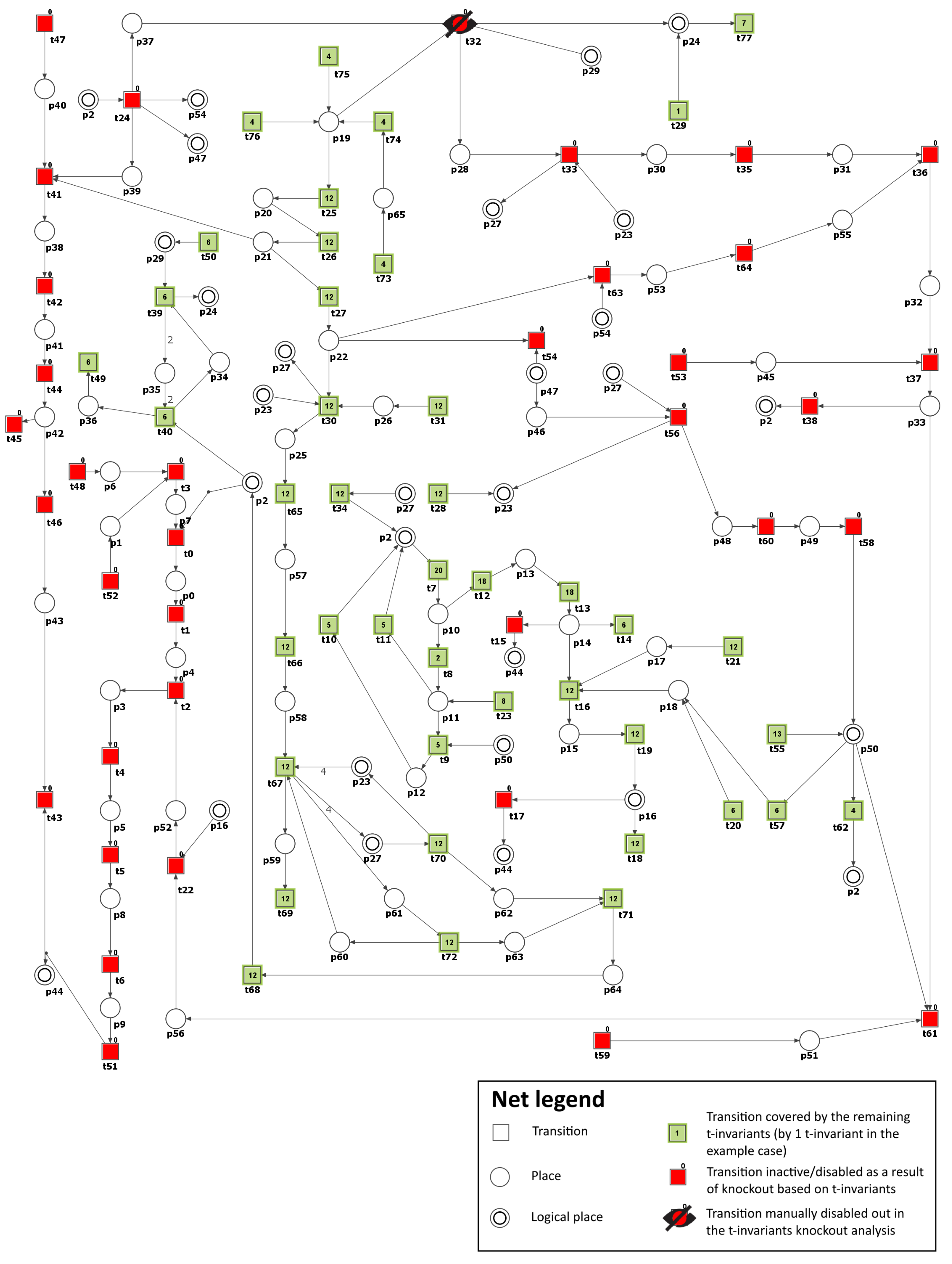
3.2. Knockout Analysis Based on t-Invariants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCT Set/Transition | Biological Function | Affected Transitions |
|---|---|---|
| Protein kinase C pathway | 69.23% (48.75%) | |
| NADP usage | 52.56% | |
| NADPH formation | 50.00% | |
| Atherosclerosis development and progression affected by ROS and oxidized LDL | 44.87% (29.51%) | |
| ROS reaction with NO | 34.62% | |
| Antioxidant defense mechanism involving glutathione-dependent enzymes | 32.05% (29.49%) | |
| Activation of the thromboxane receptor leading to apoptosis, vascular muscle cell activation, and vascular cell adhesion molecules expression | 24.36% (21.80%) | |
| Activation of proatherogenic proteins leading to atherosclerosis progression | 20.51% (19.23) | |
| Impact of peroxynitrite on nitric oxide (NO) synthesis | 15.38% (14.10%) | |
| Vasoconstriction and endothelial dysfunction induced by PGI synthase disruption and inactivation | 14.10% (12.82%) | |
| ROS production by AGE-RAGE complex | 3.85% | |
| Metabolic pathway that converts glucose into glyceraldehyde 3-phosphate | 2.56% (0%) | |
| Increased production of ROS as a consequence of mitochondrial dysfunction resulting from hyperglycemia | 1.28% (0%) | |
| Glucose uptake and transport across the cell membrane | 1.28% (0%) |
3.3. Limitations of the Study
3.4. Potential Clinical Implications for Hyperglycemic Patients with Atherosclerosis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGEs | advanced glycation end products |
| AKT | protein kinase B |
| ATP | adenosine triphosphate |
| BH4 | tetrahydrobiopterin |
| CO | carbon dioxide |
| CVD | cardiovascular disease |
| DAG | diacylglycerol |
| DHAP | dihydroxyacetone phosphate |
| DM | diabetes mellitus |
| ECM | extracellular matrix |
| ECs | endothelial cells |
| Egr-1 | early growth response protein 1 |
| ELAM-1 | endothelial-leukocyte adhesion molecule 1 |
| eNOS | endothelial nitric oxide synthase |
| ERK 1/2 | extracellular signal-regulated kinase 1/2 |
| ET-1 | endothelin 1 |
| ETC | electron transport chain |
| ER | endoplasmic reticulum |
| FAD | flavin adenine dinucleotide |
| FADH | reduced flavin adenine dinucleotide |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GFAT | catalyzed by glutamine:fructose-6-phosphate aminotransferase |
| GLUT | glucose transporter |
| GPx | glutathione peroxidase |
| GPDH | glycerol-3-phosphate dehydrogenase |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| HDAC3 | histone deacetylase 3 |
| ICAM-1 | intercellular adhesion molecule 1 |
| JAKs | Janus kinases |
| LDL | low-density lipoproteins |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein |
| MGO | methylglyoxal |
| MyD88 | differentiation factor 88 |
| NAD | nicotinamide adenine dinucleotide |
| NADH | reduced nicotinamide adenine dinucleotide |
| NADP | nicotinamide adenine dinucleotide phosphate |
| NADPH oxidase | nicotinamide adenine dinucleotide phosphate oxidase |
| NO | nitric oxide |
| NOX | NADPH oxidase |
| NF-B | nuclear factor-B |
| PGI | prostacyclin 2 |
| PGH | prostaglandin H |
| PKC | protein kinase C |
| PPAR | peroxisome proliferator-activated receptor |
| RAGE | receptor for advanced glycation end products |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SDH | succinate dehydrogenase |
| Sirt-1 | sirtuin 1 |
| STATs | signal transducer and activator of transcription proteins |
| T1DM | type 1 diabetes mellitus |
| TCA | tricarboxylic acid |
| TIR | toll-interleukin-1 receptor |
| TIRAP | toll-interleukin-1 receptor (TIR) domain-containing adaptor protein |
| TP | thromboxane receptor |
| TA | thromboxane A2 |
| UDP GlcNAc | uridine 5-diphospho-N-acetylglucosamine |
| VCAM-1 | vascular cell adhesion molecule 1 |
Appendix A
| Place | Biological Meaning | References | Place | Biological Meaning | References |
|---|---|---|---|---|---|
| oxidized LDL | [7,36] | AGE-RAGE complex | [27] | ||
| LDL | [7,36] | glutathione disulfide (GSSG) | [6,25] | ||
| ROS | [6,15,16,17,19,25,26,27,36] | glutathione (GSH) | [6,7,25] | ||
| monocytes | [7,36] | water | [6,25] | ||
| MCP-1 | [7,36] | increase glucose conversion into sorbitol | [6,26] | ||
| macrophages | [7,36] | glucosamine-6P | [6,20] | ||
| ECM | [7,36] | increase fructose-6P conversion into glucosamine-6P | [6,26] | ||
| LDL in the vessel wall | [7,36] | glutamine (Gln) | [6,18,19,20] | ||
| macrophages with scavenger receptors | [7,36] | UDP-GlcNAc | [6,20] | ||
| foam cells | [7,36] | proteins O-GlcNAcylated | [6,20] | ||
| peroxynitrite | [15,16,17] | proatherogenic proteins | [6,20] | ||
| eNOS uncoupled | [15,16,17] | atherosclerotic plaque growth | [7,36] | ||
| NO level decreased | [15,16,17] | RAGE receptor | [27] | ||
| low PGI | [15,16,17] | DHAP | [18,19] | ||
| PGH | [15,16,17] | increase glyceraldehyde conversion into DHAP | [6,26] | ||
| thromboxane A | [6,15,16,17,19] | glycerol-3-phosphate | [18,19] | ||
| TP receptor activated | [6,15,16,17,19] | DAG | [6,18,19,25] | ||
| high PGI | [15,16,17] | PKC ( and ) activated | [6,19] | ||
| TA synthase | [6,19] | NF-B | [15,16,17] | ||
| glucose | [6,11,22] | VCAM-1 and ICAM-1 | [15,16,17] | ||
| glucose-6-P | [6,11] | MGO | [6,26] | ||
| fructose-6-P | [6,11,20] | increase glyceraldehyde-3-P conversion into MGO | [6,26] | ||
| glyceraldehyde-3-P | [6,11] | AGE | [26,27] | ||
| NAD | [12,13,22] | NF-B upregulated | [15,16,17] | ||
| NADP | [6,25] | pyruvate | [6,11] | ||
| 1,3-diphospho-glycerate | [6,11] | pyruvate in mitochondia | [12,13] | ||
| GAPDH active | [6,11,20] | CO | [12,13] | ||
| NADH | [6,12,13,22,25] | FAD | [6,11,12,13] | ||
| sorbitol | [22] | FADH2 | [6,12,13] | ||
| NADPH | [6,22,25] | ETC complex I | [12,13] | ||
| fructose | [22] | ETC complex II | [12,13] | ||
| metabolites of fructose | [22] | ETC coenzyme Q | [12,13] | ||
| AGE increased | [27] | GLUT (transporter) | [6,11,22] |
| Transition | Biological Meaning | References | Transition | Biological Meaning | References |
|---|---|---|---|---|---|
| oxidation | [7,36] | reaction catalyzed by glutathione reductase | [6,25] | ||
| endothelial cells stimulation | [7,36] | reaction catalyzed by glutathione peroxidase GPx | [6,25] | ||
| attracting monocytes into subendothelial space | [7,36] | reaction catalyzed by GFAT | [6,20] | ||
| LDL and ECM interaction | [7,36] | reaction forming UDP-GlcNAc | [6,20] | ||
| transformation into macrophages | [7,36] | atherosclerosis progression | [5,6,7,20,36] | ||
| scavenger receptors expression | [7,36] | proteins O-GlcNAcylation | [6,20] | ||
| foam cells formation stimulation | [7,36] | ubiquitination degradation of atheroprotective proteins | [6,20] | ||
| ROS reaction with NO | [15,16,17] | increased transcription of proatherogenic proteins | [6,20] | ||
| ONOO interaction with eNOS cofactor BH4 | [15,16,17] | synthesis by glutamine synthetase | [6,20] | ||
| decreased NO synthesis | [15,16,17] | extracellular molecules secreted by cells | [7,36] | ||
| ROS production increased through decreased NO | [15,16,17] | water usage | [6,25] | ||
| ROS production via eNOS blocking | [15,16,17] | NADPH formation | [6,22,25] | ||
| PGI synthase disrupting | [15,16,17] | foam cells accumulation | [7,36] | ||
| PGH2 buildup | [15,16,17] | LDL formation | [7,36] | ||
| vasoconstriction | [15,16,17] | RAGE expression | [27] | ||
| endothelial dysfunction | [15,16,17] | reaction catalyzed by triose phosphate isomerase | [27] | ||
| PGI conversion by TxA2 synthase | [6,15,16,17,19] | PKC ( and ) expression | [6,19,27] | ||
| vascular smooth muscles activation | [6,15,16,17,19] | reaction catalyzed by GPDH | [18,19] | ||
| apoptosis | [6,15,16,17,19] | increase in TA | [6,19] | ||
| TP receptor activation | [6,15,16,17,19] | PKC ( and ) activation by DAG | [18,19] | ||
| TA synthase expression | NF-B expression | [6,15,16,17] | |||
| PGI synthase active | [15,16,17] | reaction forming DAG | [18,19] | ||
| adhesion molecules (ICAM-1, VCAM-1, ELAM-1) expression | [6,15,16,17] | NF-B upregulation | [6,15,16,17] | ||
| eNOS coupled | [15,16,17] | ROS production by activated NADPH dependent oxidase | [6,19] | ||
| GAPDH inactive | [6,26] | reaction forming MGO | [6,26] | ||
| reaction catalyzed by hexokinase | [6,11] | reaction forming AGE | [26,27] | ||
| reaction catalyzed by phosphoglucoisomerase | [6,11] | reaction forming pyruvate | [6,11] | ||
| reaction forming glyceraldehyde-3-P | [6,11] | pyruvate transported into mitochondria | [6,12,13] | ||
| NAD formation | [12,13,22] | pyruvate enters TCA cycle | [12,13] | ||
| NADP formation | [22] | ROS generated by increased electron donation to coenzyme Q | [12,13] | ||
| reaction catalyzed by GAPDH | [6,11] | CO usage | [12,13] | ||
| GAPDH activation | [6,11] | electrons donation to ETC complex I | [12,13] | ||
| reaction catalyzed by aldose reductase | [22] | electrons passed to ETC coenzyme Q | [12,13] | ||
| oxidation catalyzed by SDH | [22] | electrons donation to ETC complex II | [12,13] | ||
| ROS production via NADPH oxidase by increased NADH | [6,12,13,22,25] | GLUT expression | [6,11,22] | ||
| reaction forming metabolites of fructose | [22] | glucose uptake | [6,11,22] | ||
| increase in AGE formation | [6,25,26,27] | food intake | [22] | ||
| AGE binding to receptor RAGE | [27] | gluconeogenesis | [6,11,22] | ||
| ROS production by AGE-RAGE complex | [27] | NADP usage | [22] |
References
- Bhatti, J.; Sehrawat, A.; Mishra, J.; Sidhu, I.; Navik, U.; Khullar, N.; Kumar, S.; Bhatti, G.; Reddy, P. Oxidative stress in the pathophysiology of type 2 diabetes and related complications: Current therapeutics strategies and future perspectives. Free Radic. Biol. Med. 2022, 184, 114–134. [Google Scholar] [CrossRef] [PubMed]
- Sakran, N.; Graham, Y.; Pintar, T.; Yang, W.; Kassir, R.; Willigendael, E.; Singhal, R.; Kooreman, Z.; Ramnarain, D.; Mahawar, K.; et al. The many faces of diabetes. Is there a need for re-classification? A narrative review. BMC Endocr. Disord. 2022, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Lubawy, D.; Formanowicz, D. Insulin Resistance and Urolithiasis as a Challenge for a Dietitian. Int. J. Environ. Res. Public Health 2022, 19, 7160. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis: Review article. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Siracuse, J.J.; Chaikof, E.L. The Pathogenesis of Diabetic Atherosclerosis. In Diabetes and Peripheral Vascular Disease. Contemporary Diabetes; Shrikhande, G., McKinsey, J., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 13–26. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Klipp, E.; Liebermeister, W.; Wierling, C.; Kowald, A.; Lehrach, H.; Herwig, R. Systems Biology: A Textbook; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Koch, I.; Reisig, W.; Schreiber, F. (Eds.) Modeling in Systems Biology. The Petri Net Approach; Springer: London, UK, 2011. [Google Scholar]
- Formanowicz, D.; Rybarczyk, A.; Radom, M.; Tanaś, K.; Formanowicz, P. A Stochastic Petri Net-Based Model of the Involvement of Interleukin 18 in Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 8574. [Google Scholar] [CrossRef]
- Chandel, N. Carbohydrate Metabolism. Cold Spring. Harb. Perspect. Biol. 2021, 13, a040568. [Google Scholar] [CrossRef]
- Sivitz, W.; Yorek, M. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox. Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef]
- Ciccarelli, G.; Conte, S.; Cimmino, G.; Maiorano, P.; Morrione, A.; Giordano, A. Mitochondrial Dysfunction: The Hidden Player in the Pathogenesis of Atherosclerosis? Int. J. Mol. Sci. 2023, 24, 1086. [Google Scholar] [CrossRef]
- Formanowicz, D.; Kozak, A.; Formanowicz, P. A Petri net based model of oxidative stress in atherosclerosis. Found. Comput. Decis. Sci. 2012, 37, 59–78. [Google Scholar] [CrossRef]
- Hink, U.; Oelze, M.; Kolb, P.; Bachschmid, M.; Zou, M.; Daiber, A.; Mollnau, H.; August, M.; Baldus, S.; Tsilimingas, N.; et al. Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J. Am. Coll. Cardiol. 2003, 42, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.; de Oliveira, J.; da Silva Pontes, L.; de Souza, J., Jr.; Gonçalves, T.; Dantas, S.; de Almeida Feitosa, M.; Silva, A.; de Medeiros, I. ROS: Basic Concepts, Sources, Cellular Signaling, and its Implications in Aging Pathways. Oxid. Med. Cell. Longev. 2022, 19, 1225578. [Google Scholar] [CrossRef] [PubMed]
- Hinton, M.; Thliveris, J.; Hatch, G.; Dakshinamurti, S. Nitric oxide augments signaling for contraction in hypoxic pulmonary arterial smooth muscle-Implications for hypoxic pulmonary hypertension. Front. Physiol. 2023, 14, 1144574. [Google Scholar] [CrossRef] [PubMed]
- Kolczynska, K.; Loza-Valdes, A.; Hawro, I.; Sumara, G. Diacylglycerol-evoked activation of PKC and PKD isoforms in regulation of glucose and lipid metabolism: A review. Lipids Health Dis. 2020, 19, 113. [Google Scholar] [CrossRef] [PubMed]
- Lien, C.; Chen, S.; Tsai, M.; Lin, C. Potential Role of Protein Kinase C in the Pathophysiology of Diabetes-Associated Atherosclerosis. Front. Pharmacol. 2021, 12, 716332. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, S.; Choi, C. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, T.V.; Procopio, T.; Mancuso, E.; Arcidiacono, G.P.; Andreozzi, F.; Arturi, F.; Sciacqua, A.; Perticone, F.; Hribal, M.L.; Sesti, G. SRT1720 counteracts glucosamine-induced endoplasmic reticulum stress and endothelial dysfunction. Cardiovasc. Res. 2015, 107, 295–306. [Google Scholar] [CrossRef]
- Moemen, L.; Abdel Hamid, M.; Wahab, S.; Kenawy, M.; Abuelela, M.; Hassanin, O.; Fouly, M.; Abdelazeem, A.; Noweir, S.; Ismail, S.; et al. Role of advanced glycation end products and sorbitol dehydrogenase in the pathogenesis of diabetic retinopathy. Bull. Natl. Res. Cent. 2020, 44, 1–11. [Google Scholar] [CrossRef]
- Black, H.S. A Synopsis of the Associations of Oxidative Stress, ROS, and Antioxidants with Diabetes Mellitus. Antioxidants 2022, 11, 2003. [Google Scholar] [CrossRef]
- Wright, E., Jr.; Scism-Bacon, J.L.; Glass, L.C. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int. J. Clin. Pract. 2006, 60, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Physiological and Pathological Roles of Aldose Reductase. Metabolites 2021, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Wetzels, S.; Wouters, K.; Schalkwijk, C.; Vanmierlo, T.; Hendriks, J. Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 421. [Google Scholar] [CrossRef] [PubMed]
- Twarda-Clapa, A.; Olczak, A.; Białkowska, A.; Koziołkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fukami, K. RAGE signaling regulates the progression of diabetic complications. Front. Pharmacol. 2023, 14, 1128872. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2709–2729. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, Z.; Wang, L.; Wang, G.; Wang, Z.; Dong, X.; Wen, B.; Zhang, Z. The Pathogenesis of Diabetes Mellitus by Oxidative Stress and Inflammation: Its Inhibition by Berberine. Front. Pharmacol. 2018, 9, 782. [Google Scholar] [CrossRef]
- Ceriello, A.; Quagliaro, L.; Catone, B.; Pascon, R.; Piazzola, M.; Bais, B.; Marra, G.; Tonutti, L.; Taboga, C.; Motz, E. Role of Hyperglycemia in Nitrotyrosine Postprandial Generation. Diabetes Care 2002, 25, 1439–1443. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Poznyak, A.; Grechko, A.; Orekhova, V.; Khotina, V.; Ivanova, E.; Orekhov, A. NADPH Oxidases and Their Role in Atherosclerosis. Biomedicines 2020, 8, 206. [Google Scholar] [CrossRef]
- Chew, P.; Yuen, D.; Stefanovic, N.; Pete, J.; Coughlan, M.; Jandeleit-Dahm, K.; Thomas, M.; Rosenfeldt, F.; Cooper, M.; de Haan, J. Antiatherosclerotic and renoprotective effects of ebselen in the diabetic apolipoprotein E/GPx1-double knockout mouse. Diabetes 2010, 59, 3198–3207. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E. Does Elevated Glucose Promote Atherosclerosis? Pros and Cons. Circ. Res. 2016, 119, 190–193. [Google Scholar] [CrossRef] [PubMed]
- La Sala, L.; Prattichizzo, F.; Ceriello, A. The Link Between Diabetes and Atherosclerosis. Eur. J. Prev. Cardiol. 2019, 26, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Orchard, T.J.; Costacou, T.; Kretowski, A.; Nesto, R.W. Type 1 Diabetes and Coronary Artery Disease. Diabetes Care 2006, 29, 2528–2538. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus-Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed]
- Murata, T. Petri nets: Properties, analysis and applications. Proc. IEEE 1989, 77, 541–580. [Google Scholar] [CrossRef]
- David, R.; Alla, H. Discrete, Continuous, and Hybrid Petri Nets; Springer: Berlin, Heidelberg, 2010. [Google Scholar]
- Formanowicz, D.; Kozak, A.; Głowacki, T.; Radom, M.; Formanowicz, P. Hemojuvelin–hepcidin axis modeled and analyzed using Petri nets. J. Biomed. Inform. 2013, 46, 1030–1043. [Google Scholar] [CrossRef]
- Sackmann, A.; Heiner, M.; Koch, I. Application of Petri net based analysis techniques to signal transduction pathway. BMC Bioinform. 2006, 7, 482. [Google Scholar] [CrossRef]
- Formanowicz, D.; Rybarczyk, A.; Radom, M.; Formanowicz, P. A Role of Inflammation and Immunity in Essential Hypertension—Modeled and Analyzed Using Petri Nets. Int. J. Mol. Sci. 2020, 21, 3348. [Google Scholar] [CrossRef]
- Formanowicz, D.; Radom, M.; Rybarczyk, A.; Tanaś, K.; Formanowicz, P. Control of Cholesterol Metabolism Using a Systems Approach. Biology 2022, 11, 430. [Google Scholar] [CrossRef]
- Radom, M.; Rybarczyk, A.; Szawulak, B.; Andrzejewski, H.; Chabelski, P.; Kozak, A.; Formanowicz, P. Holmes: A graphical tool for development, simulation and analysis of Petri net based models of complex biological systems. Bioinformatics 2017, 33, 3822–3823. [Google Scholar] [CrossRef] [PubMed]
- Einloft, J.; Ackermann, J.; Nöthen, J.; Koch, I. MonaLisa—Visualization and analysis of functional modules in biochemical networks. Bioinformatics 2013, 29, 1469–1470. [Google Scholar] [CrossRef] [PubMed]
- Radom, M.; Szawulak, B. Holmes 1.1. User Manual. 2022. Available online: http://www.cs.put.poznan.pl/mradom/Holmes/HolmesEN_v1.10.pdf (accessed on 10 November 2023).
- Jubaidi, F.; Zainalabidin, S.; Taib, I.; Abdul Hamid, Z.; Mohamad Anuar, N.; Jalil, J.; Mohd Nor, N.; Budin, S. The Role of PKC-MAPK Signalling Pathways in the Development of Hyperglycemia-Induced Cardiovascular Complications. Int. J. Mol. Sci. 2022, 23, 8582. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, S.; Speer, A.; Ackermann, J.; Koch, I. Petri net modelling of gene regulation of the Duchenne muscular dystrophy. Biosystems 2008, 92, 189–205. [Google Scholar] [CrossRef]
- Pan, D.; Xu, L.; Guo, M. The role of protein kinase C in diabetic microvascular complications. Front. Endocrinol. 2022, 13, 973058. [Google Scholar] [CrossRef] [PubMed]
- AlQuadeib, B.T.; Aleanizy, F.S.; Alqahtani, F.Y.; Alshammari, R.A.; Aldarwesh, A.; Alsarra, I. Determination of Ruboxistaurin analysis in rat plasma utilizing LC–MS/MS technique. Saudi Pharm. J. 2023, 31, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Aruni, B.; Satish, K.S. Aldose reductase: Congenial and injurious profiles of an enigmatic enzyme. Biochem. Med. Metab. Biol. 1992, 48, 91–121. [Google Scholar] [CrossRef]
- Wai, H.T.; Jeremiah, S.; Scott, G.; Concetta, D.F.; Ettore, P.; Cristiano, F.; Stefania, T.; Marta, C.; Virgilio, E.; Giacomo, L.; et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Investig. 2011, 121, 4462–4476. [Google Scholar] [CrossRef]
- Vikramadithyan, R.K.; Hu, Y.; Noh, H.L.; Liang, C.P.; Hallam, K.; Tall, A.R.; Ramasamy, R.; Goldberg, I.J. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J. Clin. Investig. 2005, 115, 2434–2443. [Google Scholar] [CrossRef]
- Jannapureddy, S.; Sharma, M.; Yepuri, G.; Schmidt, A.M.; Ramasamy, R. Aldose Reductase: An Emerging Target for Development of Interventions for Diabetic Cardiovascular Complications. Front. Endocrinol. 2021, 12, 636267. [Google Scholar] [CrossRef]
- Ramana, K.; Srivastava, S. Aldose reductase: A novel therapeutic target for inflammatory pathologies. Int. J. Biochem. Cell Biol. 2010, 42, 17–20. [Google Scholar] [CrossRef]
- Gopal, K.; Karwi, Q.; Tabatabaei Dakhili, S.; Wagg, C.; Zhang, L.; Sun, Q.; Saed, T.; Panidarapu, S.; Perfetti, R.; Ramasamy, R.; et al. Aldose reductase inhibition alleviates diabetic cardiomyopathy and is associated with a decrease in myocardial fatty acid oxidation. Int. J. Biochem. Cell Biol. 2023, 22, 73. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Z.; Zhou, W.; Fan, S.; Ma, R.; Xue, L.; Yang, L.; Li, Y.; Tan, H.; Shao, Q.; et al. Epalrestat protects against diabetic peripheral neuropathy by alleviating oxidative stress and inhibiting polyol pathway. Neural Regen. Res. 2016, 11, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Oates, P. Aldose reductase, still a compelling target for diabetic neuropathy. Curr. Drug Targets 2008, 9, 14–36. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, P.; Pegklidou, K.; Chatzopoulou, M.; Nicolaou, I.; Demopoulos, J. Aldose Reductase Enzyme and its Implication to Major Health Problems of the 21st Century. Curr. Med. Chem. 2009, 16, 734–752. [Google Scholar] [CrossRef]
- Sarikaya, M.; Yazihan, N.; Das Evcimen, N. Relationship between aldose reductase enzyme and the signaling pathway of protein kinase C in an in vitro diabetic retinopathy model. Can. J. Physiol. Pharmacol. 2020, 98, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Burman, A.; Tripathi, A. Advanced glycation end products: Key mediator and therapeutic target of cardiovascular complications in diabetes. World J. Diabetes 2023, 14, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Testa, R.; Bonfigli, A.R.; Prattichizzo, F.; La Sala, L.; De Nigris, V.; Ceriello, A. The “Metabolic Memory” Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications. Nutrients 2017, 9, 437. [Google Scholar] [CrossRef]
- Marinos, K.; Dimitrios, D.; Phaedon, D.Z.; Christina, P.; Athanasios, G.P. Impact of advanced glycation end products (AGEs) signaling in coronary artery disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 611–619. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T. Role of Hyperglycemia-Induced Advanced Glycation End Product (AGE) Accumulation in Atherosclerosis. Ann. Vasc. Dis. 2018, 11, 253–258. [Google Scholar] [CrossRef]
- Oshitari, T. Advanced Glycation End-Products and Diabetic Neuropathy of the Retina. Int. J. Mol. Sci. 2023, 24, 2927. [Google Scholar] [CrossRef] [PubMed]
- Sourris, K.; Watson, A.; Jandeleit-Dahm, K. Inhibitors of Advanced Glycation End Product (AGE) Formation and Accumulation. Handb. Exp. Pharmacol. 2021, 264, 395–423. [Google Scholar] [CrossRef] [PubMed]
- Samuel, R.; Rama, N.; KiranKumar, Y.; Stephen, S.; Gonzales, N.; Jerry, L.N. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin. Chim. Acta 2000, 301, 65–77. [Google Scholar] [CrossRef]
- Miyata, T.; van Ypersele, D.; Ueda, Y.; Ichimori, K.; Inagi, R.; Onogi, H.; Ishikawa, N.; Nangaku, M.; Kurokawa, K. Angiotensin II receptor antagonists and angiotensin-converting enzyme inhibitors lower in vitro the formation of advanced glycation end products: Biochemical mechanisms. J. Am. Soc. Nephrol. 2002, 13, 2478–2487. [Google Scholar] [CrossRef]
- Li, X.; Zheng, T.; Sang, S.; Lv, L. Quercetin inhibits advanced glycation end product formation by trapping methylglyoxal and glyoxal. J. Agric. Food Chem. 2014, 62, 12152–12158. [Google Scholar] [CrossRef]
- Reynaert, N.L.; Vanfleteren, L.E.G.W.; Perkins, T.N. The AGE-RAGE Axis and the Pathophysiology of Multimorbidity in COPD. J. Clin. Med. 2023, 12, 3366. [Google Scholar] [CrossRef]
- Gryszczyńska, B.; Budzyń, M.; Formanowicz, D.; Wanic-Kossowska, M.; Formanowicz, P.; Majewski, W.; Iskra, M.; Kasprzak, M.P. Selected Atherosclerosis-Related Diseases May Differentially Affect the Relationship between Plasma Advanced Glycation End Products, Receptor sRAGE, and Uric Acid. J. Clin. Med. 2020, 9, 1416. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]

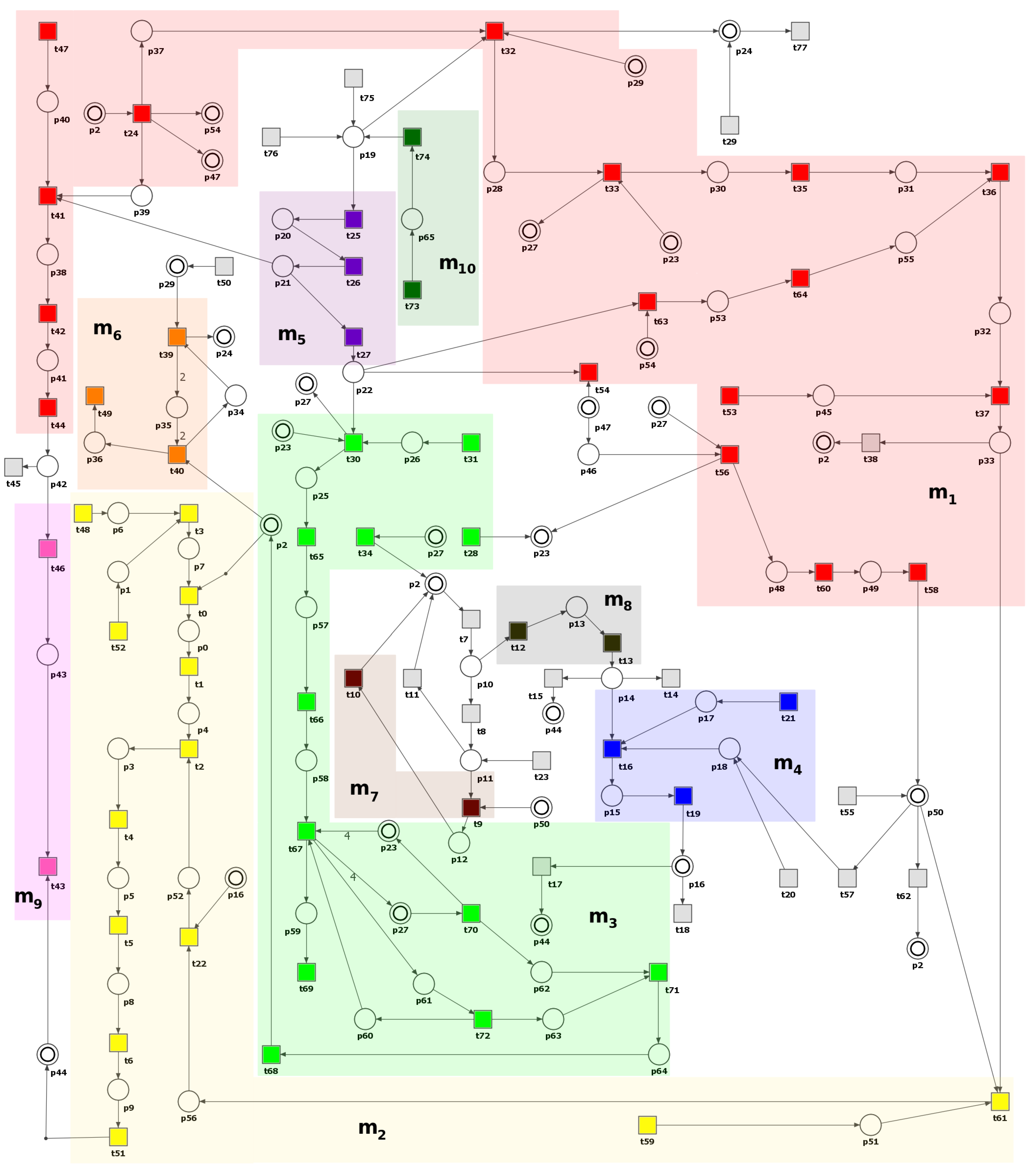
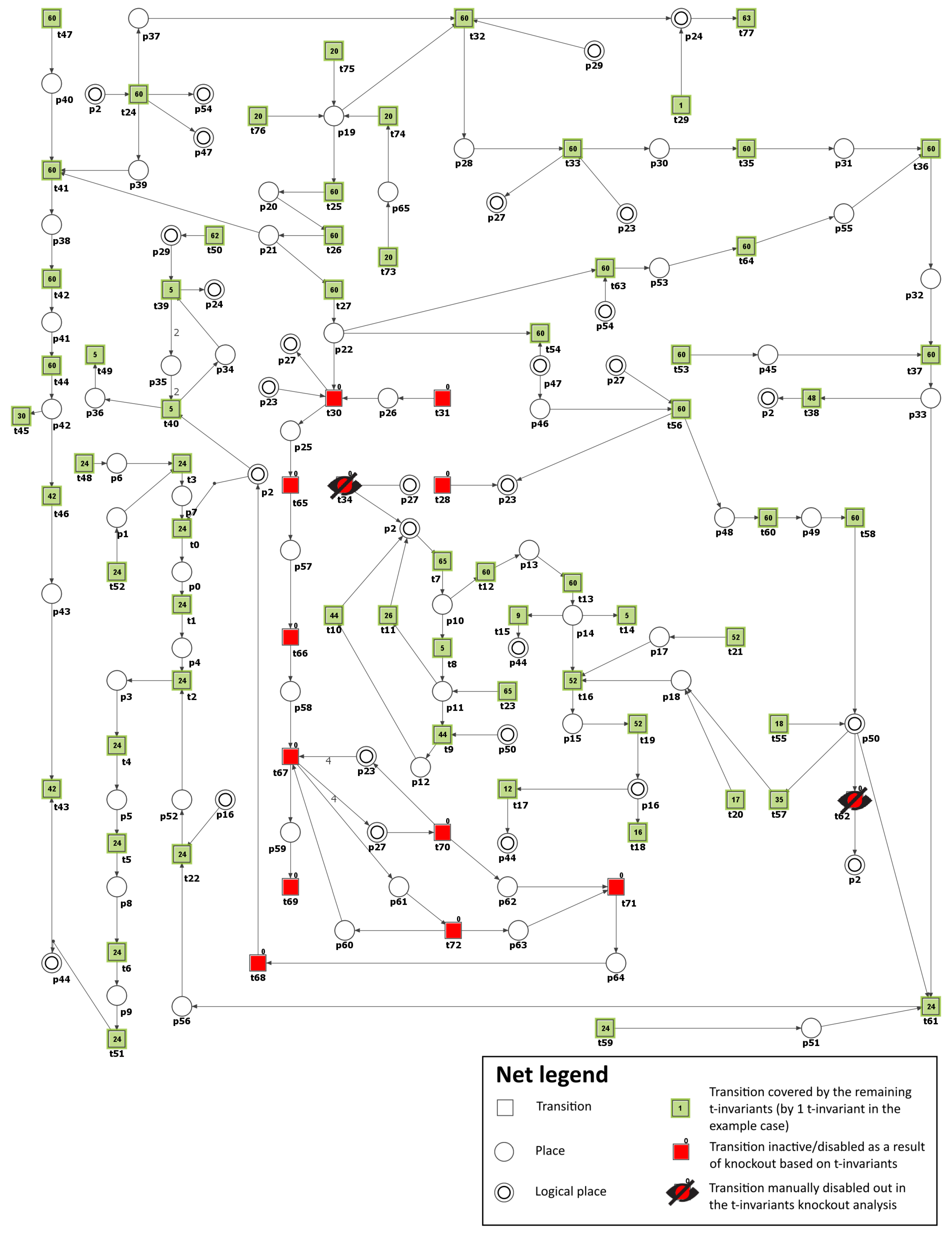
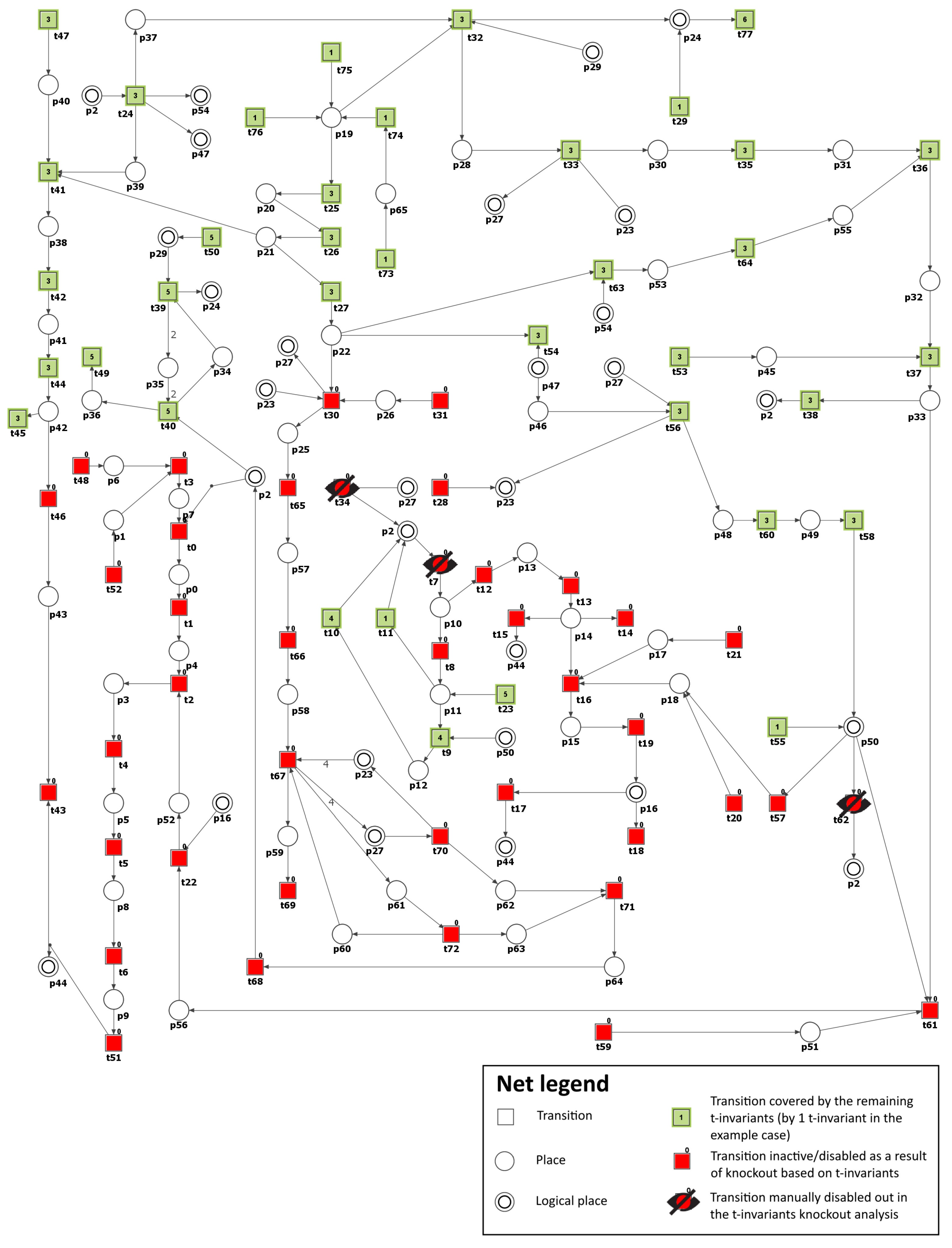

| MCT Set | Contained Transitions | Biological Interpretation |
|---|---|---|
| , , , , , , , , , , , , , , , , | Protein kinase C pathway. | |
| , , , , , , , , , , , , | Atherosclerosis development and progression affected by ROS and oxidized LDL. | |
| , , , , , , , , , , , | Increased production of ROS as a consequence of mitochondrial dysfunction resulting from hyperglycemia. | |
| , , | Activation of the thromboxane receptor (TP) leading to apoptosis, vascular muscle cell activation, and vascular cell adhesion molecules expression. | |
| , , | Metabolic pathway that converts glucose into glyceraldehyde 3-phosphate. | |
| , , | Antioxidant defense mechanism involving glutathione-dependent enzymes. | |
| , | Impact of peroxynitrite on nitric oxide (NO) synthesis. | |
| , | Vasoconstriction and endothelial dysfunction induced by PGI synthase disruption and inactivation. | |
| , | Activation of proatherogenic proteins leading to atherosclerosis progression. | |
| , | Glucose uptake and transport across the cell membrane. |
| Inhibited Process/Molecule | Knocked-Out Transitions, and MCT Sets | Disabled Transitions and MCT Sets | Number of Remaining t-Invariants (Percentage of Remaining t-Invariants) | The Number of Remaining t-Invariants That Include Transition (Atherosclerosis Progression), in Their Supports (Percentage of Remaining t-Invariants That Contain Transition in Their Supports) |
|---|---|---|---|---|
| Inhibition of PKC ( and ) pathway in glucose metabolism (see Scenario 2) | , | , , , , , , , , , , | 14 (out of 150) () | 0 (out of 90) () |
| Inhibition of polyol pathway in glucose metabolism (see Scenario 3) | , , , , , , | 27 (out of 150) () | 0 (out of 90) () | |
| Inhibition of the advanced glycation end-products (AGEs) (see Scenario 4) | , , , , , , | 27 (out of 150) () | 0 (out of 90) () | |
| Inhibition of NADPH oxidase (see Scenario 5) | , | 71 (out of 150) () | 42 (out of 90) () | |
| Inhibition of NADPH oxidase and oxidized LDL formation (see Scenario 5) | , , | , | 47 (out of 150) () | 18 (out of 90) () |
| Inhibition of NADPH oxidase and peroxynitrite formation (see Scenario 5) | , , | , , , , , , , , , , , | 6 (out of 150) () | 0 (out of 90) () |
| Inhibition of NADPH oxidase and AGE formation (see Scenario 5) | , , | , , , , , , , , , , , , | 11 (out of 150) () | 0 (out of 90) () |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybarczyk, A.; Formanowicz, D.; Formanowicz, P. Key Therapeutic Targets to Treat Hyperglycemia-Induced Atherosclerosis Analyzed Using a Petri Net-Based Model. Metabolites 2023, 13, 1191. https://doi.org/10.3390/metabo13121191
Rybarczyk A, Formanowicz D, Formanowicz P. Key Therapeutic Targets to Treat Hyperglycemia-Induced Atherosclerosis Analyzed Using a Petri Net-Based Model. Metabolites. 2023; 13(12):1191. https://doi.org/10.3390/metabo13121191
Chicago/Turabian StyleRybarczyk, Agnieszka, Dorota Formanowicz, and Piotr Formanowicz. 2023. "Key Therapeutic Targets to Treat Hyperglycemia-Induced Atherosclerosis Analyzed Using a Petri Net-Based Model" Metabolites 13, no. 12: 1191. https://doi.org/10.3390/metabo13121191
APA StyleRybarczyk, A., Formanowicz, D., & Formanowicz, P. (2023). Key Therapeutic Targets to Treat Hyperglycemia-Induced Atherosclerosis Analyzed Using a Petri Net-Based Model. Metabolites, 13(12), 1191. https://doi.org/10.3390/metabo13121191