
Neurotransmitters in Type 2 Diabetes and the Control of Systemic and Central Energy Balance

 , ,
, ,
Abstract
:1. Introduction
2. Type 2 Diabetes and Neurological Complications
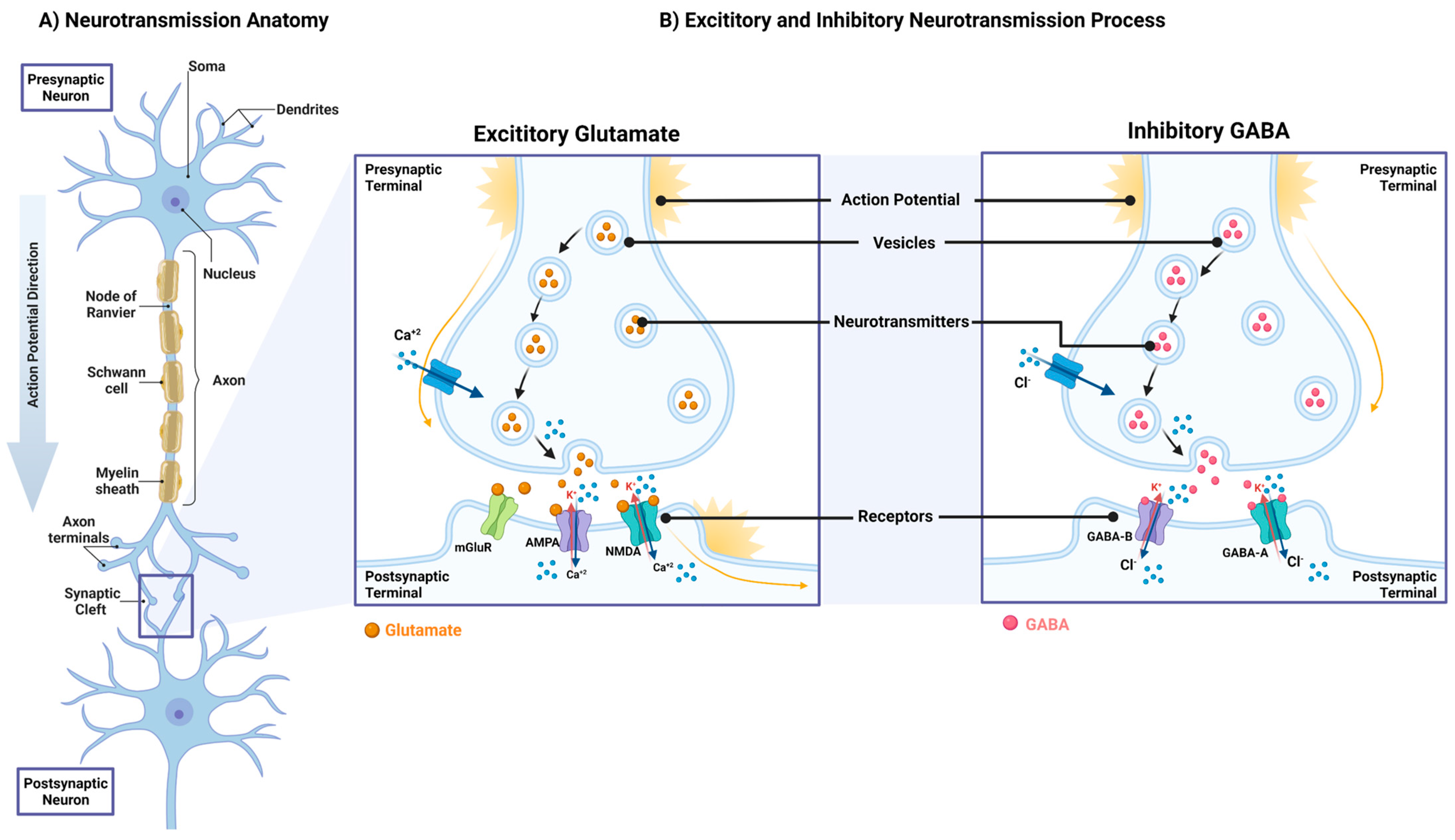
3. Neurotransmitters

{kind=link}
{kind=link}
{kind=link}
| Category | Neurotransmitter | Function |
|---|---|---|
| Amino Acids | Gamma-aminobutyric acid (GABA) | Learning, memory, locomotion, metabolism mediators, appetite regulation [63,64] |
| Glutamate (Glu) | Memory, learning, cognition, appetite regulation [48,64] | |
| Glycine (Gly) | Motor control, sensory and auditory processing, cardiovascular, and respiratory functions [65] | |
| Amines | Dopamine (DA) | Motivation, memory, attention, locomotion control [66] |
| Norepinephrine (NE) | Emotional arousal, regulating blood pressure, mood, appetite [49] | |
| Epinephrine | Boosts oxygen and glucose supply to brain and muscles, increases awareness [48] | |
| Serotonin (5-HT) | Regulate sleep–wake cycle, mood, appetite and digestion [65] | |
| Histamine | Regulate sleep–wake cycle, stress response, appetite and memory [67] | |
| Acetyl Choline | Acetylcholine (Ach) | Cognition, learning, memory, modulation of electrical, and mechanical functions of the heart [51,68] |
| Other | Nitric Oxide (NO) | Learning, memory, homeostatic functions [69] |
| Hydrogen Sulfide (HS) | Neuromodulator, smooth muscle relaxation [48] | |
| Purines (ATP) | Controls intracellular energy homoeostasis, autonomic control, sensory transduction [70,71] |
4. Effect of Metabolic Dysregulation on Neurotransmitters Functions
5. Role of The Hypothalamus in Controlling Energy Balance
5.1. Hypothalamic Neurotransmitters
5.2. Central Melanocortin System
6. Regulating CNS Energetic Demands
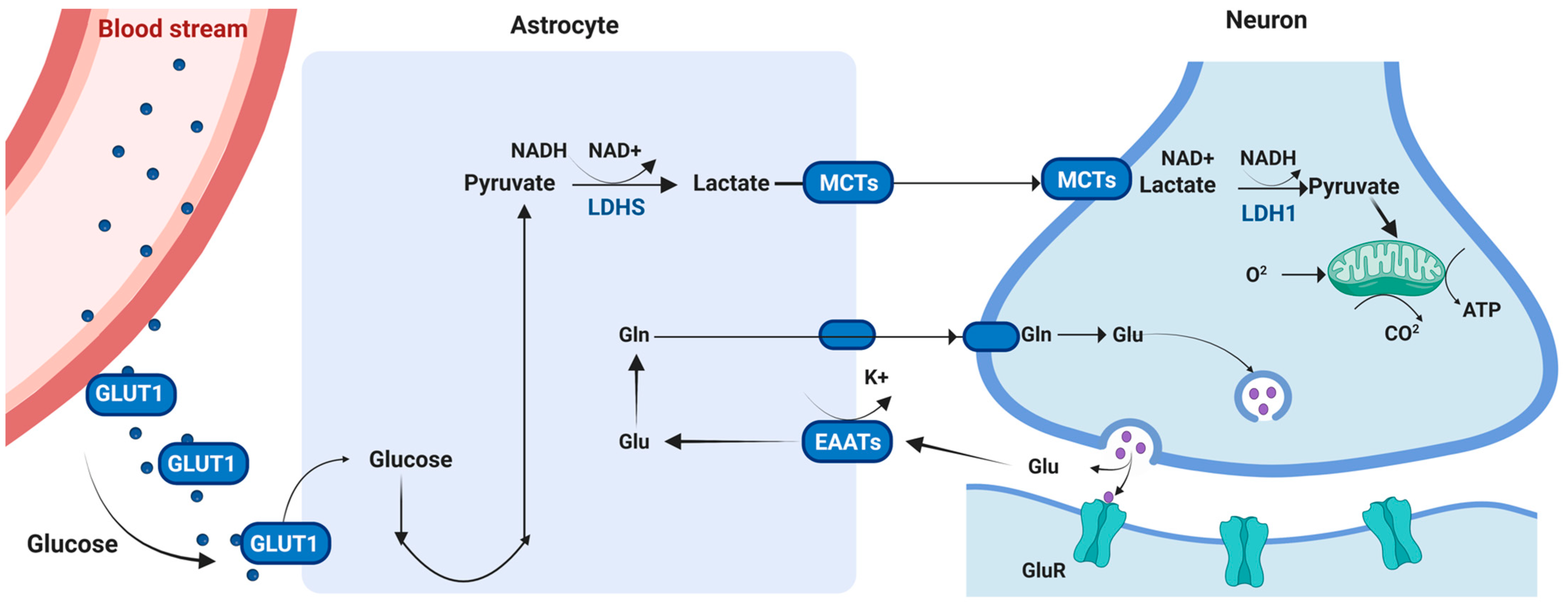
6.1. Energy Metabolism in the Brain
6.2. Energy Substrates: Glucose, Ketone Bodies and Lactate
| Glucose Transporter | Function |
|---|---|
| Glut1 | Maintaining glucose transportation into the CNS via BBB |
| Glut3 | Highly distributed in the brain and expressed in neurophil synapses activities |
| Glut4 | Regulating insulin signaling in the CNS |
| Glut8 | Supporting neuron cells glucose requirements |
6.3. Cell-Type Specific Mechanisms
6.4. Neuronal Bioenergetics and the Regulation of Neurotransmission
7. Metabolic Factors Affecting Neurotransmission
7.1. Glucose Metabolism
7.2. Glycemic Variability
7.3. Insulin Signaling
7.4. Lipid Metabolism
8. Perspectives and Future Work in the Field
8.1. Emerging Role of Inflammation in Neurotransmitter Function
8.2. Neuroinflammation
8.3. Hypothalamic Inflammation
8.4. Hypothalamic Regulation of Energy Homeostasis: The Roles of Sleep and Thermogenesis
8.5. Therapeutic Approaches for Neurological Disorders
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization (WHO). Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 16 September 2022).
- International Diabetes Federation. IDF Diabetes Atlas 2021, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021; Available online: https://diabetesatlas.org/idfawp/resource-files/2021/07/IDF_Atlas_10th_Edition_2021.pdf (accessed on 10 February 2023).
- Malone, J.I.; Hansen, B.C. Does obesity cause type 2 diabetes mellitus (T2DM)? Or is it the opposite? Pediatr. Diabetes 2019, 20, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denis, G.V.; Obin, M.S. “Metabolically healthy obesity”: Origins and implications. Mol. Aspects Med. 2013, 34, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, K.D.; Farooqi, I.S.; Friedman, J.M.; Klein, S.; Loos, R.J.F.; Mangelsdorf, D.J.; O’Rahilly, S.; Ravussin, E.; Redman, L.M.; Ryan, D.H.; et al. The energy balance model of obesity: Beyond calories in, calories out. Am. J. Clin. Nutr. 2022, 115, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.F.; Laube, H. Obesity and diabetes mellitus. Adv. Metab. Disord. 1974, 7, 243–255. [Google Scholar] [PubMed]
- Kim, K.-S.; Choi, Y.K.; Kim, M.J.; Hwang, J.W.; Min, K.; Jung, S.Y.; Kim, S.-K.; Choi, Y.-S.; Cho, Y.-W. Umbilical Cord-Mesenchymal Stem Cell-Conditioned Medium Improves Insulin Resistance in C2C12 Cell. Diabetes Metab. J. 2021, 45, 260–269. [Google Scholar] [CrossRef]
- Eo, H.; Valentine, R.J. Saturated Fatty Acid-Induced Endoplasmic Reticulum Stress and Insulin Resistance Are Prevented by Imoxin in C2C12 Myotubes. Front. Physiol. 2022, 13, 842819. [Google Scholar] [CrossRef]
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia 2021, 64, 994–1006. [Google Scholar] [CrossRef]
- Bäckdahl, J.; Franzén, L.; Massier, L.; Li, Q.; Jalkanen, J.; Gao, H.; Andersson, A.; Bhalla, N.; Thorell, A.; Rydén, M.; et al. Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab. 2021, 33, 1869–1882.e6. [Google Scholar] [CrossRef]
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 2020, 579, 279–283. [Google Scholar] [CrossRef]
- Dimitriadis, G.D.; Maratou, E.; Kountouri, A.; Board, M.; Lambadiari, V. Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach. Nutrients 2021, 13, 159. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Karamanou, M.; Protogerou, A.; Tsoucalas, G.; Androutsos, G.; Poulakou-Rebelakou, E. Milestones in the history of diabetes mellitus: The main contributors. World J. Diabetes 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Margolis, R.U.; Altszuler, N. Insulin in the Cerebrospinal Fluid. Nature 1967, 215, 1375–1376. [Google Scholar] [CrossRef]
- Devaskar, S.U.; Giddings, S.J.; Rajakumar, P.A.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 1994, 269, 8445–8454. [Google Scholar] [CrossRef]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef] [Green Version]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef]
- Cai, W.; Xue, C.; Sakaguchi, M.; Konishi, M.; Shirazian, A.; Ferris, H.A.; Li, M.E.; Yu, R.; Kleinridders, A.; Pothos, E.N.; et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Investig. 2018, 128, 2914–2926. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, R.; Dargahi, L.; Haeri, A.; Moosavi, M.; Mohamed, Z.; Ahmadiani, A. Brain Insulin Dysregulation: Implication for Neurological and Neuropsychiatric Disorders. Mol. Neurobiol. 2013, 47, 1045–1065. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.-U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of Brain Insulin Receptor in Control of Body Weight and Reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Vogt, M.C.; Brüning, J.C. CNS insulin signaling in the control of energy homeostasis and glucose metabolism—From embryo to old age. Trends Endocrinol. Metab. 2013, 24, 76–84. [Google Scholar] [CrossRef]
- Okamoto, H.; Nakae, J.; Kitamura, T.; Park, B.-C.; Dragatsis, I.; Accili, D. Transgenic rescue of insulin receptor–deficient mice. J. Clin. Investig. 2004, 114, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Erichsen, J.M.; Fadel, J.R.; Reagan, L.P. Peripheral versus central insulin and leptin resistance: Role in metabolic disorders, cognition, and neuropsychiatric diseases. Neuropharmacology 2022, 203, 108877. [Google Scholar] [CrossRef]
- Ruud, J.; Steculorum, S.M.; Brüning, J.C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 2017, 8, 15259. [Google Scholar] [CrossRef] [Green Version]
- Grote, C.W.; Groover, A.L.; Ryals, J.M.; Geiger, P.C.; Feldman, E.L.; Wright, D.E. Peripheral nervous system insulin resistance in ob/ob mice. Acta Neuropathol. Commun. 2013, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Palta, P.; Carlson, M.C.; Crum, R.M.; Colantuoni, E.; Sharrett, A.R.; Yasar, S.; Nahin, R.L.; DeKosky, S.T.; Snitz, B.; Lopez, O.; et al. Diabetes and Cognitive Decline in Older Adults: The Ginkgo Evaluation of Memory Study. J. Gerontol. Ser. A 2018, 73, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Spauwen, P.J.J.; Köhler, S.; Verhey, F.R.J.; Stehouwer, C.D.A.; van Boxtel, M.P.J. Effects of Type 2 Diabetes on 12-Year Cognitive Change. Diabetes Care 2013, 36, 1554–1561. [Google Scholar] [CrossRef] [Green Version]
- Tuligenga, R.H.; Dugravot, A.; Tabák, A.G.; Elbaz, A.; Brunner, E.J.; Kivimäki, M.; Singh-Manoux, A. Midlife type 2 diabetes and poor glycaemic control as risk factors for cognitive decline in early old age: A post-hoc analysis of the Whitehall II cohort study. Lancet Diabetes Endocrinol. 2014, 2, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Serrano, A.M.; Duarte, J.M.N. Brain Metabolism Alterations in Type 2 Diabetes: What Did We Learn from Diet-Induced Diabetes Models? Front. Neurosci. 2020, 14, 229. [Google Scholar] [CrossRef] [Green Version]
- Karvani, M.; Simos, P.; Stavrakaki, S.; Kapoukranidou, D. Neurocognitive impairment in type 2 diabetes mellitus. Hormones 2019, 18, 523–534. [Google Scholar] [CrossRef]
- Wang, K.-C.; Woung, L.-C.; Tsai, M.-T.; Liu, C.-C.; Su, Y.-H.; Li, C.-Y. Risk of Alzheimer’s Disease in Relation to Diabetes: A Population-Based Cohort Study. Neuroepidemiology 2012, 38, 237–244. [Google Scholar] [CrossRef]
- González, A.; Calfío, C.; Churruca, M.; Maccioni, R.B. Glucose metabolism and AD: Evidence for a potential diabetes type 3. Alzheimer’s Res. Ther. 2022, 14, 56. [Google Scholar] [CrossRef]
- Thomas, K.R.; Bangen, K.J.; Weigand, A.J.; Edmonds, E.C.; Sundermann, E.; Wong, C.G.; Eppig, J.; Werhane, M.L.; Delano-Wood, L.; Bondi, M.W. Type 2 Diabetes Interacts with Alzheimer Disease Risk Factors to Predict Functional Decline. Alzheimer Dis. Assoc. Disord. 2020, 34, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Mechanistic Insight into Oxidative Stress-Triggered Signaling Pathways and Type 2 Diabetes. Molecules 2022, 27, 950. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J. Neurol. Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Wang, F.; Wang, J.; Liu, C.; Zhou, Y.; Xu, Z.; Zhang, C.; Sun, B.; Guan, Y. Pathological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: The Receptor for Advanced Glycation End Products (RAGE). Front. Aging Neurosci. 2020, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Momeni, Z.; Neapetung, J.; Pacholko, A.; Kiir, T.A.B.; Yamamoto, Y.; Bekar, L.K.; Campanucci, V.A. Hyperglycemia induces RAGE-dependent hippocampal spatial memory impairments. Physiol. Behav. 2021, 229, 113287. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Taboada, I.; Alberquilla, S.; Martín, E.D.; Anand, R.; Vietti-Michelina, S.; Tebeka, N.N.; Cantley, J.; Cragg, S.J.; Moratalla, R.; Vallejo, M. Diabetes Causes Dysfunctional Dopamine Neurotransmission Favoring Nigrostriatal Degeneration in Mice. Mov. Disord. 2020, 35, 1636–1648. [Google Scholar] [CrossRef]
- Gaspar, J.M.; Baptista, F.I.; Macedo, M.P.; Ambrósio, A.F. Inside the Diabetic Brain: Role of Different Players Involved in Cognitive Decline. ACS Chem. Neurosci. 2016, 7, 131–142. [Google Scholar] [CrossRef]
- Moon, J.-M.; Thapliyal, N.; Hussain, K.K.; Goyal, R.N.; Shim, Y.-B. Conducting polymer-based electrochemical biosensors for neurotransmitters: A review. Biosens. Bioelectron. 2018, 102, 540–552. [Google Scholar] [CrossRef]
- Tavakolian-Ardakani; Hosu; Cristea; Mazloum-Ardakani; Marrazza Latest Trends in Electrochemical Sensors for Neurotransmitters: A Review. Sensors 2019, 19, 2037. [CrossRef] [Green Version]
- Da, Y.; Luo, S.; Tian, Y. Real-Time Monitoring of Neurotransmitters in the Brain of Living Animals. ACS Appl. Mater. Interfaces 2022, 15, 138–157. [Google Scholar] [CrossRef]
- Intachai, K.; Chattipakorn, S.C.; Chattipakorn, N.; Shinlapawittayatorn, K. Revisiting the Cardioprotective Effects of Acetylcholine Receptor Activation against Myocardial Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 2466. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S. “Sick-but-not-dead”: Multiple paths to catecholamine deficiency in Lewy body diseases. Stress 2020, 23, 633–637. [Google Scholar] [CrossRef]
- Brennenstuhl, H.; Jung-Klawitter, S.; Assmann, B.; Opladen, T. Inherited Disorders of Neurotransmitters: Classification and Practical Approaches for Diagnosis and Treatment. Neuropediatrics 2019, 50, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Potier, B.; Lallemant, L.; Parrot, S.; Huguet-Lachon, A.; Gourdon, G.; Dutar, P.; Gomes-Pereira, M. DM1 Transgenic Mice Exhibit Abnormal Neurotransmitter Homeostasis and Synaptic Plasticity in Association with RNA Foci and Mis-Splicing in the Hippocampus. Int. J. Mol. Sci. 2022, 23, 592. [Google Scholar] [CrossRef]
- Teleanu, R.I.; Niculescu, A.-G.; Roza, E.; Vladâcenco, O.; Grumezescu, A.M.; Teleanu, D.M. Neurotransmitters—Key Factors in Neurological and Neurodegenerative Disorders of the Central Nervous System. Int. J. Mol. Sci. 2022, 23, 5954. [Google Scholar] [CrossRef]
- Xu, Y.; Tong, Q. Expanding neurotransmitters in the hypothalamic neurocircuitry for energy balance regulation. Protein Cell 2011, 2, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Satarker, S.; Bojja, S.L.; Gurram, P.C.; Mudgal, J.; Arora, D.; Nampoothiri, M. Astrocytic Glutamatergic Transmission and Its Implications in Neurodegenerative Disorders. Cells 2022, 11, 1139. [Google Scholar] [CrossRef]
- Bonansco, C.; Fuenzalida, M. Plasticity of Hippocampal Excitatory-Inhibitory Balance: Missing the Synaptic Control in the Epileptic Brain. Neural Plast. 2016, 2016, 8607038. [Google Scholar] [CrossRef] [Green Version]
- Hampe, C.S.; Mitoma, H.; Manto, M. GABA and Glutamate: Their Transmitter Role in the CNS and Pancreatic Islets. In GABA and Glutamate—New Developments in Neurotransmission Research; Samardzic, J., Ed.; InTechOpen: London, UK, 2018; ISBN 978-953-51-3821-1. Available online: http://www.intechopen.com/books/gaba-and-glutamate-new-developments-in-neurotransmission-research/gaba-and-glutamate-their-transmitter-role-in-the-cns-and-pancreatic-islets (accessed on 10 February 2023).
- Bi, D.; Wen, L.; Wu, Z.; Shen, Y. GABAergic dysfunction in excitatory and inhibitory (E/I) imbalance drives the pathogenesis of Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 1312–1329. [Google Scholar] [CrossRef]
- Aosaki, T.; Miura, M.; Suzuki, T.; Nishimura, K.; Masuda, M. Acetylcholine-dopamine balance hypothesis in the striatum: An update: Acetylcholine-dopamine balance hypothesis. Geriatr. Gerontol. Int. 2010, 10, S148–S157. [Google Scholar] [CrossRef]
- André, V.M.; Cepeda, C.; Levine, M.S. Dopamine and Glutamate in Huntington’s Disease: A Balancing Act: Dopamine-Glutamate Balance in Huntington’s Disease. CNS Neurosci. Ther. 2010, 16, 163–178. [Google Scholar] [CrossRef]
- Lee, S.-E.; Lee, Y.; Lee, G.H. The regulation of glutamic acid decarboxylases in GABA neurotransmission in the brain. Arch. Pharm. Res. 2019, 42, 1031–1039. [Google Scholar] [CrossRef]
- Delgado, T.C. Glutamate and GABA in Appetite Regulation. Front. Endocrinol. 2013, 4, 103. Available online: http://journal.frontiersin.org/article/10.3389/fendo.2013.00103/abstract (accessed on 2 September 2022). [CrossRef] [PubMed] [Green Version]
- Niyonambaza, S.D.; Kumar, P.; Xing, P.; Mathault, J.; De Koninck, P.; Boisselier, E.; Boukadoum, M.; Miled, A. A Review of Neurotransmitters Sensing Methods for Neuro-Engineering Research. Appl. Sci. 2019, 9, 4719. [Google Scholar] [CrossRef] [Green Version]
- Gowrishankar, R.; Hahn, M.K.; Blakely, R.D. Good riddance to dopamine: Roles for the dopamine transporter in synaptic function and dopamine-associated brain disorders. Neurochem. Int. 2014, 73, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Nakamura, T.; Yanai, K. Histamine N-Methyltransferase in the Brain. Int. J. Mol. Sci. 2019, 20, 737. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef]
- Huang, Z.; Xie, N.; Illes, P.; Di Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.-G.; Huang, C.; et al. From purines to purinergic signalling: Molecular functions and human diseases. Signal Transduct. Target. Ther. 2021, 6, 162. [Google Scholar] [CrossRef]
- Arumugasamy, S.K.; Chellasamy, G.; Gopi, S.; Govindaraju, S.; Yun, K. Current advances in the detection of neurotransmitters by nanomaterials: An update. TrAC Trends Anal. Chem. 2020, 123, 115766. [Google Scholar] [CrossRef]
- Lasseigne, A.M.; Echeverry, F.A.; Ijaz, S.; Michel, J.C.; Martin, E.A.; Marsh, A.J.; Trujillo, E.; Marsden, K.C.; Pereda, A.E.; Miller, A.C. Electrical synaptic transmission requires a postsynaptic scaffolding protein. eLife 2021, 10, e66898. [Google Scholar] [CrossRef]
- Belousov, A.B.; Fontes, J.D. Neuronal gap junctions: Making and breaking connections during development and injury. Trends Neurosci. 2013, 36, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Totland, M.Z.; Rasmussen, N.L.; Knudsen, L.M.; Leithe, E. Regulation of gap junction intercellular communication by connexin ubiquitination: Physiological and pathophysiological implications. Cell. Mol. Life Sci. 2020, 77, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.I.; Li, W.; Hertzberg, E.L.; Marotta, C.A. Elevated connexin43 immunoreactivity at sites of amyloid plaques in alzheimer’s disease. Brain Res. 1996, 717, 173–178. [Google Scholar] [CrossRef]
- Choudhury, S.P.; Bano, S.; Sen, S.; Suchal, K.; Kumar, S.; Nikolajeff, F.; Dey, S.K.; Sharma, V. Altered neural cell junctions and ion-channels leading to disrupted neuron communication in Parkinson’s disease. npj Park. Dis. 2022, 8, 66. [Google Scholar] [CrossRef]
- Charvériat, M.; Mouthon, F.; Rein, W.; Verkhratsky, A. Connexins as therapeutic targets in neurological and neuropsychiatric disorders. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2021, 1867, 166098. [Google Scholar] [CrossRef]
- Ahmadian, E.; Eftekhari, A.; Samiei, M.; Maleki Dizaj, S.; Vinken, M. The role and therapeutic potential of connexins, pannexins and their channels in Parkinson’s disease. Cell. Signal. 2019, 58, 111–118. [Google Scholar] [CrossRef]
- Güemes, A.; Georgiou, P. Review of the role of the nervous system in glucose homoeostasis and future perspectives towards the management of diabetes. Bioelectron. Med. 2018, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Pignalosa, F.C.; Desiderio, A.; Mirra, P.; Nigro, C.; Perruolo, G.; Ulianich, L.; Formisano, P.; Beguinot, F.; Miele, C.; Napoli, R.; et al. Diabetes and Cognitive Impairment: A Role for Glucotoxicity and Dopaminergic Dysfunction. Int. J. Mol. Sci. 2021, 22, 12366. [Google Scholar] [CrossRef]
- Sickmann, H.M.; Waagepetersen, H.S.; Schousboe, A.; Benie, A.J.; Bouman, S.D. Obesity and Type 2 Diabetes in Rats are Associated with Altered Brain Glycogen and Amino-Acid Homeostasis. J. Cereb. Blood Flow Metab. 2010, 30, 1527–1537. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Biessels, G.-J.; Gispen, W.H.; Ramakers, G.M.J. Synaptic transmission changes in the pyramidal cells of the hippocampus in streptozotocin-induced diabetes mellitus in rats. Brain Res. 2006, 1073–1074, 276–280. [Google Scholar] [CrossRef]
- Huang, X.-T.; Li, C.; Peng, X.-P.; Guo, J.; Yue, S.-J.; Liu, W.; Zhao, F.-Y.; Han, J.-Z.; Huang, Y.-H.; Cheng, Q.-M.; et al. An excessive increase in glutamate contributes to glucose-toxicity in β-cells via activation of pancreatic NMDA receptors in rodent diabetes. Sci. Rep. 2017, 7, 44120. [Google Scholar] [CrossRef]
- d’Almeida, O.C.; Violante, I.R.; Quendera, B.; Moreno, C.; Gomes, L.; Castelo-Branco, M. The neurometabolic profiles of GABA and Glutamate as revealed by proton magnetic resonance spectroscopy in type 1 and type 2 diabetes. PLoS ONE 2020, 15, e0240907. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, G.K.; Ball, K.K.; Cruz, N.F.; Dienel, G.A. Hyperglycaemia and Diabetes Impair Gap Junctional Communication among Astrocytes. ASN Neuro 2010, 2, AN20090048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, W.S.; Orseth, M.L.; Nunemaker, C.S.; Satin, L.S.; Piston, D.W.; Benninger, R.K.P. Connexin-36 Gap Junctions Regulate In Vivo First- and Second-Phase Insulin Secretion Dynamics and Glucose Tolerance in the Conscious Mouse. Diabetes 2012, 61, 1700–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St. Clair, J.R.; Westacott, M.J.; Farnsworth, N.L.; Kravets, V.; Schleicher, W.E.; Miranda, J.; Heintz, A.; Ludin, N.W.; Benninger, R.K. Restoring Connexin-36 Function in Diabetogenic Environments Precludes Mouse and Human Islet Dysfunction. bioRxiv. 2020. Available online: http://biorxiv.org/lookup/doi/10.1101/2020.11.03.366179 (accessed on 21 January 2023).
- Das, U.N. Obesity: Genes, brain, gut, and environment. Nutrition 2010, 26, 459–473. [Google Scholar] [CrossRef]
- Wallace, C.W.; Fordahl, S.C. Obesity and dietary fat influence dopamine neurotransmission: Exploring the convergence of metabolic state, physiological stress, and inflammation on dopaminergic control of food intake. Nutr. Res. Rev. 2021, 35, 236–251. [Google Scholar] [CrossRef]
- Wang, G.-J.; Volkow, N.D.; Logan, J.; Pappas, N.R.; Wong, C.T.; Zhu, W.; Netusll, N.; Fowler, J.S. Brain dopamine and obesity. Lancet 2001, 357, 354–357. [Google Scholar] [CrossRef]
- Mehay, D.; Silberman, Y.; Arnold, A.C. The Arcuate Nucleus of the Hypothalamus and Metabolic Regulation: An Emerging Role for Renin–Angiotensin Pathways. Int. J. Mol. Sci. 2021, 22, 7050. [Google Scholar] [CrossRef]
- Sa, M.; Park, M.G.; Lee, C.J. Role of Hypothalamic Reactive Astrocytes in Diet-Induced Obesity. Mol. Cells 2022, 45, 65–75. [Google Scholar] [CrossRef]
- Schneeberger, M.; Gomis, R.; Claret, M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 2014, 220, T25–T46. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, M.O.; Horvath, T.L. Hypothalamic control of energy balance: Insights into the role of synaptic plasticity. Trends Neurosci. 2013, 36, 65–73. [Google Scholar] [CrossRef]
- Jarvie, B.C.; Hentges, S.T. Expression of GABAergic and glutamatergic phenotypic markers in hypothalamic proopiomelanocortin neurons. J. Comp. Neurol. 2012, 520, 3863–3876. [Google Scholar] [CrossRef] [Green Version]
- Kiss, J.; Csaba, Z.; Csáki, Á.; Halász, B. Glutamatergic innervation of neuropeptide Y and pro-opiomelanocortin-containing neurons in the hypothalamic arcuate nucleus of the rat. Eur. J. Neurosci. 2005, 21, 2111–2119. [Google Scholar] [CrossRef]
- Jones, G.L.; Wittmann, G.; Yokosawa, E.B.; Yu, H.; Mercer, A.J.; Lechan, R.M.; Low, M.J. Selective Restoration of Pomc Expression in Glutamatergic POMC Neurons: Evidence for a Dynamic Hypothalamic Neurotransmitter Network. Eneuro 2019, 6, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Trotta, M.; Bello, E.P.; Alsina, R.; Tavella, M.B.; Ferrán, J.L.; Rubinstein, M.; Bumaschny, V.F. Hypothalamic Pomc expression restricted to GABAergic neurons suppresses Npy overexpression and restores food intake in obese mice. Mol. Metab. 2020, 37, 100985. [Google Scholar] [CrossRef]
- Wu, Q.; Boyle, M.P.; Palmiter, R.D. Loss of GABAergic Signaling by AgRP Neurons to the Parabrachial Nucleus Leads to Starvation. Cell 2009, 137, 1225–1234. [Google Scholar] [CrossRef] [Green Version]
- Krashes, M.J.; Shah, B.P.; Koda, S.; Lowell, B.B. Rapid versus Delayed Stimulation of Feeding by the Endogenously Released AgRP Neuron Mediators GABA, NPY, and AgRP. Cell Metab. 2013, 18, 588–595. [Google Scholar] [CrossRef] [Green Version]
- Suyama, S.; Yada, T. New insight into GABAergic neurons in the hypothalamic feeding regulation. J. Physiol. Sci. 2018, 68, 717–722. [Google Scholar] [CrossRef]
- Zhang, X.; van den Pol, A.N. Hypothalamic arcuate nucleus tyrosine hydroxylase neurons play orexigenic role in energy homeostasis. Nat. Neurosci. 2016, 19, 1341–1347. [Google Scholar] [CrossRef]
- Meguid, M.M.; Fetissov, S.O.; Varma, M.; Sato, T.; Zhang, L.; Laviano, A.; Rossi-Fanelli, F. Hypothalamic dopamine and serotonin in the regulation of food intake. Nutrition 2000, 16, 843–857. [Google Scholar] [CrossRef]
- Jeong, J.H.; Lee, D.K.; Jo, Y.-H. Cholinergic neurons in the dorsomedial hypothalamus regulate food intake. Mol. Metab. 2017, 6, 306–312. [Google Scholar] [CrossRef]
- Herman, A.M.; Ortiz-Guzman, J.; Kochukov, M.; Herman, I.; Quast, K.B.; Patel, J.M.; Tepe, B.; Carlson, J.C.; Ung, K.; Selever, J.; et al. A cholinergic basal forebrain feeding circuit modulates appetite suppression. Nature 2016, 538, 253–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thinnes, A.; Klein, J. Food-Induced Increase of Acetylcholine in Mouse Hypothalamus. ACS Chem. Neurosci. 2019, 10, 1892–1899. [Google Scholar] [CrossRef]
- Girardet, C.; Butler, A.A. Neural melanocortin receptors in obesity and related metabolic disorders. Biochim. Biophys. Acta 2014, 1842, 482–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dib, L.; San-Jose, L.M.; Ducrest, A.-L.; Salamin, N.; Roulin, A. Selection on the Major Color Gene Melanocortin-1-Receptor Shaped the Evolution of the Melanocortin System Genes. Int. J. Mol. Sci. 2017, 18, 2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.-X. The melanocortin-4 receptor: Physiology, pharmacology, and pathophysiology. Endocr. Rev. 2010, 31, 506–543. [Google Scholar] [CrossRef] [Green Version]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 1997, 278, 135–138. [Google Scholar] [CrossRef]
- Yang, Z.; Tao, Y.-X. Biased signaling initiated by agouti-related peptide through human melanocortin-3 and -4 receptors. Biochim. Biophys. Acta 2016, 1862, 1485–1494. [Google Scholar] [CrossRef]
- Yang, L.-K.; Tao, Y.-X. Biased signaling at neural melanocortin receptors in regulation of energy homeostasis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2486–2495. [Google Scholar] [CrossRef]
- Newman, E.A.; Chai, B.-X.; Zhang, W.; Li, J.-Y.; Ammori, J.B.; Mulholland, M.W. Activation of the melanocortin-4 receptor mobilizes intracellular free calcium in immortalized hypothalamic neurons. J. Surg. Res. 2006, 132, 201–207. [Google Scholar] [CrossRef]
- Chai, B.; Li, J.-Y.; Zhang, W.; Ammori, J.B.; Mulholland, M.W. Melanocortin-3 receptor activates MAP kinase via PI3 kinase. Regul. Pept. 2007, 139, 115–121. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.-B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- Vongs, A.; Lynn, N.M.; Rosenblum, C.I. Activation of MAP kinase by MC4-R through PI3 kinase. Regul. Pept. 2004, 120, 113–118. [Google Scholar] [CrossRef]
- Sutton, G.M.; Duos, B.; Patterson, L.M.; Berthoud, H.-R. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology 2005, 146, 3739–3747. [Google Scholar] [CrossRef] [Green Version]
- Chai, B.; Li, J.-Y.; Zhang, W.; Wang, H.; Mulholland, M.W. Melanocortin-4 receptor activation inhibits c-Jun N-terminal kinase activity and promotes insulin signaling. Peptides 2009, 30, 1098–1104. [Google Scholar] [CrossRef] [Green Version]
- Damm, E.; Buech, T.R.H.; Gudermann, T.; Breit, A. Melanocortin-induced PKA activation inhibits AMPK activity via ERK-1/2 and LKB-1 in hypothalamic GT1-7 cells. Mol. Endocrinol. 2012, 26, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Huszar, D.; Lynch, C.A.; Fairchild-Huntress, V.; Dunmore, J.H.; Fang, Q.; Berkemeier, L.R.; Gu, W.; Kesterson, R.A.; Boston, B.A.; Cone, R.D.; et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997, 88, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.S.; Marsh, D.J.; Trumbauer, M.E.; Frazier, E.G.; Guan, X.M.; Yu, H.; Rosenblum, C.I.; Vongs, A.; Feng, Y.; Cao, L.; et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat. Genet. 2000, 26, 97–102. [Google Scholar] [CrossRef]
- Fan, W.; Dinulescu, D.M.; Butler, A.A.; Zhou, J.; Marks, D.L.; Cone, R.D. The central melanocortin system can directly regulate serum insulin levels. Endocrinology 2000, 141, 3072–3079. [Google Scholar] [CrossRef]
- Obici, S.; Feng, Z.; Tan, J.; Liu, L.; Karkanias, G.; Rossetti, L. Central melanocortin receptors regulate insulin action. J. Clin. Investig. 2001, 108, 1079–1085. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Keogh, J.M.; Yeo, G.S.H.; Lank, E.J.; Cheetham, T.; O’Rahilly, S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 2003, 348, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Thearle, M.S.; Muller, Y.L.; Hanson, R.L.; Mullins, M.; AbdusSamad, M.; Tran, J.; Knowler, W.C.; Bogardus, C.; Krakoff, J.; Baier, L.J. Greater Impact of Melanocortin-4 Receptor Deficiency on Rates of Growth and Risk of Type 2 Diabetes During Childhood Compared With Adulthood in Pima Indians. Diabetes 2012, 61, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, B.; Takeuchi, F.; Chandak, G.R.; Kato, N.; Pan, H.W.; Zhou, D.H.; Pan, H.Y.; Mi, J.; AGEN-T2D Consortium. Common polymorphism near the MC4R gene is associated with type 2 diabetes: Data from a meta-analysis of 123,373 individuals. Diabetologia 2012, 55, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Nogueiras, R.; Wiedmer, P.; Perez-Tilve, D.; Veyrat-Durebex, C.; Keogh, J.M.; Sutton, G.M.; Pfluger, P.T.; Castaneda, T.R.; Neschen, S.; Hofmann, S.M.; et al. The central melanocortin system directly controls peripheral lipid metabolism. J. Clin. Investig. 2007, 117, 3475–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enriori, P.J.; Chen, W.; Garcia-Rudaz, M.C.; Grayson, B.E.; Evans, A.E.; Comstock, S.M.; Gebhardt, U.; Müller, H.L.; Reinehr, T.; Henry, B.A.; et al. α-Melanocyte stimulating hormone promotes muscle glucose uptake via melanocortin 5 receptors. Mol. Metab. 2016, 5, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Shpakov, A.O.; Derkach, K.V.; Berstein, L.M. Brain signaling systems in the Type 2 diabetes and metabolic syndrome: Promising target to treat and prevent these diseases. Future Sci. OA 2015, 1, FSO25. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, D.; Mioni, C.; Altavilla, D.; Leone, S.; Bazzani, C.; Minutoli, L.; Bitto, A.; Cainazzo, M.-M.; Marini, H.; Zaffe, D.; et al. Both early and delayed treatment with melanocortin 4 receptor-stimulating melanocortins produces neuroprotection in cerebral ischemia. Endocrinology 2006, 147, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, D.; Bitto, A.; Galantucci, M.; Zaffe, D.; Ottani, A.; Irrera, N.; Neri, L.; Cavallini, G.M.; Altavilla, D.; Botticelli, A.R.; et al. Melanocortins protect against progression of Alzheimer’s disease in triple-transgenic mice by targeting multiple pathophysiological pathways. Neurobiol. Aging 2014, 35, 537–547. [Google Scholar] [CrossRef]
- Giuliani, D.; Galantucci, M.; Neri, L.; Canalini, F.; Calevro, A.; Bitto, A.; Ottani, A.; Vandini, E.; Sena, P.; Sandrini, M.; et al. Melanocortins protect against brain damage and counteract cognitive decline in a transgenic mouse model of moderate Alzheimer׳s disease. Eur. J. Pharmacol. 2014, 740, 144–150. [Google Scholar] [CrossRef]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005, 123, 493–505. [Google Scholar] [CrossRef]
- Fani, L.; Bak, S.; Delhanty, P.; van Rossum, E.F.C.; van den Akker, E.L.T. The melanocortin-4 receptor as target for obesity treatment: A systematic review of emerging pharmacological therapeutic options. Int. J. Obes. 2005 2014, 38, 163–169. [Google Scholar] [CrossRef]
- Markham, A. Setmelanotide: First Approval. Drugs 2021, 81, 397–403. [Google Scholar] [CrossRef]
- Kievit, P.; Halem, H.; Marks, D.L.; Dong, J.Z.; Glavas, M.M.; Sinnayah, P.; Pranger, L.; Cowley, M.A.; Grove, K.L.; Culler, M.D. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes 2013, 62, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Kühnen, P.; Clément, K.; Wiegand, S.; Blankenstein, O.; Gottesdiener, K.; Martini, L.L.; Mai, K.; Blume-Peytavi, U.; Grüters, A.; Krude, H. Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist. N. Engl. J. Med. 2016, 375, 240–246. [Google Scholar] [CrossRef]
- Clemmensen, C.; Finan, B.; Fischer, K.; Tom, R.Z.; Legutko, B.; Sehrer, L.; Heine, D.; Grassl, N.; Meyer, C.W.; Henderson, B.; et al. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol. Med. 2015, 7, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Pellerin, L. Food for thought: The importance of glucose and other energy substrates for sustaining brain function under varying levels of activity. Diabetes Metab. 2010, 36, S59–S63. [Google Scholar] [CrossRef]
- Killeen, P.R.; Russell, V.A.; Tannock, R. Neuroenergetics. Curr. Dir. Psychol. Sci. 2016, 25, 124–129. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, H.M.; Hashemiaghdam, A.; Ashrafi, G.; Harbauer, A.B. Mitochondria in Neuronal Health: From Energy Metabolism to Parkinson’s Disease. Adv. Biol. 2021, 5, 2100663. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Brain Energy and Metabolism. In Neuroscience in the 21st Century; Pfaff, D.W., Volkow, N.D., Eds.; Springer: New York, NY, USA, 2016; pp. 1879–1909. ISBN 978-1-4939-3473-7. Available online: http://link.springer.com/10.1007/978-1-4939-3474-4_56 (accessed on 11 September 2022).
- Sifat, A.E.; Nozohouri, S.; Archie, S.R.; Chowdhury, E.A.; Abbruscato, T.J. Brain Energy Metabolism in Ischemic Stroke: Effects of Smoking and Diabetes. Int. J. Mol. Sci. 2022, 23, 8512. [Google Scholar] [CrossRef]
- Martin, W.R.W.; Wieler, M.; Hanstock, C.C. Is brain lactate increased in Huntington’s disease? J. Neurol. Sci. 2007, 263, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Schurr, A. Lactate, glucose and energy metabolism in the ischemic brain (Review). Int. J. Mol. Med. 2002, 10, 131–136. Available online: http://www.spandidos-publications.com/10.3892/ijmm.10.2.131 (accessed on 11 September 2022). [CrossRef] [PubMed]
- Margineanu, M.B.; Mahmood, H.; Fiumelli, H.; Magistretti, P.J. L-Lactate Regulates the Expression of Synaptic Plasticity and Neuroprotection Genes in Cortical Neurons: A Transcriptome Analysis. Front. Mol. Neurosci. 2018, 11, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, B.; Mantha, O.L.; Schor, J.; Pascual, A.; Plaçais, P.-Y.; Pavlowsky, A.; Preat, T. Glia fuel neurons with locally synthesized ketone bodies to sustain memory under starvation. Nat. Metab. 2022, 4, 213–224. [Google Scholar] [CrossRef]
- Chung, J.Y.; Kim, O.Y.; Song, J. Role of ketone bodies in diabetes-induced dementia: Sirtuins, insulin resistance, synaptic plasticity, mitochondrial dysfunction, and neurotransmitter. Nutr. Rev. 2022, 80, 774–785. [Google Scholar] [CrossRef]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; Murtagh, D.K.J.; Gilbertson, L.J.; Asztely, F.J.S.; Lynch, C.D.P. Low-fat versus ketogenic diet in Parkinson’s disease: A pilot randomized controlled trial: Low-Fat Versus Ketogenic Diet in PD. Mov. Disord. 2018, 33, 1306–1314. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Chen, Y.; Waagepetersen, H.S. Effects of ketone bodies in Alzheimer’s disease in relation to neural hypometabolism, β-amyloid toxicity, and astrocyte function. J. Neurochem. 2015, 134, 7–20. [Google Scholar] [CrossRef]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic Diet Metabolites Reduce Firing in Central Neurons by Opening KATP Channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef] [Green Version]
- Mujica-Parodi, L.R.; Amgalan, A.; Sultan, S.F.; Antal, B.; Sun, X.; Skiena, S.; Lithen, A.; Adra, N.; Ratai, E.-M.; Weistuch, C.; et al. Diet modulates brain network stability, a biomarker for brain aging, in young adults. Proc. Natl. Acad. Sci. USA 2020, 117, 6170–6177. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T.A.; Mathew, T.C.; Dashti, A.A.; Asfar, S.; Al-Zaid, N.; Dashti, H.M. Effect of low-calorie versus low-carbohydrate ketogenic diet in type 2 diabetes. Nutrition 2012, 28, 1016–1021. [Google Scholar] [CrossRef]
- Carneiro, L.; Geller, S.; Hébert, A.; Repond, C.; Fioramonti, X.; Leloup, C.; Pellerin, L. Hypothalamic sensing of ketone bodies after prolonged cerebral exposure leads to metabolic control dysregulation. Sci. Rep. 2016, 6, 34909. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, L.; Geller, S.; Fioramonti, X.; Hébert, A.; Repond, C.; Leloup, C.; Pellerin, L. Evidence for hypothalamic ketone body sensing: Impact on food intake and peripheral metabolic responses in mice. Am. J. Physiol.-Endocrinol. Metab. 2016, 310, E103–E115. [Google Scholar] [CrossRef] [Green Version]
- Reagan, L.P.; Rosell, D.R.; Alves, S.E.; Hoskin, E.K.; McCall, A.L.; Charron, M.J.; McEwen, B.S. GLUT8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Res. 2002, 932, 129–134. [Google Scholar] [CrossRef]
- Ren, H.; Yan, S.; Zhang, B.; Lu, T.Y.; Arancio, O.; Accili, D. Glut4 expression defines an insulin-sensitive hypothalamic neuronal population. Mol. Metab. 2014, 3, 452–459. [Google Scholar] [CrossRef]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2022, 12, 825816. [Google Scholar] [CrossRef]
- Mason, S. Lactate Shuttles in Neuroenergetics—Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. Available online: http://journal.frontiersin.org/article/10.3389/fnins.2017.00043/full (accessed on 9 September 2022). [CrossRef] [Green Version]
- Staricha, K.; Meyers, N.; Garvin, J.; Liu, Q.; Rarick, K.; Harder, D.; Cohen, S. Effect of high glucose condition on glucose metabolism in primary astrocytes. Brain Res. 2020, 1732, 146702. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Descalzi, G.; Gao, V.; Steinman, M.Q.; Suzuki, A.; Alberini, C.M. Lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Commun. Biol. 2019, 2, 247. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K. Lactate Supply from Astrocytes to Neurons and its Role in Ischemic Stroke-induced Neurodegeneration. Neuroscience 2022, 481, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Sheng, Z.-H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022, 23, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Brand, M.D.; Gerencser, A.A. Mitochondrial bioenergetics and neuronal survival modelled in primary neuronal culture and isolated nerve terminals. J. Bioenerg. Biomembr. 2015, 47, 63–74. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.Y.; Roy, J.; Fung, M.L.; Heng, B.C.; Zhang, C.; Lim, L.W. Relationships between Mitochondrial Dysfunction and Neurotransmission Failure in Alzheimer’s Disease. Aging Dis. 2020, 11, 1291. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Tovar, S. Editorial: Crosstalk of Mitochondria with Brain Insulin and Leptin Signaling. Front. Endocrinol. 2018, 9, 761. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Seiça, R.; Oliveira, C.R. Increased Vulnerability of Brain Mitochondria in Diabetic (Goto-Kakizaki) Rats With Aging and Amyloid-β Exposure. Diabetes 2003, 52, 1449–1456. [Google Scholar] [CrossRef] [Green Version]
- Petrov, D.; Pedrós, I.; Artiach, G.; Sureda, F.X.; Barroso, E.; Pallàs, M.; Casadesús, G.; Beas-Zarate, C.; Carro, E.; Ferrer, I.; et al. High-fat diet-induced deregulation of hippocampal insulin signaling and mitochondrial homeostasis deficiences contribute to Alzheimer disease pathology in rodents. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2015, 1852, 1687–1699. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.-T.; Chen, K.-Y.; Wang, W.; Chiu, J.-Y.; Wu, D.; Chao, T.-Y.; Hu, C.-J.; Chau, K.-Y.; Bamodu, O. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells 2020, 9, 740. [Google Scholar] [CrossRef] [Green Version]
- Głuchowska, K.; Pliszka, M.; Szablewski, L. Expression of glucose transporters in human neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2021, 540, 8–15. [Google Scholar] [CrossRef]
- Van Bussel, F.C.G.; Backes, W.H.; Hofman, P.A.M.; Puts, N.A.J.; Edden, R.A.E.; van Boxtel, M.P.J.; Schram, M.T.; Stehouwer, C.D.A.; Wildberger, J.E.; Jansen, J.F.A. Increased GABA concentrations in type 2 diabetes mellitus are related to lower cognitive functioning. Medicine 2016, 95, e4803. [Google Scholar] [CrossRef]
- Sickmann, H.M.; Waagepetersen, H.S.; Schousboe, A.; Benie, A.J.; Bouman, S.D. Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem. Int. 2012, 60, 267–275. [Google Scholar] [CrossRef]
- Thielen, J.; Gancheva, S.; Hong, D.; Rohani Rankouhi, S.; Chen, B.; Apostolopoulou, M.; Anadol-Schmitz, E.; Roden, M.; Norris, D.G.; Tendolkar, I. Higher GABA concentration in the medial prefrontal cortex of Type 2 diabetes patients is associated with episodic memory dysfunction. Hum. Brain Mapp. 2019, 40, 4287–4295. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Lammers, N.M.; Trinko, R.; Opland, D.M.; Figee, M.; Ackermans, M.T.; Booij, J.; van den Munckhof, P.; Schuurman, P.R.; Fliers, E.; et al. Striatal dopamine regulates systemic glucose metabolism in humans and mice. Sci. Transl. Med. 2018, 10, eaar3752. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-J.; Shen, Z.; Cui, R.-X.; Huang, Y.; Xu, D.-L.; Zhao, F.-L.; Pan, J.; Shi, A.-M.; Liu, T.; Yu, Y.-L. Thioredoxin-Interacting Protein (TXNIP) Regulates Parkin/PINK1-mediated Mitophagy in Dopaminergic Neurons under High-glucose Conditions: Implications for Molecular Links Between Parkinson’s Disease and Diabetes. Neurosci. Bull. 2020, 36, 346–358. [Google Scholar] [CrossRef]
- Renaud, J.; Bournival, J.; Zottig, X.; Martinoli, M.-G. Resveratrol Protects DAergic PC12 Cells from High Glucose-Induced Oxidative Stress and Apoptosis: Effect on p53 and GRP75 Localization. Neurotox. Res. 2014, 25, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Lacković, Z.; Šlković, M.; Kuci, Z.; Relja, M. Effect of Long-Lasting Diabetes Mellitus on Rat and Human Brain Monoamines. J. Neurochem. 1990, 54, 143–147. [Google Scholar] [CrossRef]
- Zemdegs, J.; Quesseveur, G.; Jarriault, D.; Pénicaud, L.; Fioramonti, X.; Guiard, B.P. High-fat diet-induced metabolic disorders impairs 5-HT function and anxiety-like behavior in mice: Correlation between metabolic disorders and anxiety. Br. J. Pharmacol. 2016, 173, 2095–2110. [Google Scholar] [CrossRef] [Green Version]
- Derkach, K.V.; Bondareva, V.M.; Chistyakova, O.V.; Berstein, L.M.; Shpakov, A.O. The Effect of Long-Term Intranasal Serotonin Treatment on Metabolic Parameters and Hormonal Signaling in Rats with High-Fat Diet/Low-Dose Streptozotocin-Induced Type 2 Diabetes. Int. J. Endocrinol. 2015, 2015, 245459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, S.; Kumar, T.P.; Mathew, J.; Anju, T.; Paulose, C. Hypoglycemia induced changes in cholinergic receptor expression in the cerebellum of diabetic rats. J. Biomed. Sci. 2010, 17, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Cao, K.; Guo, B.; Xiang, J.; Dong, Y.-T.; Qi, X.-L.; Yu, W.-F.; Xiao, Y.; Guan, Z.-Z. Lowered levels of nicotinic acetylcholine receptors and elevated apoptosis in the hippocampus of brains from patients with type 2 diabetes mellitus and db/db mice. Aging 2020, 12, 14205–14218. [Google Scholar] [CrossRef] [PubMed]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent High Glucose Enhances Apoptosis Related to Oxidative Stress in Human Umbilical Vein Endothelial Cells. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Sohn, J.-H.; Jang, M.U.; Kim, S.-H.; Choi, M.-G.; Ryu, O.-H.; Lee, S.; Choi, H.-C. Association between Visit-to-Visit Glucose Variability and Cognitive Function in Aged Type 2 Diabetic Patients: A Cross-Sectional Study. PLoS ONE 2015, 10, e0132118. [Google Scholar] [CrossRef] [Green Version]
- Minami, T.; Ito, Y.; Yamada, M.; Furuta, R.; Minagawa, F.; Kamata, K.; Kameda, A.; Terauchi, Y. The effect of long-term past glycemic control on executive function among patients with type 2 diabetes mellitus. Diabetol. Int. 2020, 11, 114–120. [Google Scholar] [CrossRef]
- Li, T.-C.; Yang, C.-P.; Tseng, S.-T.; Li, C.-I.; Liu, C.-S.; Lin, W.-Y.; Hwang, K.-L.; Yang, S.-Y.; Chiang, J.-H.; Lin, C.-C. Visit-to-Visit Variations in Fasting Plasma Glucose and HbA1c Associated With an Increased Risk of Alzheimer Disease: Taiwan Diabetes Study. Diabetes Care 2017, 40, 1210–1217. [Google Scholar] [CrossRef] [Green Version]
- Quincozes-Santos, A.; Bobermin, L.D.; de Assis, A.M.; Gonçalves, C.-A.; Souza, D.O. Fluctuations in glucose levels induce glial toxicity with glutamatergic, oxidative and inflammatory implications. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2017, 1863, 1–14. [Google Scholar] [CrossRef]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline. Front. Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef] [Green Version]
- Havrankova, J.; Roth, J.; Brownstein, M.J. Concentrations of Insulin and of Insulin Receptors in the Brain are Independent of Peripheral Insulin Levels. J. Clin. Investig. 1979, 64, 636–642. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef] [Green Version]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimer’s Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Dubey, S.K.; Lakshmi, K.K.; Krishna, K.V.; Agrawal, M.; Singhvi, G.; Saha, R.N.; Saraf, S.; Saraf, S.; Shukla, R.; Alexander, A. Insulin mediated novel therapies for the treatment of Alzheimer’s disease. Life Sci. 2020, 249, 117540. [Google Scholar] [CrossRef]
- Duarte, A.I.; Santos, M.S.; Seiça, R.; Oliveira, C.R. de Insulin affects synaptosomal GABA and glutamate transport under oxidative stress conditions. Brain Res. 2003, 977, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.-Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef]
- Bruce, K.D.; Zsombok, A.; Eckel, R.H. Lipid Processing in the Brain: A Key Regulator of Systemic Metabolism. Front. Endocrinol. 2017, 8, 60. Available online: http://journal.frontiersin.org/article/10.3389/fendo.2017.00060/full (accessed on 9 October 2022). [CrossRef] [Green Version]
- Barber, C.N.; Raben, D.M. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front. Cell. Neurosci. 2019, 13, 212. [Google Scholar] [CrossRef] [Green Version]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Low, Y.L.; Jin, L.; Morris, E.R.; Pan, Y.; Nicolazzo, J.A. Pioglitazone Increases Blood–Brain Barrier Expression of Fatty Acid-Binding Protein 5 and Docosahexaenoic Acid Trafficking into the Brain. Mol. Pharm. 2020, 17, 873–884. [Google Scholar] [CrossRef]
- Watanabe, A.; Toyota, T.; Owada, Y.; Hayashi, T.; Iwayama, Y.; Matsumata, M.; Ishitsuka, Y.; Nakaya, A.; Maekawa, M.; Ohnishi, T.; et al. Fabp7 Maps to a Quantitative Trait Locus for a Schizophrenia Endophenotype. PLoS Biol. 2007, 5, e297. [Google Scholar] [CrossRef] [Green Version]
- Sandoval-Salazar, C.; Ramírez-Emiliano, J.; Trejo-Bahena, A.; Oviedo-Solís, C.I.; Solís-Ortiz, M.S. A high-fat diet decreases GABA concentration in the frontal cortex and hippocampus of rats. Biol. Res. 2016, 49, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizarbe, B.; Cherix, A.; Duarte, J.M.N.; Cardinaux, J.-R.; Gruetter, R. High-fat diet consumption alters energy metabolism in the mouse hypothalamus. Int. J. Obes. 2019, 43, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D. Guardians of the brain: How a special immune system protects our grey matter. Nature 2022, 606, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Appel, S.H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 15558–15563. [Google Scholar] [CrossRef] [Green Version]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Morales, I.; Guzmán-Martà nez, L.; Cerda-Troncoso, C.; Farías, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 112. Available online: http://journal.frontiersin.org/article/10.3389/fncel.2014.00112/abstract (accessed on 10 October 2022). [CrossRef] [Green Version]
- Wang, D.-S.; Zurek, A.A.; Lecker, I.; Yu, J.; Abramian, A.M.; Avramescu, S.; Davies, P.A.; Moss, S.J.; Lu, W.-Y.; Orser, B.A. Memory Deficits Induced by Inflammation Are Regulated by α5-Subunit-Containing GABAA Receptors. Cell Rep. 2012, 2, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Richetto, J.; Calabrese, F.; Riva, M.A.; Meyer, U. Prenatal Immune Activation Induces Maturation-Dependent Alterations in the Prefrontal GABAergic Transcriptome. Schizophr. Bull. 2014, 40, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Schwab, C.; Mcgeer, P.L. Astrocytes are GABAergic cells that modulate microglial activity. Glia 2011, 59, 152–165. [Google Scholar] [CrossRef]
- Pribiag, H.; Stellwagen, D. TNF- Downregulates Inhibitory Neurotransmission through Protein Phosphatase 1-Dependent Trafficking of GABAA Receptors. J. Neurosci. 2013, 33, 15879–15893. [Google Scholar] [CrossRef] [Green Version]
- Khare, P.; Datusalia, A.K.; Sharma, S.S. Parthenolide, an NF-κB Inhibitor Ameliorates Diabetes-Induced Behavioural Deficit, Neurotransmitter Imbalance and Neuroinflammation in Type 2 Diabetes Rat Model. Neuromol. Med. 2017, 19, 101–112. [Google Scholar] [CrossRef]
- Le Thuc, O.; Stobbe, K.; Cansell, C.; Nahon, J.-L.; Blondeau, N.; Rovère, C. Hypothalamic Inflammation and Energy Balance Disruptions: Spotlight on Chemokines. Front. Endocrinol. 2017, 8, 197. [Google Scholar] [CrossRef] [Green Version]
- Dionysopoulou, S.; Charmandari, E.; Bargiota, A.; Vlahos, N.F.; Mastorakos, G.; Valsamakis, G. The Role of Hypothalamic Inflammation in Diet-Induced Obesity and Its Association with Cognitive and Mood Disorders. Nutrients 2021, 13, 498. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Chabowski, A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H. Hypothalamic GABAergic neurocircuitry in the regulation of energy homeostasis and sleep/wake control. Med. Rev. 2022, 2, 531–540. Available online: https://www.degruyter.com/document/doi/10.1515/mr-2022-0022/html (accessed on 30 September 2022). [CrossRef]
- Li, L.; Zhang, M.-Q.; Sun, X.; Liu, W.-Y.; Huang, Z.-L.; Wang, Y.-Q. Role of Dorsomedial Hypothalamus GABAergic Neurons in Sleep–Wake States in Response to Changes in Ambient Temperature in Mice. Int. J. Mol. Sci. 2022, 23, 1270. [Google Scholar] [CrossRef]
- Fonseca-Santos, B.; Chorilli, M. The uses of resveratrol for neurological diseases treatment and insights for nanotechnology based-drug delivery systems. Int. J. Pharm. 2020, 589, 119832. [Google Scholar] [CrossRef]
- Han, Y.-S.; Zheng, W.-H.; Bastianetto, S.; Chabot, J.-G.; Quirion, R. Neuroprotective effects of resveratrol against β -amyloid-induced neurotoxicity in rat hippocampal neurons: Involvement of protein kinase C: Neuroprotective effect of resveratrol. Br. J. Pharmacol. 2004, 141, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Li, Z.; Chen, H.; Xu, X.; Li, Y.; Gui, Y. Neuroprotective Effect of Trans-Resveratrol in Mild to Moderate Alzheimer Disease: A Randomized, Double-Blind Trial. Neurol. Ther. 2021, 10, 905–917. [Google Scholar] [CrossRef]
- Salehi, B.; Calina, D.; Docea, A.; Koirala, N.; Aryal, S.; Lombardo, D.; Pasqua, L.; Taheri, Y.; Marina Salgado Castillo, C.; Martorell, M.; et al. Curcumin’s Nanomedicine Formulations for Therapeutic Application in Neurological Diseases. J. Clin. Med. 2020, 9, 430. [Google Scholar] [CrossRef] [Green Version]
- Mandel, S.A.; Amit, T.; Kalfon, L.; Reznichenko, L.; Youdim, M.B.H. Targeting Multiple Neurodegenerative Diseases Etiologies with Multimodal-Acting Green Tea Catechins. J. Nutr. 2008, 138, 1578S–1583S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, S.; Muhammad, P.; Chesworth, R.; Rehman, F.U.; Qian, R.; Zheng, M.; Shi, B. Nanomedicine-based immunotherapy for central nervous system disorders. Acta Pharmacol. Sin. 2020, 41, 936–953. [Google Scholar] [CrossRef] [PubMed]
- Yavarpour-Bali, H.; Ghasemi-Kasman, M.; Pirzadeh, M. Curcumin-loaded nanoparticles: A novel therapeutic strategy in treatment of central nervous system disorders. Int. J. Nanomed. 2019, 14, 4449–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Rocha Lindner, G.; Bonfanti Santos, D.; Colle, D.; Gasnhar Moreira, E.L.; Daniel Prediger, R.; Farina, M.; Khalil, N.M.; Mara Mainardes, R. Improved neuroprotective effects of resveratrol-loaded polysorbate 80-coated poly(lactide) nanoparticles in MPTP-induced Parkinsonism. Nanomedicine 2015, 10, 1127–1138. [Google Scholar] [CrossRef]
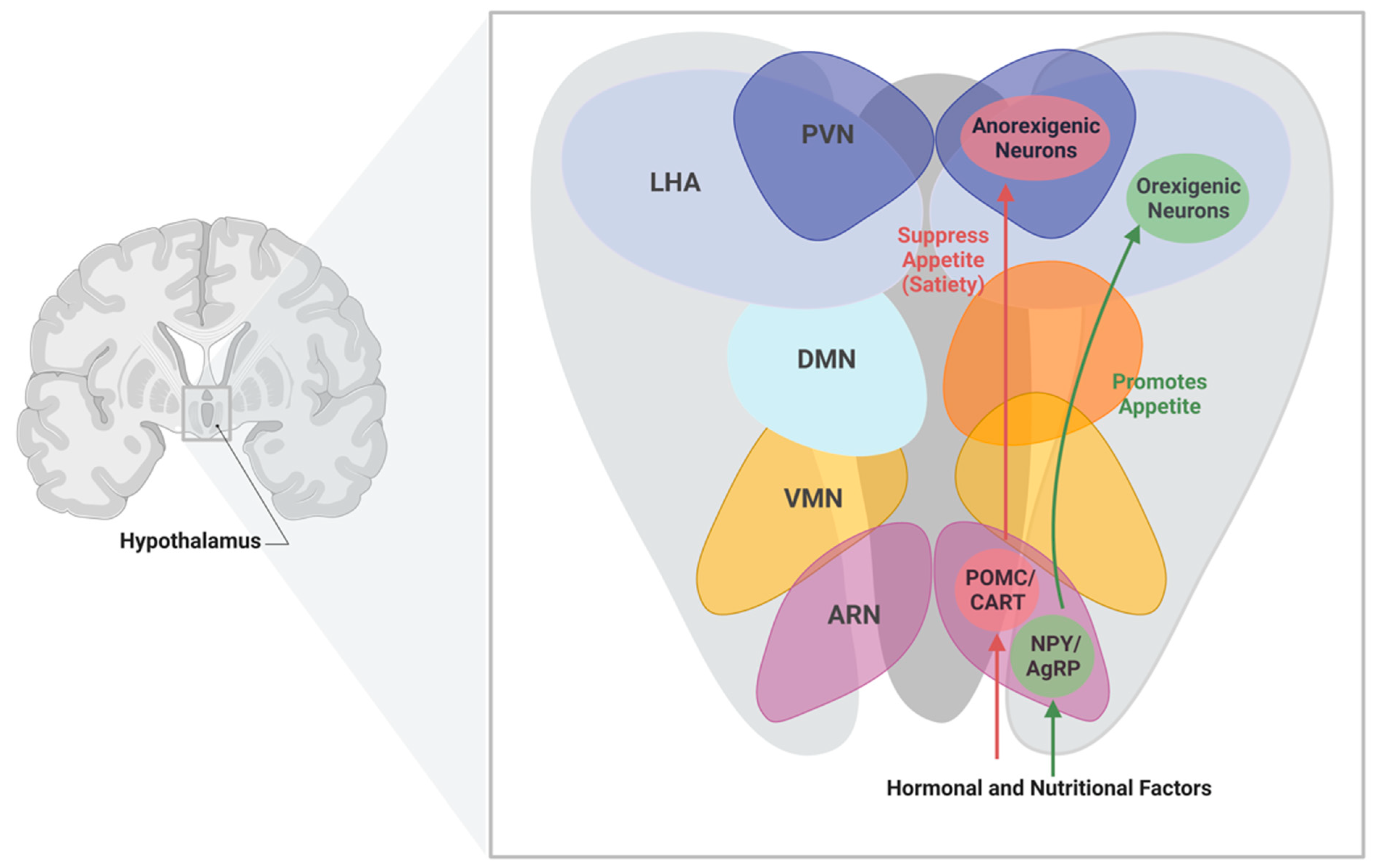
| Nuclei | Function |
|---|---|
| Arcuate Nucleus (ARN) | Regulates energy balance by sensing alterations in hormonal and nutritional factors in the blood stream |
| Paraventricular Nucleus (PVN) | Maintains whole body energy homeostasis through the regulation of food intake by neuropeptides |
| Dorsal Medial Nucleus (DMN) | Controls physiological processes and circadian rhythms |
| Lateral Hypothalamus Area (LHA) | Mediate orexigenic, behavioral, and physiological responses |
| Ventromedial Nucleus (VMN) | Regulates energy balance and thermogenesis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Sayyar, A.; Hammad, M.M.; Williams, M.R.; Al-Onaizi, M.; Abubaker, J.; Alzaid, F. Neurotransmitters in Type 2 Diabetes and the Control of Systemic and Central Energy Balance. Metabolites 2023, 13, 384. https://doi.org/10.3390/metabo13030384
Al-Sayyar A, Hammad MM, Williams MR, Al-Onaizi M, Abubaker J, Alzaid F. Neurotransmitters in Type 2 Diabetes and the Control of Systemic and Central Energy Balance. Metabolites. 2023; 13(3):384. https://doi.org/10.3390/metabo13030384
Chicago/Turabian StyleAl-Sayyar, Amnah, Maha M. Hammad, Michayla R. Williams, Mohammed Al-Onaizi, Jehad Abubaker, and Fawaz Alzaid. 2023. "Neurotransmitters in Type 2 Diabetes and the Control of Systemic and Central Energy Balance" Metabolites 13, no. 3: 384. https://doi.org/10.3390/metabo13030384
APA StyleAl-Sayyar, A., Hammad, M. M., Williams, M. R., Al-Onaizi, M., Abubaker, J., & Alzaid, F. (2023). Neurotransmitters in Type 2 Diabetes and the Control of Systemic and Central Energy Balance. Metabolites, 13(3), 384. https://doi.org/10.3390/metabo13030384