

Effect of Cow-Calf Supplementation on Gene Expression, Processes, and Pathways Related to Adipogenesis and Lipogenesis in Longissimus thoracis Muscle of F1 Angus × Nellore Cattle at Weaning

 , , , ,
, , , ,  ,
,  and
and
Abstract
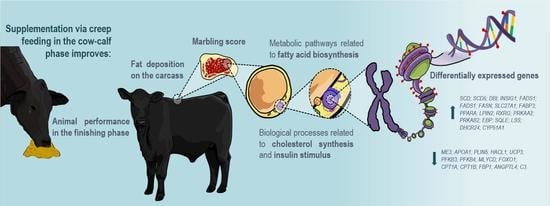
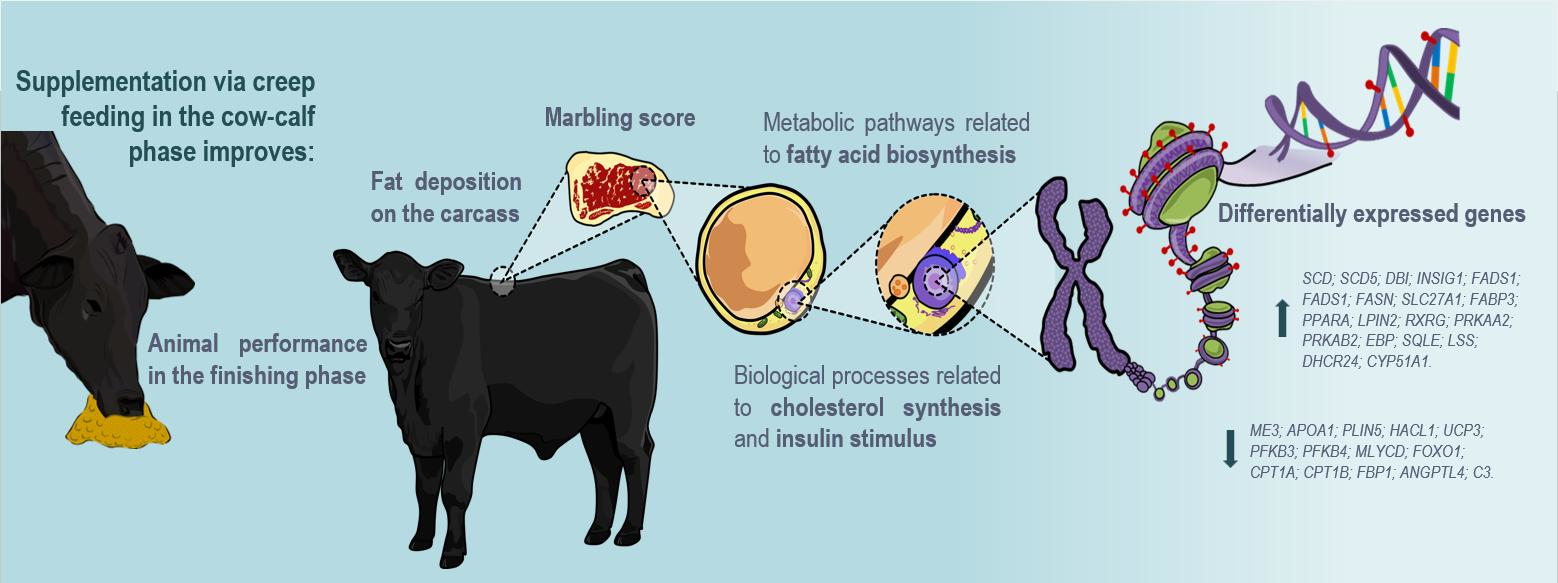
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Collection of Muscle Tissue at Weaning and Slaughter
2.3. Analysis of Meat Quality
2.4. Statistical Analysis of Weight, Weight Gain, Carcass, and Meat Data
2.5. Analysis of Differential Gene Expression
2.5.1. RNA Extraction and Sequencing
2.5.2. Mapping of Sequences to the Reference Genome and Identification of Differentially Expressed Genes
2.5.3. Functional Analysis of Differentially Expressed Genes
3. Results
3.1. Pre- and Post-Weaning Performance, Carcass and Meat Quality
3.2. Analysis of Differential Gene Expression
3.2.1. Concentration and Integrity of Total RNA
3.2.2. RNA Sequencing and Mapping of Reads to the Reference Genome
3.2.3. Identification of Differentially Expressed Genes
3.2.4. Functional Enrichment Analysis of Differentially Expressed Genes
4. Discussion
4.1. Pre- and Post-Weaning Performance, Carcass and Meat Quality
4.2. Differentially Expressed Genes and Alterations in Biological Processes and Metabolic Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheffler, J.; McCann, M.; Greiner, S.; Jiang, H.; Hanigan, M.; Bridges, G.; Lake, S.; Gerrard, D. Early metabolic imprinting events increase marbling scores in fed cattle. J. Anim. Sci. 2014, 92, 320–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantas, C.C.O.; de Mattos Negrão, F.; Geron, L.J.V.; Mexia, A.A. O uso da técnica do Creep-feeding na suplementação de bezerros. PUBVET 2010, 4, 899–904. [Google Scholar]
- Stewart, L. Creep feeding beef calves. UGA Coop. Ext. Bull. 2017, 1315, 1–8. [Google Scholar]
- Zhang, N. Epigenetic modulation of DNA methylation by nutrition and its mechanisms in animals. Anim. Nutr. 2015, 1, 144–151. [Google Scholar] [CrossRef]
- McKay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta Physiol. 2011, 202, 103–118. [Google Scholar] [CrossRef]
- Farias, N.; Ho, N.; Butler, S.; Delaney, L.; Morrison, J.; Shahrzad, S.; Coomber, B.L. The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J. Nutr. Biochem. 2015, 26, 818–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, S.A.; Befekadu, R.; Hahn-Strömberg, V.; Nilsson, T.K. DNA methylation and expression of the folate transporter genes in colorectal cancer. Tumor Biol. 2015, 36, 5581–5590. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Kennedy, A.J.; Sweatt, J.D. DNA methylation and its implications and accessibility for neuropsychiatric therapeutics. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 591. [Google Scholar] [CrossRef] [Green Version]
- Vucetic, Z.; Kimmel, J.; Totoki, K.; Hollenbeck, E.; Reyes, T.M. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology 2010, 151, 4756–4764. [Google Scholar] [CrossRef] [Green Version]
- Bogdarina, I.; Haase, A.; Langley-Evans, S.; Clark, A.J. Glucocorticoid effects on the programming of AT1b angiotensin receptor gene methylation and expression in the rat. PLoS ONE 2010, 5, e9237. [Google Scholar] [CrossRef] [Green Version]
- Jousse, C.; Parry, L.; Lambert-Langlais, S.; Maurin, A.C.; Averous, J.; Bruhat, A.; Carraro, V.; Tost, J.; Letteron, P.; Chen, P. Perinatal undernutrition affects the methylation and expression of the leptin gene in adults: Implication for the understanding of metabolic syndrome. FASEB J. 2011, 25, 3271–3278. [Google Scholar] [CrossRef] [PubMed]
- Dudley, K.J.; Sloboda, D.M.; Connor, K.L.; Beltrand, J.; Vickers, M.H. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 2011, 6, e21662. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.; Murani, E.; Schwerin, M.; Metges, C.C.; Wimmers, K.; Ponsuksili, S. Somatic cytochrome c (CYCS) gene expression and promoter-specific DNA methylation in a porcine model of prenatal exposure to maternal dietary protein excess and restriction. Br. J. Nutr. 2012, 107, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.B.; Crouse, J.D. Relative contributions of acetate, lactate and glucose to lipogenesis in bovine intramuscular and subcutaneous adipose tissue. J. Nutr. 1984, 114, 792–800. [Google Scholar] [CrossRef]
- Baldassini, W.A.; Ferreira, M.S.; Santiago, B.M.; Chardulo, L.A.L.; Curi, R.A.; Lanna, D.P.D.; Ribeiro, R.V.; Martins, C.L.; Arrigoni, M.; Neto, O.R.M. Intake, performance, meat quality and fatty acid profile of Nellore bulls finished in feedlot with diets containing dry corn gluten feed. Livest. Sci. 2021, 253, 104715. [Google Scholar] [CrossRef]
- Lopez, R.; Krehbiel, C.R.; Thomas, M.G.; Hallford, D.M.; Keisler, D.H.; Obeidat, B.S.; Hernandez, J.A.; Bryant, W.D.; Garcia, M.; Flores, R. Effect of increasing level of dietary protein on serum concentrations of metabolic hormones and mammary development in Holstein heifers consuming a moderate-energy diet. J. Dairy Sci. 2001, 84, 161. [Google Scholar]
- ABIEC. Beef Report. 2022. Available online: https://www.abiec.com.br/publicacoes/beef-report-2022/ (accessed on 19 September 2022).
- Hocquette, J.-F.; Gondret, F.; Baeza, E.; Medale, F.; Jurie, C.; Pethick, D. Intramuscular fat content in meat-producing animals: Development, genetic and nutritional control, and identification of putative markers. Animal 2010, 4, 303–319. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Ford, S.P.; Zhu, M.-J. Optimizing livestock production efficiency through maternal nutritional management and fetal developmental programming. Anim. Front. 2017, 7, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Lanna, D.; Barioni, L.; Nepomuceno, N.; Almeida, R.; Tedeschi, L. Ração de Lucro Máximo—RLM, Version 3.2.1; RLM: Piracicaba, Brazil, 2011.
- AUS-MEAT Limited. Handbook of Australian Beef Processing; AUS-MEAT Limited: Murarrie, Australia, 2018. [Google Scholar]
- Shackelford, S.; Wheeler, T.; Koohmaraie, M. Evaluation of slice shear force as an objective method of assessing beef longissimus tenderness. J. Anim. Sci. 1999, 77, 2693–2699. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 2 June 2022).
- Saini, H.K.; Griffiths-Jones, S.; Enright, A.J. Genomic analysis of human microRNA transcripts. Proc. Natl. Acad. Sci. USA 2007, 104, 17719–17724. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 June 2022).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 2 June 2022).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, gkw937. [Google Scholar] [CrossRef]
- Costa e Silva, L.F.; Engle, T.E.; de Valadares Filho, S.; Rotta, P.P.; Villadiego, F.A.C.; Silva, F.A.S.; Martins, E.C.; Silva, L.H.R.; Paulino, M.F. Nellore cows and their calves during the lactation period: Performance, intake, milk composition, and total apparent digestibility. Trop. Anim. Health Prod. 2015, 47, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.A.; Valadares Filho, S.D.C.; Henriques, L.T.; Paulino, P.V.R.; Detmann, E.; Fonseca, E.A.; Benedeti, P.D.B.; Silva, L.D.D. Nutritional requirements of nursing Nellore calves. Rev. Bras. Zootec. 2012, 41, 1212–1221. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, V.V.; Paulino, M.F.; Detmann, E.; Valadares Filho, S.C.; Lopes, S.A.; Rennó, L.N.; Sampaio, C.B.; Silva, A.G. A meta-analysis of the effects of creep-feeding supplementation on performance and nutritional characteristics by beef calves grazing on tropical pastures. Livest. Sci. 2019, 227, 175–182. [Google Scholar] [CrossRef]
- Cremin, J., Jr.; Faulkner, D.; Merchen, N.; Fahey, G., Jr.; Fernando, R.; Willms, C. Digestion criteria in nursing beef calves supplemented with limited levels of protein and energy. J. Anim. Sci. 1991, 69, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, D.; Hummel, D.; Buskirk, D.; Berger, L.; Parrett, D.; Cmarik, G. Performance and nutrient metabolism by nursing calves supplemented with limited or unlimited corn or soyhulls. J. Anim. Sci. 1994, 72, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Lopes, S.A.; Paulino, M.F.; Detmann, E.; Valente, É.E.L.; Rennó, L.N.; Valadares, R.F.D.; Cardenas, J.E.G.; de Almeida, D.M.; de Moura, F.H.; Oliveira, C.A.S. Evaluation of supplementation plans for suckling beef calves managed on tropical pasture. Semin. Ciênc. Agrár. 2017, 38, 1027–1039. [Google Scholar] [CrossRef] [Green Version]
- Asher, A.; Shabtay, A.; Cohen-Zinder, M.; Aharoni, Y.; Miron, J.; Agmon, R.; Halachmi, I.; Orlov, A.; Haim, A.; Tedeschi, L. Consistency of feed efficiency ranking and mechanisms associated with inter-animal variation among growing calves. J. Anim. Sci. 2018, 96, 990–1009. [Google Scholar] [CrossRef]
- Gerrits, W.J.; France, J.; Dijkstra, J.; Bosch, M.W.; Tolman, G.H.; Tamminga, S. Evaluation of a model integrating protein and energy metabolism in preruminant calves. J. Nutr. 1997, 127, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Hunt, M.; Garmyn, A.; O’Quinn, T.; Corbin, C.; Legako, J.; Rathmann, R.; Brooks, J.; Miller, M. Consumer assessment of beef palatability from four beef muscles from USDA Choice and Select graded carcasses. Meat Sci. 2014, 98, 1–8. [Google Scholar] [CrossRef]
- Du, M.; Huang, Y.; Das, A.; Yang, Q.; Duarte, M.; Dodson, M.; Zhu, M.-J. Meat Science and Muscle Biology Symposium: Manipulating mesenchymal progenitor cell differentiation to optimize performance and carcass value of beef cattle. J. Anim. Sci. 2013, 91, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Peng, T.; Shang, H.; Shang, X.; Zhao, X.; Qu, M.; Song, X. RNA-Seq Analysis Reveals the Potential Molecular Mechanisms of Puerarin on Intramuscular Fat Deposition in Heat-Stressed Beef Cattle. Front. Nutr. 2022, 9, 817557. [Google Scholar] [CrossRef]
- Brown, J.D.; Plutzky, J. Peroxisome proliferator–activated receptors as transcriptional nodal points and therapeutic targets. Circulation 2007, 115, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.M.; Lazar, M.A. Peroxisome proliferator-activated receptor γ in diabetes and metabolism. Trends Pharmacol. Sci. 2004, 25, 331–336. [Google Scholar] [CrossRef]
- Ladeira, M.; Schoonmaker, J.; Gionbelli, M.; Dias, J.; Gionbelli, T.; Carvalho, J.R.; Teixeira, P. Nutrigenomics and beef quality: A review about lipogenesis. Int. J. Mol. Sci. 2016, 17, 918. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.; Woodward, B.; Otter, N.; Doran, O. Relationship between the expression of key lipogenic enzymes, fatty acid composition, and intramuscular fat content of Limousin and Aberdeen Angus cattle. Livest. Sci. 2010, 127, 22–29. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Nara, T.Y. Structure, function, and dietary regulation of (delta) 6,(delta) 5, and (delta) 9 desaturases. Annu. Rev. Nutr. 2004, 24, 345. [Google Scholar] [CrossRef]
- Bionaz, M.; Chen, S.; Khan, M.J.; Loor, J.J. Functional Role of PPARs in Ruminants: Potential Targets for Fine-Tuning Metabolism during Growth and Lactation. PPAR Res. 2013, 2013, 28. [Google Scholar] [CrossRef] [Green Version]
- Ibeagha-Awemu, E.M.; Akwanji, K.A.; Beaudoin, F.; Zhao, X. Associations between variants of FADS genes and omega-3 and omega-6 milk fatty acids of Canadian Holstein cows. BMC Genet. 2014, 15, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladeira, M.M.; de Oliveira, D.M.; Schoonmaker, J.P.; Chizzotti, M.L.; Barreto, H.G.; Paiva, L.V.; Coelho, T.C.; Neto, O.R.M.; Gionbelli, M.P.; Chalfun-Junior, A. Expression of lipogenic genes in the muscle of beef cattle fed oilseeds and vitamin E. Agri Gene 2020, 15, 100097. [Google Scholar] [CrossRef]
- Duckett, S.; Gillis, M. Effects of oil source and fish oil addition on ruminal biohydrogenation of fatty acids and conjugated linoleic acid formation in beef steers fed finishing diets. J. Anim. Sci. 2010, 88, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Schroyen, M.; Li, B.; Arevalo Sureda, E.; Zhang, Y.; Leblois, J.; Deforce, D.; Van Nieuwerburgh, F.; Wavreille, J.; Everaert, N. Pre-Weaning Inulin Supplementation Alters the Ileal Transcriptome in Pigs Regarding Lipid Metabolism. Vet. Sci. 2021, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Mangaraj, M.; Nanda, R.; Panda, S. Apolipoprotein AI: A molecule of diverse function. Indian J. Clin. Biochem. 2016, 31, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poudyal, H.; Panchal, S.K.; Diwan, V.; Brown, L. Omega-3 fatty acids and metabolic syndrome: Effects and emerging mechanisms of action. Prog. Lipid Res. 2011, 50, 372–387. [Google Scholar] [CrossRef]
- Fox, J.; McGill Jr, H.; Carey, K.; Getz, G. In vivo regulation of hepatic LDL receptor mRNA in the baboon. Differential effects of saturated and unsaturated fat. J. Biol. Chem. 1987, 262, 7014–7020. [Google Scholar] [CrossRef]
- Blædel, T.; Holm, J.B.; Sundekilde, U.K.; Schmedes, M.S.; Hess, A.L.; Lorenzen, J.K.; Kristiansen, K.; Dalsgaard, T.K.; Astrup, A.; Larsen, L.H. A randomised, controlled, crossover study of the effect of diet on angiopoietin-like protein 4 (ANGPTL4) through modification of the gut microbiome. J. Nutr. Sci. 2016, 5, E45. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Kim, S.S. Probiotics and prebiotics: Present status and future perspectives on metabolic disorders. Nutrients 2016, 8, 173. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.B.; Wang, Z.S.; Peng, Q.H.; Tan, C.; Zou, H.W. Effects of different levels of protein supplementary diet on gene expressions related to intramuscular deposition in early-weaned yaks. Anim. Sci. J. 2014, 85, 411–419. [Google Scholar] [CrossRef]
- Turcotte, L.P.; Swenberger, J.R.; Yee, A.J. High carbohydrate availability increases LCFA uptake and decreases LCFA oxidation in perfused muscle. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E177–E183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, A.J.; Turcotte, L.P. Insulin fails to alter plasma LCFA metabolism in muscle perfused at similar glucose uptake. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E73–E77. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zheng, D.; Peng, Z.; Zhu, Y.; Li, R.; Wu, Q.; Li, Y.; Li, H.; Xu, W.; Zhang, M. Identification of differentially expressed genes and lipid metabolism signaling pathways between muscle and fat tissues in broiler chickens. J. Poult. Sci. 2021, 58, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Graugnard, D.E.; Piantoni, P.; Bionaz, M.; Berger, L.L.; Faulkner, D.B.; Loor, J.J. Adipogenic and energy metabolism gene networks in longissimus lumborum during rapid post-weaning growth in Angus and Angus× Simmental cattle fed high-starch or low-starch diets. BMC Genom. 2009, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpkin, J.C.; Yellon, D.M.; Davidson, S.M.; Lim, S.Y.; Wynne, A.M.; Smith, C.C. Apelin-13 and apelin-36 exhibit direct cardioprotective activity against ischemiareperfusion injury. Basic Res. Cardiol. 2007, 102, 518–528. [Google Scholar] [CrossRef]
- Castan-Laurell, I.; Dray, C.; Knauf, C.; Kunduzova, O.; Valet, P. Apelin, a promising target for type 2 diabetes treatment? Trends Endocrinol. Metab. 2012, 23, 234–241. [Google Scholar] [CrossRef]
- Ruderman, N.; Prentki, M. AMP kinase and malonyl-CoA: Targets for therapy of the metabolic syndrome. Nat. Rev. Drug Discov. 2004, 3, 340–351. [Google Scholar] [CrossRef]
- Bouzakri, K.; Austin, R.; Rune, A.; Lassman, M.E.; Garcia-Roves, P.M.; Berger, J.P.; Krook, A.; Chibalin, A.V.; Zhang, B.B.; Zierath, J.R. Malonyl CoenzymeA decarboxylase regulates lipid and glucose metabolism in human skeletal muscle. Diabetes 2008, 57, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- Dyck, J.R.; Hopkins, T.A.; Bonnet, S.; Michelakis, E.D.; Young, M.E.; Watanabe, M.; Kawase, Y.; Jishage, K.-I.; Lopaschuk, G.D. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 2006, 114, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Weyer, C.; Tataranni, P.A.; Pratley, R.E. Insulin action and insulinemia are closely related to the fasting complement C3, but not acylation stimulating protein concentration. Diabetes Care 2000, 23, 779–785. [Google Scholar] [CrossRef] [Green Version]
- Barbu, A.; Hamad, O.A.; Lind, L.; Ekdahl, K.N.; Nilsson, B. The role of complement factor C3 in lipid metabolism. Mol. Immunol. 2015, 67, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Picard, B.; Gagaoua, M. Muscle fiber properties in cattle and their relationships with meat qualities: An overview. J. Agric. Food Chem. 2020, 68, 6021–6039. [Google Scholar] [CrossRef] [PubMed]
- Aragão, R.D.S.; Guzmán-Quevedo, O.; Pérez-García, G.; Manhaes-de-Castro, R.; Bolanos-Jiménez, F. Maternal protein restriction impairs the transcriptional metabolic flexibility of skeletal muscle in adult rat offspring. Br. J. Nutr. 2014, 112, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Hariharan, K.; TidAng, J.; Frangioudakis, G.; Beale, S.M.; Wright, L.E.; Zeng, X.Y.; Leslie, S.J.; Li, J.-Y.; Kraegen, E.W. Enhancement of muscle mitochondrial oxidative capacity and alterations in insulin action are lipid species dependent: Potent tissue-specific effects of medium-chain fatty acids. Diabetes 2009, 58, 2547–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandstetter, A.M.; Picard, B.; Geay, Y. Muscle fibre characteristics in four muscles of growing bulls: I. Postnatal differentiation. Livest. Prod. Sci. 1998, 53, 15–23. [Google Scholar] [CrossRef]
- Huang, J.; Zhu, R.; Shi, D. The role of FATP1 in lipid accumulation: A review. Mol. Cell. Biochem. 2021, 476, 1897–1903. [Google Scholar] [CrossRef]
- Jeong, J.; Kwon, E.G.; Im, S.K.; Seo, K.S.; Baik, M. Expression of fat deposition and fat removal genes is associated with intramuscular fat content in longissimus dorsi muscle of Korean cattle steers. J. Anim. Sci. 2012, 90, 2044–2053. [Google Scholar] [CrossRef]
- Chen, X.; Luo, Y.; Wang, R.; Zhou, B.; Huang, Z.; Jia, G.; Zhao, H.; Liu, G. Effects of fatty acid transport protein 1 on proliferation and differentiation of porcine intramuscular preadipocytes. Anim. Sci. J. 2017, 88, 731–738. [Google Scholar] [CrossRef]
- Qi, R.; Long, D.; Wang, J.; Wang, Q.; Huang, X.; Cao, C.; Gao, G.; Huang, J. MicroRNA-199a targets the fatty acid transport protein 1 gene and inhibits the adipogenic trans-differentiation of C2C12 myoblasts. Cell. Physiol. Biochem. 2016, 39, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Xie, L.; Ma, J.-E.; Luo, W.; Zhang, L.; Chao, Z.; Chen, S.; Nie, Q.; Lin, Z.; Zhang, X. Lower expression of SLC27A1 enhances intramuscular fat deposition in chicken via down-regulated fatty acid oxidation mediated by CPT1A. Front. Physiol. 2017, 8, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitart, M.; Osorio-Conles, Ó.; Pentinat, T.; Cebrià, J.; García-Villoria, J.; Sala, D.; Sebastián, D.; Zorzano, A.; Ribes, A.; Jiménez-Chillarón, J.C. Fatty acid transport protein 1 (FATP1) localizes in mitochondria in mouse skeletal muscle and regulates lipid and ketone body disposal. PLoS ONE 2014, 9, e98109. [Google Scholar] [CrossRef]
- Lobo, S.; Wiczer, B.M.; Smith, A.J.; Hall, A.M.; Bernlohr, D.A. Fatty acid metabolism in adipocytes: Functional analysis of fatty acid transport proteins 1 and 4. J. Lipid Res. 2007, 48, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.; Schoonjans, K.; Lefebvre, A.-M.; Staels, B.; Auwerx, J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARα and PPARγ activators. J. Biol. Chem. 1997, 272, 28210–28217. [Google Scholar] [CrossRef] [Green Version]
- Qi, R.; Feng, M.; Tan, X.; Gan, L.; Yan, G.; Sun, C. FATP1 silence inhibits the differentiation and induces the apoptosis in chicken preadipocytes. Mol. Biol. Rep. 2013, 40, 2907–2914. [Google Scholar] [CrossRef]
- Liu, X.; Li, S.; Wang, L.; Zhang, W.; Wang, Y.; Gui, L.; Zan, L.; Zhao, C. The effect of FATP1 on adipocyte differentiation in Qinchuan beef cattle. Animals 2021, 11, 2789. [Google Scholar] [CrossRef]
- La Cour Poulsen, L.; Siersbæk, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell. Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Kitamura, T.; Kitamura, Y.; Biggs III, W.H.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell 2003, 4, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Ioannilli, L.; Ciccarone, F.; Ciriolo, M.R. Adipose tissue and FoxO1: Bridging physiology and mechanisms. Cells 2020, 9, 849. [Google Scholar] [CrossRef] [Green Version]
- Dowell, P.; Otto, T.C.; Adi, S.; Lane, M.D. Convergence of peroxisome proliferator-activated receptor γ and Foxo1 signaling pathways. J. Biol. Chem. 2003, 278, 45485–45491. [Google Scholar] [CrossRef] [Green Version]
- Attie, A.D. Insig: A significant integrator of nutrient and hormonal signals. J. Clin. Investig. 2004, 113, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Kast-Woelbern, H.R.; Dana, S.L.; Cesario, R.M.; Sun, L.; de Grandpre, L.Y.; Brooks, M.E.; Osburn, D.L.; Reifel-Miller, A.; Klausing, K.; Leibowitz, M.D. Rosiglitazone induction of Insig-1 in white adipose tissue reveals a novel interplay of peroxisome proliferator-activated receptor γ and sterol regulatory element-binding protein in the regulation of adipogenesis. J. Biol. Chem. 2004, 279, 23908–23915. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Takaishi, K.; Cook, W.; McCorkle, S.K.; Unger, R.H. Insig-1 “brakes” lipogenesis in adipocytes and inhibits differentiation of preadipocytes. Proc. Natl. Acad. Sci. USA 2003, 100, 9476–9481. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, P.D.; Oliveira, D.M.; Chizzotti, M.L.; Chalfun-Junior, A.; Coelho, T.C.; Gionbelli, M.; Paiva, L.V.; Carvalho, J.R.R.; Ladeira, M.M. Subspecies and diet affect the expression of genes involved in lipid metabolism and chemical composition of muscle in beef cattle. Meat Sci. 2017, 133, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Reue, K. The lipin family: Mutations and metabolism. Curr. Opin. Lipidol. 2009, 20, 165. [Google Scholar] [CrossRef]
- Zhang, P.; Reue, K. Lipin proteins and glycerolipid metabolism: Roles at the ER membrane and beyond. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1583–1595. [Google Scholar] [CrossRef]
- Phan, J.; Reue, K. Lipin, a lipodystrophy and obesity gene. Cell Metab. 2005, 1, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Reue, K.; Donkor, J. Lipin: A determinant of adiposity, insulin sensitivity and energy balance. Future Lipidol. 2006, 1, 91–101. [Google Scholar] [CrossRef]
- Péterfy, M.; Phan, J.; Xu, P.; Reue, K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet. 2001, 27, 121–124. [Google Scholar] [CrossRef]
- Reue, K.; Xu, P.; Wang, X.-P.; Slavin, B.G. Adipose tissue deficiency, glucose intolerance, and increased atherosclerosis result from mutation in the mouse fatty liver dystrophy (fld) gene. J. Lipid Res. 2000, 41, 1067–1076. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BWi (kg) | WW (kg) | ADG1 (kg) | BWf (kg) | ADG2 (kg) | HCW (kg) | |
|---|---|---|---|---|---|---|
| G1 1 | 61.29 ± 2.41 | 228.92 ± 5.07 b | 0.93 ± 0.02 b | 484.64 ± 5.96 | 1.36 ± 0.02 | 269.22 ± 8.23 |
| G2 1 | 57.55 ± 2.61 | 243.57 ± 5.70 a | 1.03 ± 0.03 a | 491.85 ± 7.85 | 1.32 ± 0.03 | 273.62 ± 9.28 |
| BFT (mm) | IMF (%) | MS | REA (cm2) | WBSF7 (kg) | WBSF14 (kg) | |
|---|---|---|---|---|---|---|
| G1 1 | 10.61 ± 0.42 b | 4.95 ± 0.20 b | 321.50 ± 13.65 b | 67.94 ± 1.16 | 4.52 ± 0.11 | 3.45 ± 0.11 |
| G2 1 | 12.96 ± 0.83 a | 5.80 ± 0.23 a | 366.11 ± 12.39 a | 65.50 ± 0.93 | 4.28 ± 0.12 | 3.42 ± 0.09 |
| Animal/Sample | No. of Generated PE Reads | No. of Mapped PE Reads | No. of Uniquely Mapped PE Reads | % of Uniquely Mapped PE Reads |
|---|---|---|---|---|
| 1/RC1 | 11,679,440 | 11,506,606 | 11,203,820 | 95.93 |
| 2/RC2 | 10,814,468 | 10,674,013 | 10,406,095 | 96.22 |
| 3/RC3 | 9,853,062 | 9,675,863 | 9,402,096 | 95.42 |
| 4/RC4 | 10,773,912 | 10,497,727 | 10,223,070 | 94.89 |
| 5/RC5 | 10,407,984 | 10,276,901 | 10,003,083 | 96.11 |
| 6/RC6 | 9,827,847 | 9,684,304 | 9,411,731 | 95.77 |
| 7/RC7 | 10,068,925 | 9,867,642 | 9,628,738 | 95.63 |
| 8/RC8 | 9,866,300 | 9,636,103 | 9,384,361 | 95.12 |
| 9/RC9 | 9,846,398 | 9,631,769 | 9,327,987 | 94.74 |
| 10/RC10 | 9,854,664 | 9,722,289 | 9,476,470 | 96.16 |
| 11/RC11 | 9,880,030 | 9,722,399 | 9,479,344 | 95.94 |
| 12/RC12 | 12,482,586 | 12,229,115 | 11,918,101 | 95.48 |
| Mean | 10,446,301 | 10,260,394 | 9,988,741 | 95.62 |
| Animal/Sample | No. of Generated PE Reads | No. of Mapped PE Reads | No. of Uniquely Mapped PE Reads | % of Uniquely Mapped PE Reads |
|---|---|---|---|---|
| 13/RC13 | 9,479,645 | 9,250,146 | 9,033,774 | 95.30 |
| 14/RC14 | 10,640,508 | 10,341,889 | 10,068,523 | 94.62 |
| 15/RC15 | 9,553,313 | 9,373,266 | 9,141,017 | 95.68 |
| 16/RC16 | 9,333,592 | 9,207,412 | 8,986,543 | 96.28 |
| 17/RC17 | 9,129,520 | 9,022,713 | 8,783,636 | 96.21 |
| 18/RC18 | 10,213,502 | 10,046,519 | 9,797,829 | 95.93 |
| 19/RC19 | 10,152,415 | 10,000,483 | 9,757,874 | 96.11 |
| 20/RC20 | 10,134,139 | 9,981,276 | 9,736,555 | 96.08 |
| 21/RC21 | 9,651,481 | 9,260,150 | 9,018,951 | 93.45 |
| 22/RC22 | 8,606,966 | 8,434,895 | 8,233,201 | 95.66 |
| 23/RC23 | 9,538,157 | 9,359,896 | 9,132,297 | 95.74 |
| 24/RC24 | 9,435,252 | 9,215,591 | 8,995,317 | 95.34 |
| Mean | 9,655,708 | 9,457,853 | 9,223,793 | 95.53 |
| Gene ID Ensembl | Gene Symbol | Regulated 1 | log2 FC 2 | FDR 3 |
|---|---|---|---|---|
| ENSBTAG00000017280 | C3 | Down | −2.88 | 2.35 × 10−19 |
| ENSBTAG00000046307 | CEBPD | Down | −2.56 | 2.31 × 10−16 |
| ENSBTAG00000021672 | RGS1 | Down | −2.51 | 2.70 × 10−16 |
| ENSBTAG00000048501 | ST8SIA2 | Up | 2.37 | 8.32 × 10−15 |
| ENSBTAG00000011121 | CLCN4 | Up | 1.02 | 1.52 × 10−14 |
| ENSBTAG00000014069 | PDK4 | Down | −3.34 | 1.52 × 10−14 |
| ENSBTAG00000004248 | MLYCD | Down | −1.89 | 2.63 × 10−14 |
| ENSBTAG00000013242 | MYMK | Up | 1.47 | 2.67 × 10−14 |
| ENSBTAG00000002834 | CCDC69 | Up | 1.13 | 6.93 × 10−14 |
| ENSBTAG00000022989 | FAM174B | Up | 1.12 | 8.62 × 10−14 |
| ENSBTAG00000013631 | GLUL | Down | −1.54 | 1.08 × 10−13 |
| ENSBTAG00000017956 | KCTD15 | Up | 1.02 | 1.27 × 10−13 |
| ENSBTAG00000013860 | GADD45A | Down | −1.96 | 1.75 × 10−13 |
| ENSBTAG00000002783 | PCYOX1 | Up | 0.83 | 6.15 × 10−13 |
| ENSBTAG00000007890 | SEC14L5 | Up | 2.07 | 6.85 × 10−13 |
| ENSBTAG00000021999 | CPT1A | Down | −1.70 | 7.95 × 10−13 |
| ENSBTAG00000004118 | ALAS1 | Up | 0.74 | 1.13 × 10−12 |
| ENSBTAG00000046548 | ST6GALNAC4 | Up | 0.86 | 8.42 × 10−12 |
| ENSBTAG00000007578 | SHTN1 | Down | −1.69 | 2.05 × 10−11 |
| ENSBTAG00000050158 | ---------- | Up | 0.86 | 2.05 × 10−11 |
| ENSBTAG00000011909 | ACVR1 | Up | 0.62 | 3.64 × 10−11 |
| ENSBTAG00000012314 | LDLR | Up | 2.71 | 4.10 × 10−11 |
| ENSBTAG00000015942 | DNAJA4 | Up | 1.33 | 4.11 × 10−11 |
| ENSBTAG00000014265 | SREBF2 | Up | 0.90 | 4.15 × 10−11 |
| ENSBTAG00000050852 | CXCL9 | Down | −2.34 | 4.23 × 10−11 |
| ENSBTAG00000000163 | DDIT4 | Down | −1.16 | 5.15 × 10−11 |
| ENSBTAG00000011437 | ---------- | Down | −1.54 | 6.69 × 10−11 |
| ENSBTAG00000016819 | FABP3 | Up | 1.41 | 7.36 × 10−11 |
| ENSBTAG00000048728 | ---------- | Up | 1.36 | 7.55 × 10−11 |
| ENSBTAG00000017280 | PMEPA1 | Up | 0.90 | 1.19 × 10−10 |
| Term (KEGG) | Up/Down | No. of Genes | p-Value | Genes |
|---|---|---|---|---|
| PPAR signaling pathway | Up | 9 | <0.001 | FADS2, FABP3, SLC27A1, SCD, SCD5, AQP7, DBI, PPARA, RXRG |
| Steroid biosynthesis | Up | 5 | <0.001 | SQLE, EBP, CYP51A1, DHCR24, LSS |
| Biosynthesis of unsaturated fatty acids | Up | 4 | 0.029 | FADS2, SCD, SCD5, FADS1 |
| Apelin signaling pathway | Up | 8 | 0.039 | PRKAB2, SMAD3, PRKAA2, CCND1, MYL2, MYL3, APLNR, CALM3 |
| Fatty acid metabolism | Up | 5 | 0.041 | FADS2, SCD, FASN, SCD5, FADS1 |
| AMPK signaling pathway | Down | 8 | 0.023 | PFKFB4, CPT1A, PFKFB3, EIF4EBP1, MLYCD, CPT1B, FBP1, FOXO1 |
| Glucagon signaling pathway | Down | 7 | 0.031 | CPT1A, GCGR, SIK1, CPT1B, FBP1, PLCB2, FOXO1 |
| PPAR signaling pathway | Down | 6 | 0.044 | CPT1A, APOA1, ME3, ANGPTL4, CPT1B, PLIN5 |
| Term (GO_BP) | Up/Down | No. of Genes | p-Value | Genes |
|---|---|---|---|---|
| Cholesterol biosynthetic process | Up | 5 | 0.002 | EBP, INSIG1, CYP51A1, DHCR24, LSS |
| Unsaturated fatty acid biosynthetic process | Up | 4 | 0.003 | FADS2, SCD, SCD5, FADS1 |
| Sterol biosynthetic process | Up | 3 | 0.021 | SQLE, EBP, INSIG1 |
| Cellular response to insulin stimulus | Up | 5 | 0.032 | GOT1, INSIG1, INHBB, LPIN2, HDAC9 |
| Positive regulation of lipid storage | Down | 4 | <0.001 | C3, IKBKE, FAM71F2, PLIN5 |
| Fatty acid metabolic process | Down | 6 | 0.005 | C3, CPT1A, ACOT7, UCP3, HACL1, CPT1B |
| Negative regulation of glycolytic process | Down | 3 | 0.022 | DDIT4, NUPR1, FBP1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-Zamudio, G.D.; Ganga, M.J.G.; Pereira, G.L.; Nociti, R.P.; Chiaratti, M.R.; Cooke, R.F.; Chardulo, L.A.L.; Baldassini, W.A.; Machado-Neto, O.R.; Curi, R.A. Effect of Cow-Calf Supplementation on Gene Expression, Processes, and Pathways Related to Adipogenesis and Lipogenesis in Longissimus thoracis Muscle of F1 Angus × Nellore Cattle at Weaning. Metabolites 2023, 13, 160. https://doi.org/10.3390/metabo13020160
Ramírez-Zamudio GD, Ganga MJG, Pereira GL, Nociti RP, Chiaratti MR, Cooke RF, Chardulo LAL, Baldassini WA, Machado-Neto OR, Curi RA. Effect of Cow-Calf Supplementation on Gene Expression, Processes, and Pathways Related to Adipogenesis and Lipogenesis in Longissimus thoracis Muscle of F1 Angus × Nellore Cattle at Weaning. Metabolites. 2023; 13(2):160. https://doi.org/10.3390/metabo13020160
Chicago/Turabian StyleRamírez-Zamudio, Germán Darío, Maria Júlia Generoso Ganga, Guilherme Luis Pereira, Ricardo Perecin Nociti, Marcos Roberto Chiaratti, Reinaldo Fernandes Cooke, Luis Artur Loyola Chardulo, Welder Angelo Baldassini, Otávio Rodrigues Machado-Neto, and Rogério Abdallah Curi. 2023. "Effect of Cow-Calf Supplementation on Gene Expression, Processes, and Pathways Related to Adipogenesis and Lipogenesis in Longissimus thoracis Muscle of F1 Angus × Nellore Cattle at Weaning" Metabolites 13, no. 2: 160. https://doi.org/10.3390/metabo13020160