
In Silico Prediction of Metabolic Reaction Catalyzed by Human Aldehyde Oxidase
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
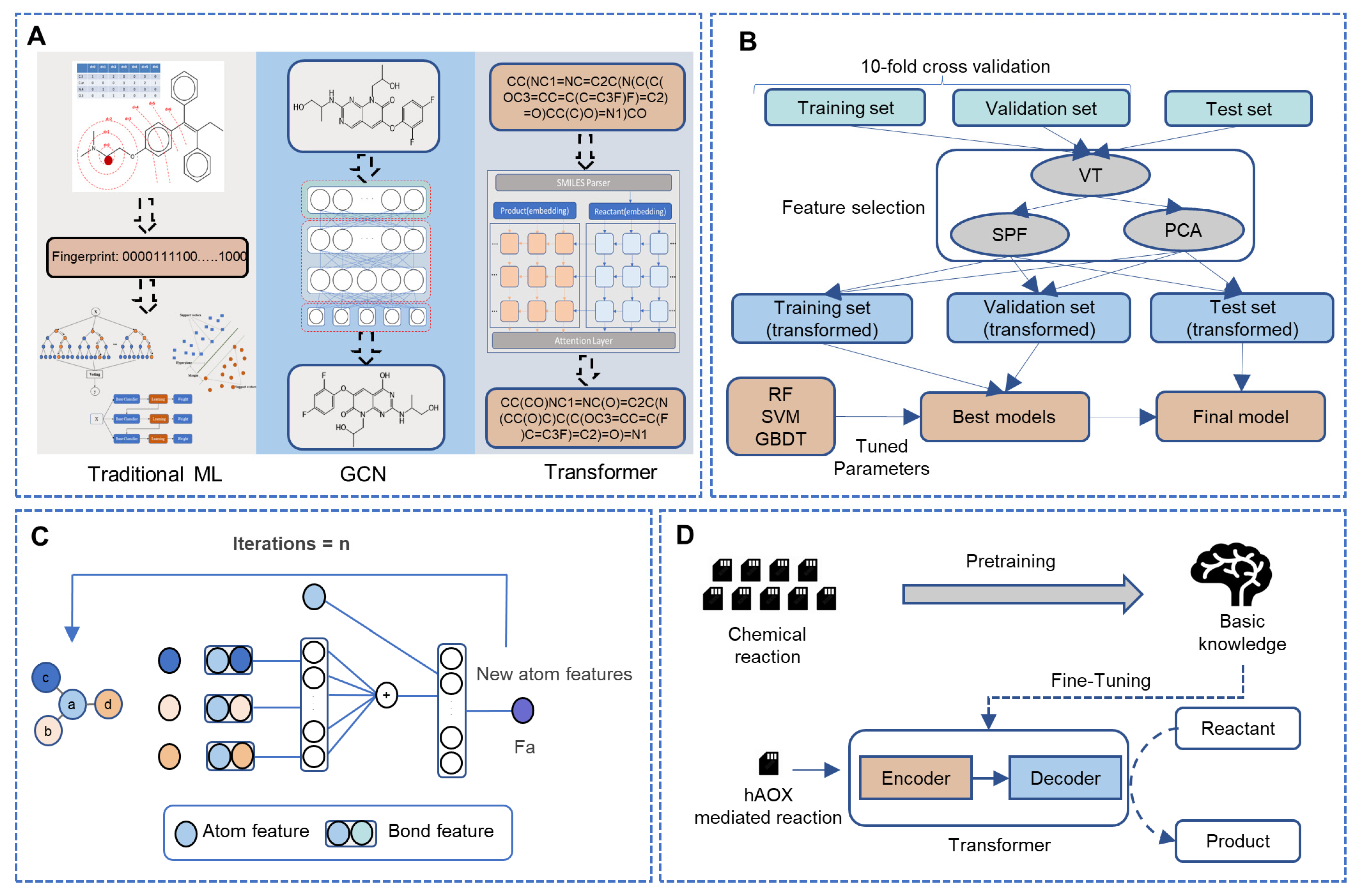
2.2. Fingerprint-Based Method
2.2.1. Potential SOMs and Atom Environment Fingerprints
2.2.2. Feature Selection
2.2.3. Model Building
2.3. Weisfeiler-Lehman Network
2.3.1. Data Preprocessing
2.3.2. Model Building
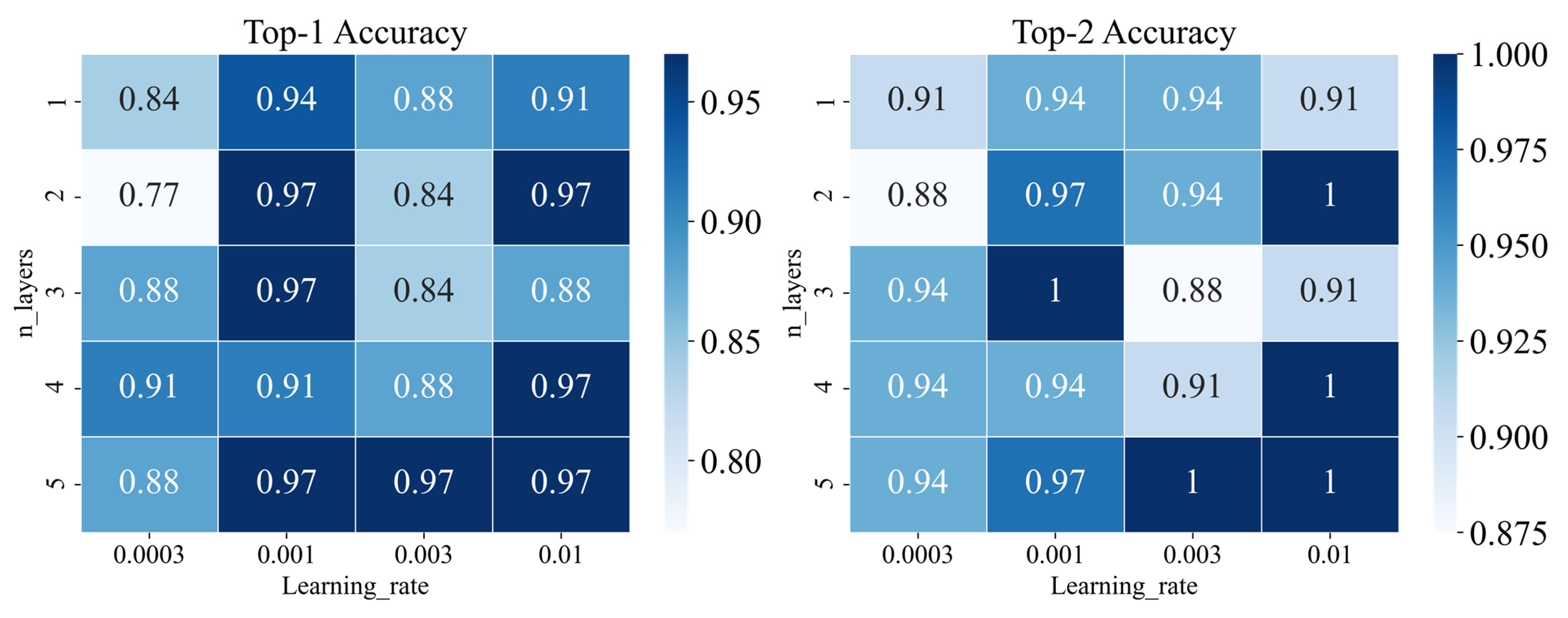
2.3.3. Tuning Parameters for WLN
2.4. Transformer
2.4.1. Data Preprocessing
2.4.2. Model Building
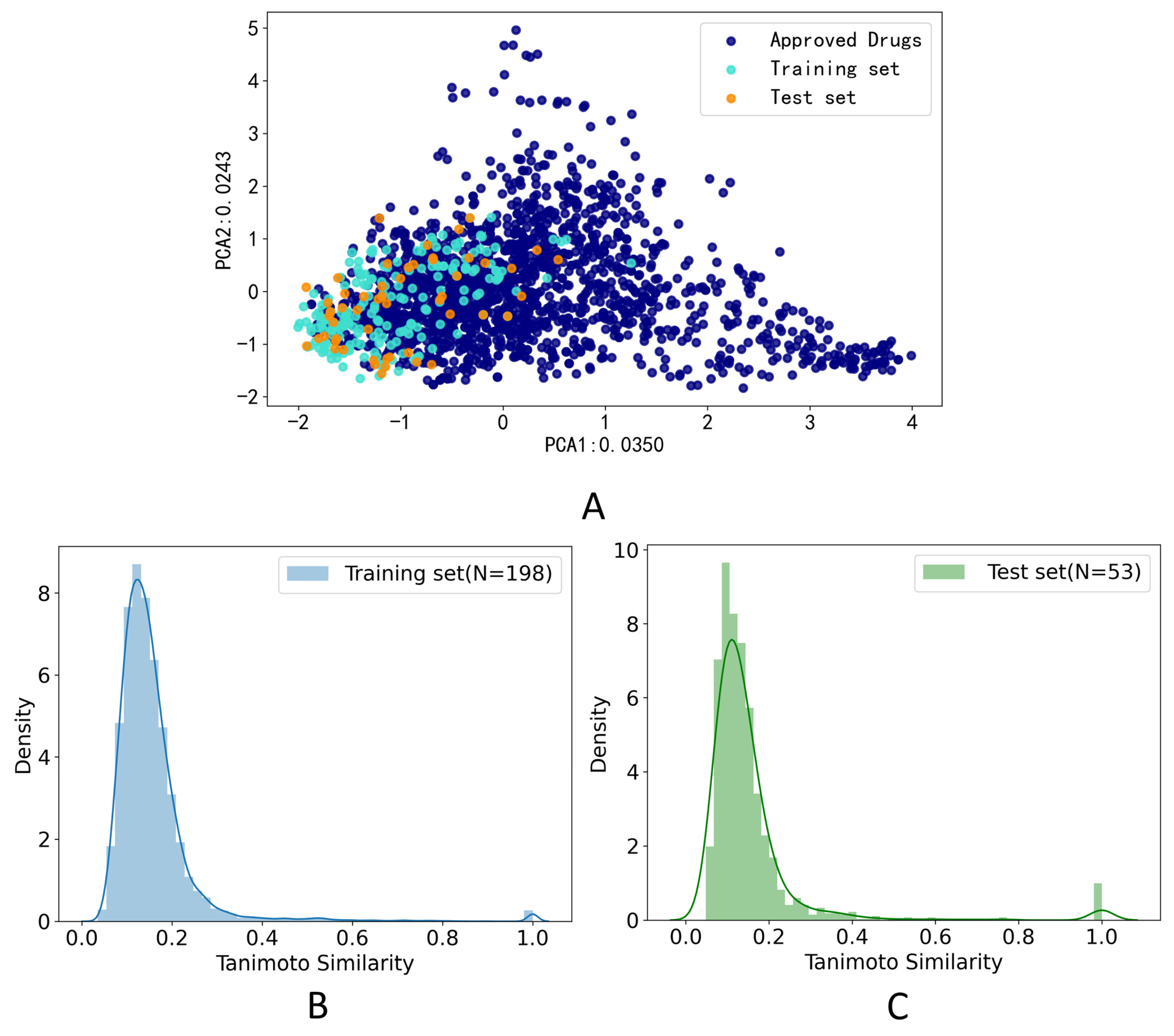
2.5. Validation of Model Performance
2.6. Comparison with Published Works
3. Results
3.1. Data Set Analysis
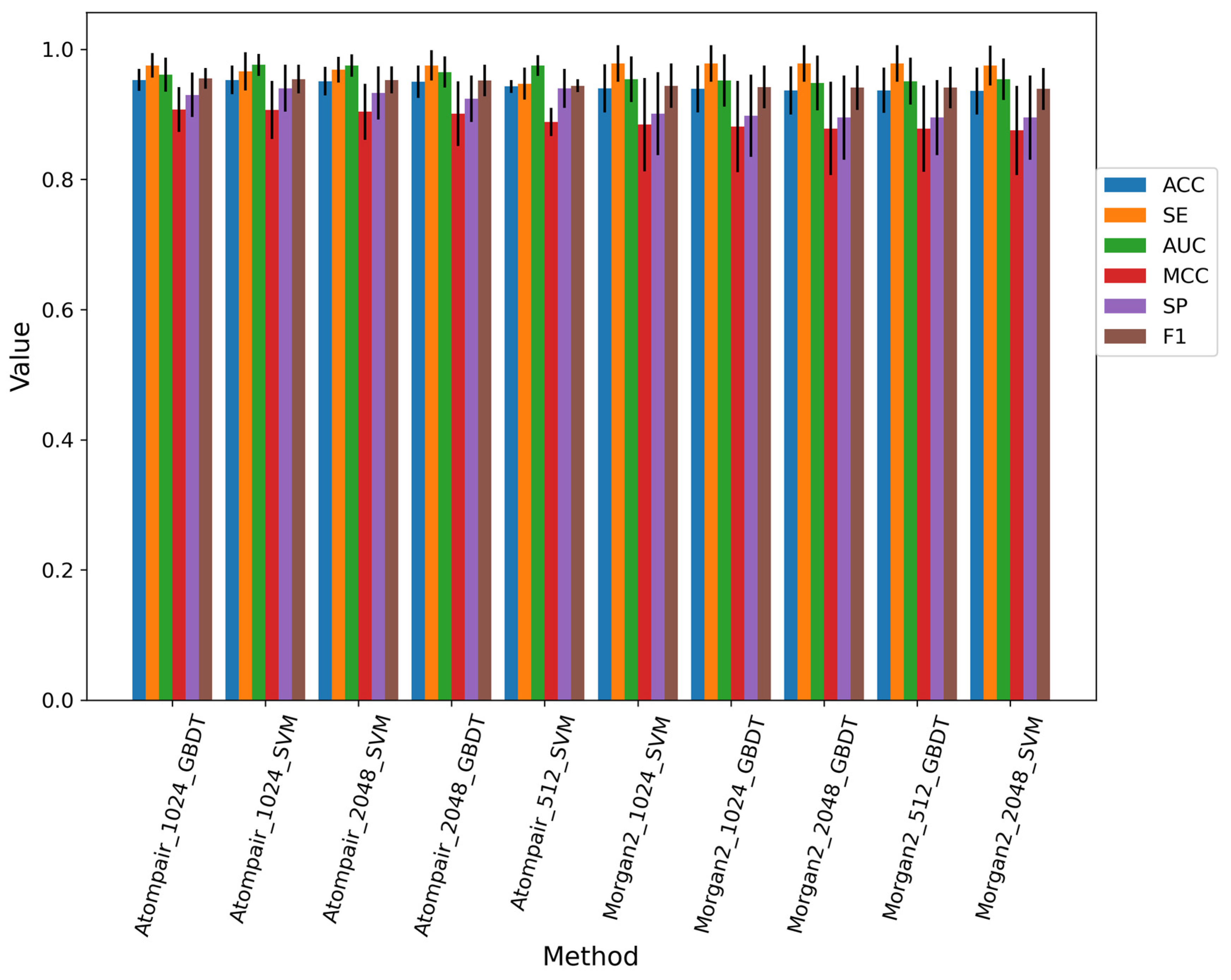
3.2. The Performance of Three Machine Learning Methods
3.2.1. Performance of the Fingerprint-Based Method
3.2.2. Performance of the Graph-Based Method
3.2.3. Performance of the Sequence-Based Method
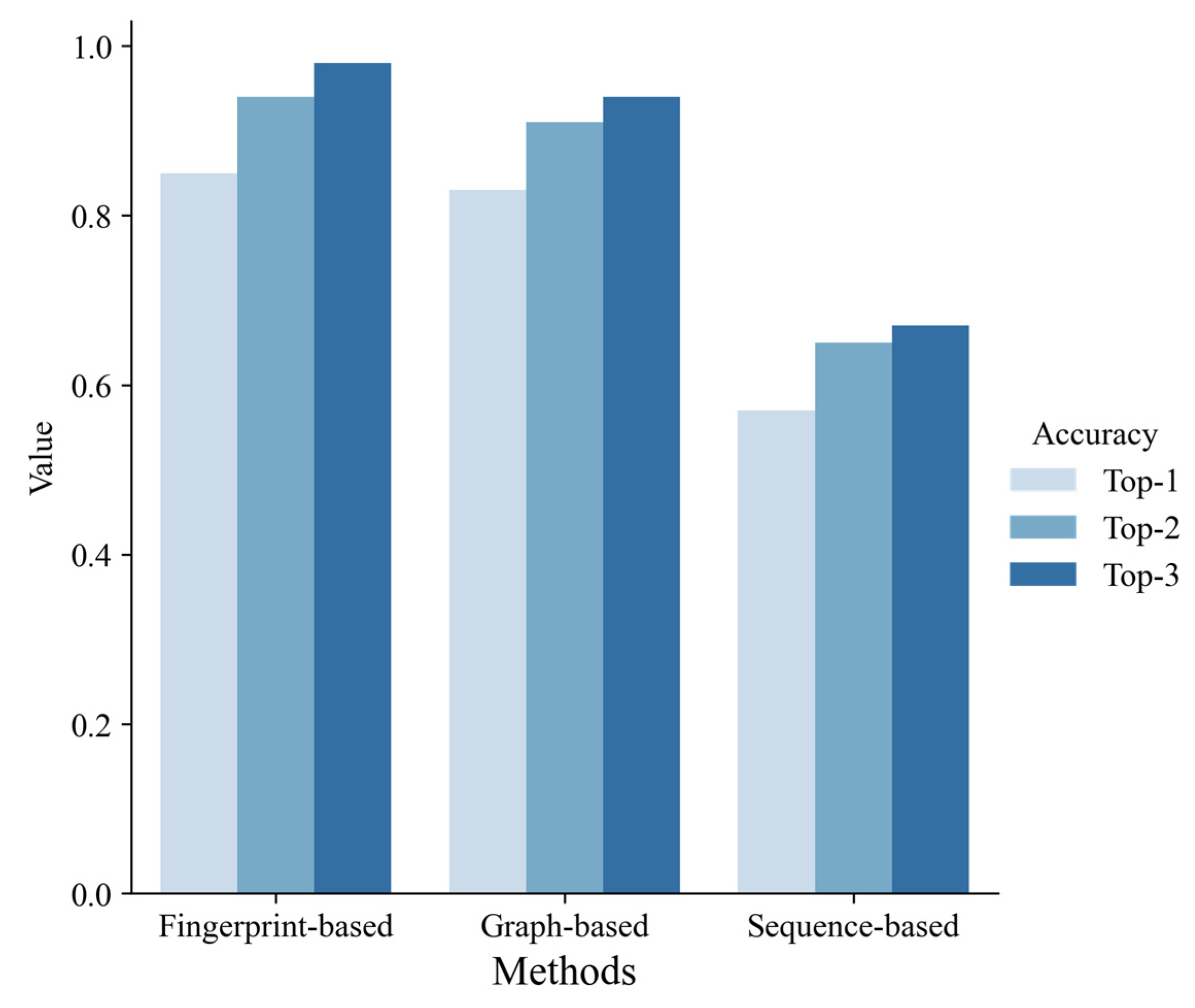
3.3. Comparison of the Methods Each Other and with Others
4. Discussion
4.1. Data Analysis
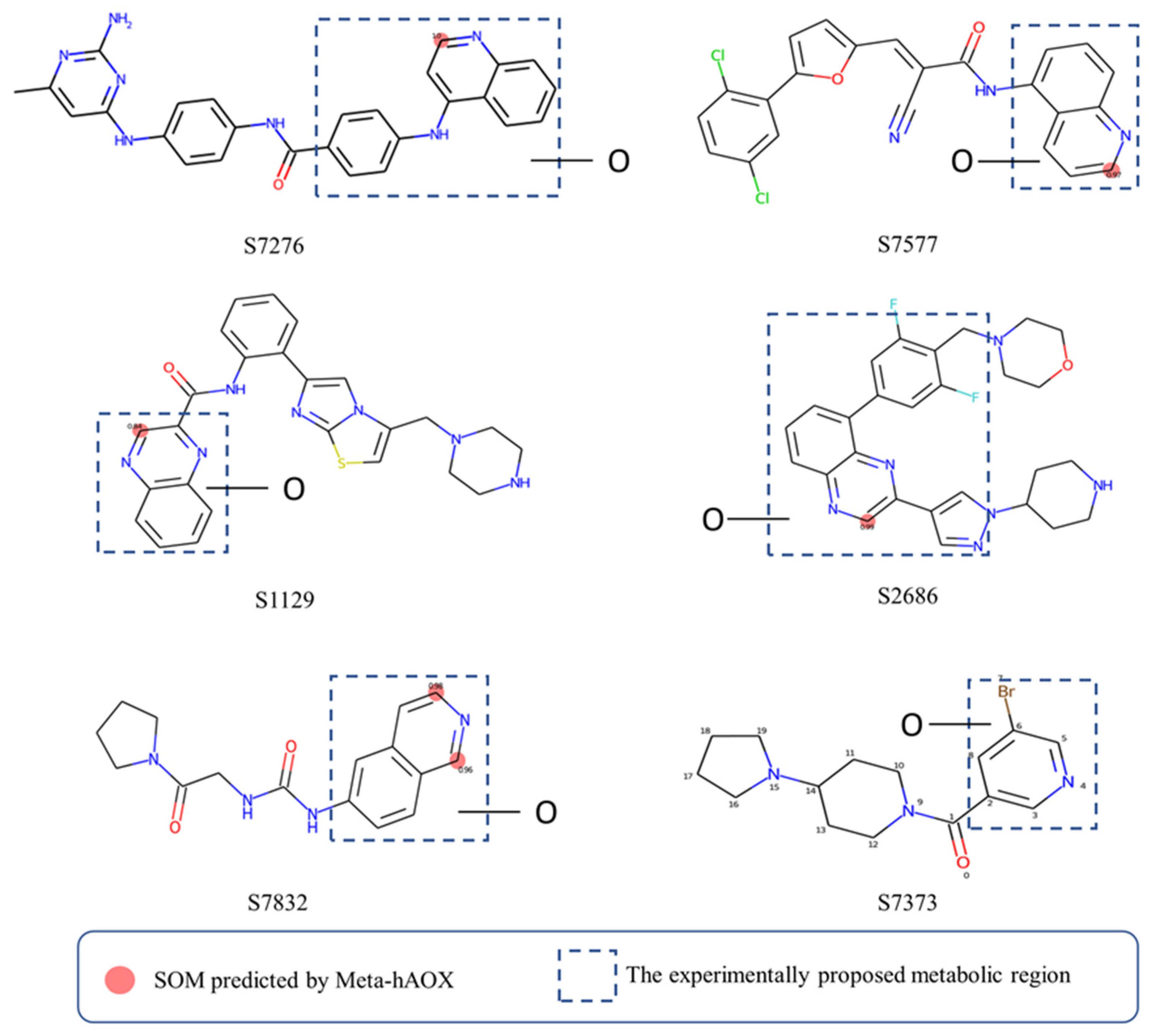
4.2. The Analysis of Our Models
4.3. Comparison of Our Model with Others
5. Conclusions
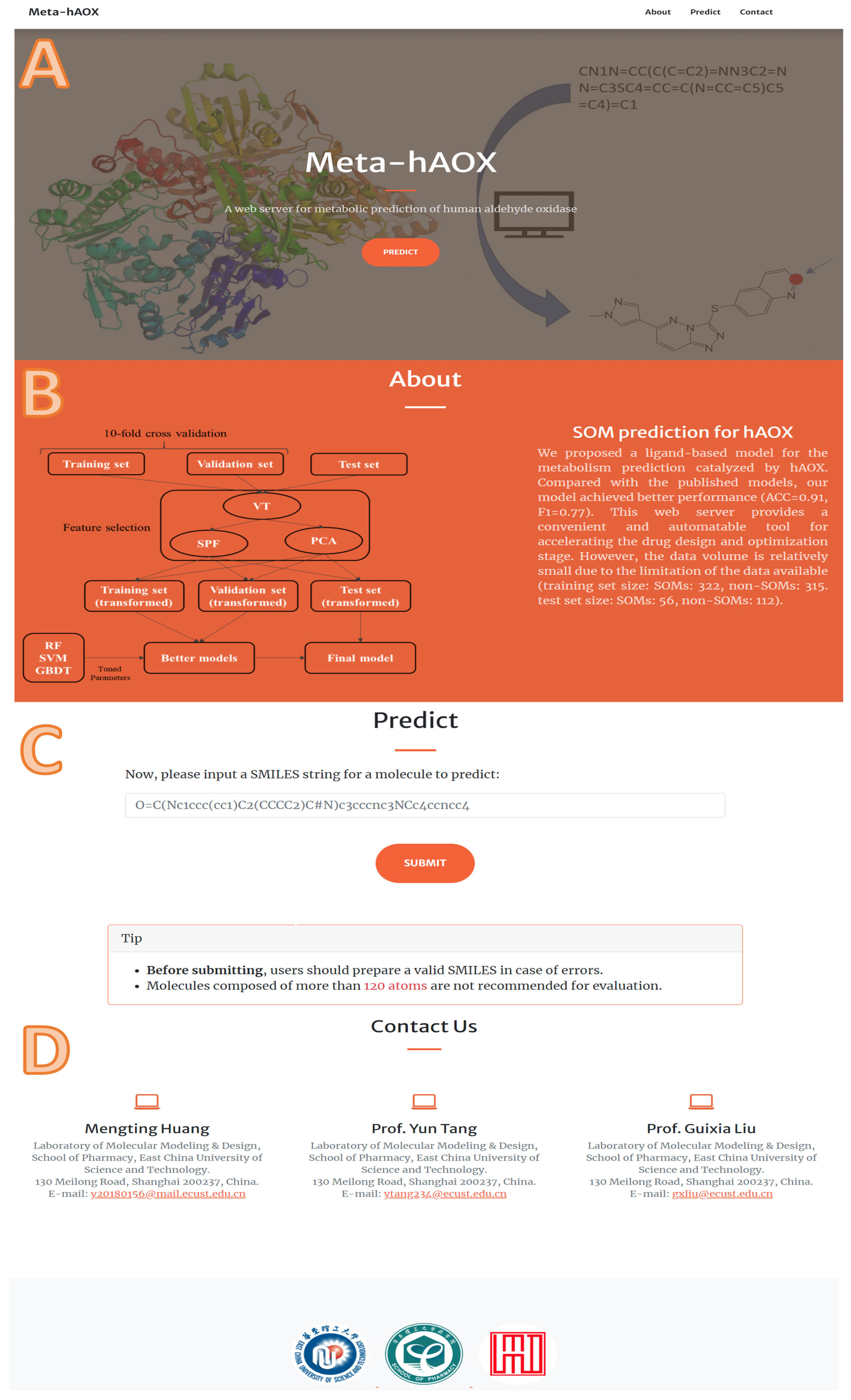
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rashidi, M.R.; Soltani, S. An overview of aldehyde oxidase: An enzyme of emerging importance in novel drug discovery. Expert Opin. Drug Discov. 2017, 12, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Pryde, D.C.; Dalvie, D.; Hu, Q.; Jones, P.; Obach, R.S.; Tran, T.D. Aldehyde Oxidase: An Enzyme of Emerging Importance in Drug Discovery. J. Med. Chem. 2010, 53, 8441–8460. [Google Scholar] [CrossRef]
- Sanoh, S.; Tayama, Y.; Sugihara, K.; Kitamura, S.; Ohta, S. Significance of aldehyde oxidase during drug development: Effects on drug metabolism, pharmacokinetics, toxicity, and efficacy. Drug Metab. Pharm. 2015, 30, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Akabane, T.; Tanaka, K.; Irie, M.; Terashita, S.; Teramura, T. Case report of extensive metabolism by aldehyde oxidase in humans: Pharmacokinetics and metabolite profile of FK3453 in rats, dogs, and humans. Xenobiotica 2011, 41, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Sanoh, S.; Nozaki, K.; Murai, H.; Terashita, S.; Teramura, T.; Ohta, S. Prediction of human metabolism of FK3453 by aldehyde oxidase using chimeric mice transplanted with human or rat hepatocytes. Drug Metab. Dispos. 2012, 40, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, H.; Weller, P.; Zheng, M.; Tao, W.; Wang, J.; Liao, G.; Monshouwer, M.; Peltz, G. In silico and in vitro pharmacogenetics: Aldehyde oxidase rapidly metabolizes a p38 kinase inhibitor. Pharm. J. 2011, 11, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.R.; Rugg, T.; Gordon, M.; Rooney, I.; Rosen, L.; Zeh, K.; Liu, R.; Burris, H.A.; Ramanathan, R.K. Unexpected renal toxicity associated with SGX523, a small molecule inhibitor of MET. Investig. N. Drugs 2013, 31, 363–369. [Google Scholar] [CrossRef]
- Lolkema, M.P.; Bohets, H.H.; Arkenau, H.T.; Lampo, A.; Barale, E.; de Jonge, M.J.A.; van Doorn, L.; Hellemans, P.; de Bono, J.S.; Eskens, F.A.L.M. The c-Met Tyrosine Kinase Inhibitor JNJ-38877605 Causes Renal Toxicity through Species-Specific Insoluble Metabolite Formation. Clin. Cancer Res. 2015, 21, 2297–2304. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.A.; Korzekwa, K.R.; McMasters, D.R.; Fandozzi, C.M.; Jones, J.P. Use of density functional calculations to predict the regioselectivity of drugs and molecules metabolized by aldehyde oxidase. J. Med. Chem. 2007, 50, 4642–4647. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, J.F.; Jones, J.P. Studies on the Mechanism of Aldehyde Oxidase and Xanthine Oxidase. J. Org. Chem. 2008, 73, 9469–9472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montefiori, M.; Lyngholm-Kjaerby, C.; Long, A.; Olsen, L.; Jorgensen, F.S. Fast Methods for Prediction of Aldehyde Oxidase-Mediated Site-of-Metabolism. Comput. Struct. Biotechnol. J. 2019, 17, 345–351. [Google Scholar] [CrossRef]
- Zhao, J.; Cui, R.; Wang, L.; Chen, Y.; Fu, Z.; Ding, X.; Cui, C.; Yang, T.; Li, X.; Xu, Y.; et al. Revisiting Aldehyde Oxidase Mediated Metabolism in Drug-like Molecules: An Improved Computational Model. J. Med. Chem. 2020, 63, 6523–6537. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, L.; Wang, Y.L.; Xing, J.; Zhou, L.; Zhong, D.; Luo, X.; Jiang, H.L.; Chen, K.; Zheng, M.; et al. Aldehyde Oxidase Mediated Metabolism in Drug-like Molecules: A Combined Computational and Experimental Study. J. Med. Chem. 2017, 60, 2973–2982. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yang, H.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. Computational Prediction of Site of Metabolism for UGT-Catalyzed Reactions. J. Chem. Inf. Model. 2019, 59, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cai, Y.; Li, W.; Liu, G.; Tang, Y. In Silico Prediction of Metabolic Epoxidation for Drug-like Molecules via Machine Learning Methods. Mol. Inform. 2020, 39, e1900178. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Wang, L.; Duan, H. A graph-convolutional neural network for addressing small-scale reaction prediction. Chem. Commun. 2021, 57, 4114–4117. [Google Scholar] [CrossRef]
- Huang, M.; Lou, C.; Wu, Z.; Li, W.; Lee, P.W.; Tang, Y.; Liu, G. In silico prediction of UGT-mediated metabolism in drug-like molecules via graph neural network. J. Cheminf. 2022, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Wu, Z.; Hsieh, C.Y.; Chen, G.; Liao, B.; Wang, Z.; Shen, C.; Cao, D.S.; Wu, J.A.; Hou, T.J. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminf. 2021, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, L.; Wang, X.; Zhang, C.; Ge, J.; Tang, J.; Su, A.; Duan, H. Data augmentation and transfer learning strategies for reaction prediction in low chemical data regimes. Org. Chem. Front. 2021, 8, 1415–1423. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, C.Y.; Bai, R.R.; Li, J.J.; Duan, H.L. Heck reaction prediction using a transformer model based on a transfer learning strategy. Chem. Commun. 2020, 56, 9368–9371. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar] [CrossRef]
- Li, M.; Zhou, J.; Hu, J.; Fan, W.; Zhang, Y.; Gu, Y.; Karypis, G. DGL-LifeSci: An Open-Source Toolkit for Deep Learning on Graphs in Life Science. Acs Omega 2021, 6, 27233–27238. [Google Scholar] [CrossRef] [PubMed]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.M.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. In Advances in Neural Information Processing Systems 32 (Nips 2019); ACM: New York, NY, USA, 2019; Volume 32. [Google Scholar]
- Cruciani, G.; Milani, N.; Benedetti, P.; Lepri, S.; Cesarini, L.; Baroni, M.; Spyrakis, F.; Tortorella, S.; Mosconi, E.; Goracci, L. From Experiments to a Fast Easy-to-Use Computational Methodology to Predict Human Aldehyde Oxidase Selectivity and Metabolic Reactions. J. Med. Chem. 2018, 61, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Nilakantan, R.; Bauman, N.; Dixon, J.S. Topological torsion: A new molecular descriptor for SAR applications. Comparison with other descriptors. J. Chem. Inf. Comput. Sci. 1987, 27, 82–85. [Google Scholar] [CrossRef]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom pairs as molecular features in structure-activity studies: Definition and applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Danishuddin; Khan, A.U. Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Scholkopf, B. An introduction to support vector machines. In Recent Advances and Trends in Nonparametric Statistics; Elsevier: Amsterdam, The Netherlands, 2003; pp. 3–17. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Coley, C.W.; Jin, W.; Rogers, L.; Jamison, T.F.; Jaakkola, T.S.; Green, W.H.; Barzilay, R.; Jensen, K.F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 2019, 10, 370–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.A.; Torrance, G.; Baldacci, L.; Cuesta, S.M.; Fenninger, F.; Gopal, N.; Choudhary, S.; May, J.W.; Holliday, G.L.; Steinbeck, C.; et al. Reaction Decoder Tool (RDT): Extracting features from chemical reactions. Bioinformatics 2016, 32, 2065–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shervashidze, N.; Schweitzer, P.; van Leeuwen, E.J.; Mehlhorn, K.; Borgwardt, K.M. Weisfeiler-Lehman Graph Kernels. J. Mach. Learn. Res. 2011, 12, 2539–2561. [Google Scholar]
- Cai, C.; Wang, S.; Xu, Y.; Zhang, W.; Tang, K.; Ouyang, Q.; Lai, L.; Pei, J. Transfer Learning for Drug Discovery. J. Med. Chem. 2020, 63, 8683–8694. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Zhang, C.; Wang, L.; Yao, C.; Ge, J.; Duan, H. Transfer Learning: Making Retrosynthetic Predictions Based on a Small Chemical Reaction Dataset Scale to a New Level. Molecules 2020, 25, 2357. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.M. Extraction of Chemical Structures and Reactions from the Literature. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2012. [Google Scholar] [CrossRef]
- Schwaller, P.; Gaudin, T.; Lanyi, D.; Bekas, C.; Laino, T. “Found in Translation”: Predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 2018, 9, 6091–6098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.D.; Gomes, J.; Nguyen, Q.L.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent. Sci. 2017, 3, 1103–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pihan, E.; Colliandre, L.; Guichou, J.F.; Douguet, D. e-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. Bioinformatics 2012, 28, 1540–1541. [Google Scholar] [CrossRef] [Green Version]
- Cerny, M.A. Prevalence of Non-Cytochrome P450-Mediated Metabolism in Food and Drug Administration-Approved Oral and Intravenous Drugs: 2006–2015. Drug Metab. Dispos. 2016, 44, 1246–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litsa, E.E.; Das, P.; Kavraki, L.E. Prediction of drug metabolites using neural machine translation. Chem. Sci. 2020, 11, 12777–12788. [Google Scholar] [CrossRef] [PubMed]
- Lepri, S.; Ceccarelli, M.; Milani, N.; Tortorella, S.; Cucco, A.; Valeri, A.; Goracci, L.; Brink, A.; Cruciani, G. Structure-metabolism relationships in human-AOX: Chemical insights from a large database of aza-aromatic and amide compounds. Proc. Natl. Acad. Sci. USA 2017, 114, E3178–E3187. [Google Scholar] [CrossRef] [Green Version]
- Manevski, N.; King, L.; Pitt, W.R.; Lecomte, F.; Toselli, F. Metabolism by Aldehyde Oxidase: Drug Design and Complementary Approaches to Challenges in Drug Discovery. J. Med. Chem. 2019, 62, 10955–10994. [Google Scholar] [CrossRef] [PubMed]
- Diamond, S.; Boer, J.; Maduskuie, T.P.; Falahatpisheh, N.; Li, Y.; Yeleswaram, S. Species-Specific Metabolism of SGX523 by Aldehyde Oxidase and the Toxicological Implications. Drug Metab. Dispos. 2010, 38, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zientek, M.; Jiang, Y.; Youdim, K.; Obach, R.S. In Vitro-In Vivo Correlation for Intrinsic Clearance for Drugs Metabolized by Human Aldehyde Oxidase. Drug Metab. Dispos. 2010, 38, 1322–1327. [Google Scholar] [CrossRef] [Green Version]
- Krenitsky, T.A.; Neil, S.M.; Elion, G.B.; Hitchings, G.H. A comparison of the specificities of xanthine oxidase and aldehyde oxidase. Arch. Biochem. Biophys. 1972, 150, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Choughule, K.V.; Barnaba, C.; Joswig-Jones, C.A.; Jones, J.P. In Vitro Oxidative Metabolism of 6-Mercaptopurine in Human Liver: Insights into the Role of the Molybdoflavoenzymes Aldehyde Oxidase, Xanthine Oxidase, and Xanthine Dehydrogenase. Drug Metab. Dispos. 2014, 42, 1334–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.D. Comparison of levels of aldehyde oxidase with cytochrome P450 activities in human liver in vitro. Biochem. Pharmacol. 1994, 48, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Hutzler, J.M.; Yang, Y.S.; Albaugh, D.; Fullenwider, C.L.; Schmenk, J.; Fisher, M.B. Characterization of aldehyde oxidase enzyme activity in cryopreserved human hepatocytes. Drug Metab. Dispos. 2012, 40, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubley, C.; Stell, J.G.; Mathieson, D.W. The oxidation of azaheterocycles with mammalian liver aldehyde oxidase. Xenobiotica 1979, 9, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Choughule, K.V.; Joswig-Jones, C.A.; Jones, J.P. Interspecies differences in the metabolism of methotrexate: An insight into the active site differences between human and rabbit aldehyde oxidase. Biochem. Pharmacol. 2015, 96, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Behera, D.; Pattem, R.; Gudi, G. Effect of commonly used organic solvents on Aldehyde oxidase-mediated Vanillin, Phthalazine and Methotrexate oxidation in human, rat and mouse liver subcellular fractions. Xenobiotica 2014, 44, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Klecker, R.W.; Cysyk, R.L.; Collins, J.M. Zebularine metabolism by aldehyde oxidase in hepatic cytosol from humans, monkeys, dogs, rats, and mice: Influence of sex and inhibitors. Bioorg. Med. Chem. 2006, 14, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Dalvie, D.; Sun, H.; Xiang, C.; Hu, Q.; Jiang, Y.; Kang, P. Effect of structural variation on aldehyde oxidase-catalyzed oxidation of zoniporide. Drug Metab. Dispos. 2012, 40, 1575–1587. [Google Scholar] [CrossRef] [Green Version]
- Dick, R.A. Refinement of In Vitro Methods for Identification of Aldehyde Oxidase Substrates Reveals Metabolites of Kinase Inhibitors. Drug Metab. Dispos. 2018, 46, 846–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, N.E.; Baldwin, S.J.; Cutler, L.; Deeks, N.; Kelly, P.J.; Nash, M.; Shardlow, C.E.; Stemp, G.; Thewlis, K.; Ayrton, A.; et al. Pharmacokinetics of the novel, high-affinity and selective dopamine D3 receptor antagonist SB-277011 in rat, dog and monkey: In vitro/in vivo correlation and the role of aldehyde oxidase. Xenobiotica 2001, 31, 677–686. [Google Scholar] [CrossRef]
- Jia, H.; Dai, G.; Weng, J.; Zhang, Z.; Wang, Q.; Zhou, F.; Jiao, L.; Cui, Y.; Ren, Y.; Fan, S.; et al. Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2, 3]triazolo[4,5-b]pyrazine (volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for treatment of cancer. J. Med. Chem. 2014, 57, 7577–7589. [Google Scholar] [CrossRef]
- Jensen, K.G.; Jacobsen, A.M.; Bundgaard, C.; Nilausen, D.O.; Thale, Z.; Chandrasena, G.; Jorgensen, M. Lack of Exposure in a First-in-Man Study Due to Aldehyde Oxidase Metabolism: Investigated by Use of C-14-microdose, Humanized Mice, Monkey Pharmacokinetics, and In Vitro Methods. Drug Metab. Dispos. 2017, 45, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Katyayan, K.; Yi, P.; Monk, S.; Cassidy, K. Excretion, Mass Balance, and Metabolism of [(14)C]LY3202626 in Humans: An Interplay of Microbial Reduction, Reabsorption, and Aldehyde Oxidase Oxidation That Leads to an Extended Excretion Profile. Drug Metab. Dispos. 2020, 48, 698–707. [Google Scholar] [CrossRef]
- Zetterberg, C.; Maltais, F.; Laitinen, L.; Liao, S.; Tsao, H.; Chakilam, A.; Hariparsad, N. VX-509 (Decernotinib)-Mediated CYP3A Time-Dependent Inhibition: An Aldehyde Oxidase Metabolite as a Perpetrator of Drug-Drug Interactions. Drug Metab. Dispos. 2016, 44, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Crouch, R.D.; Blobaum, A.L.; Felts, A.S.; Conn, P.J.; Lindsley, C.W. Species-Specific Involvement of Aldehyde Oxidase and Xanthine Oxidase in the Metabolism of the Pyrimidine-Containing mGlu5-Negative Allosteric Modulator VU0424238 (Auglurant). Drug Metab. Dispos. 2017, 45, 1245–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adusumalli, S.; Jamwal, R.; Obach, R.S.; Ryder, T.F.; Leggio, L.; Akhlaghi, F. Role of Molybdenum-Containing Enzymes in the Biotransformation of the Novel Ghrelin Receptor Inverse Agonist PF-5190457: A Reverse Translational Bed-to-Bench Approach. Drug Metab. Dispos. 2019, 47, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Stell, J.G.; Wheelhouse, R.T.; Wright, C.W. Metabolism of cryptolepine and 2-fluorocryptolepine by aldehyde oxidase. J. Pharm. Pharmacol. 2012, 64, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Nejabati, H.R.; Samadi, N.; Shahnazi, V.; Mihanfar, A.; Fattahi, A.; Latifi, Z.; Bahrami-Asl, Z.; Roshangar, L.; Nouri, M. Nicotinamide and its metabolite N1-Methylnicotinamide alleviate endocrine and metabolic abnormalities in adipose and ovarian tissues in rat model of Polycystic Ovary Syndrome. Chem. Biol. Interact. 2020, 324, 109093. [Google Scholar] [CrossRef] [PubMed]
- Beedham, C.; Critchley, D.J.; Rance, D.J. Substrate specificity of human liver aldehyde oxidase toward substituted quinazolines and phthalazines: A comparison with hepatic enzyme from guinea pig, rabbit, and baboon. Arch. Biochem. Biophys. 1995, 319, 481–490. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | SMARTS | Descriptions | Example |
|---|---|---|---|
| A | [$(cR;H]:[nX2R])] | The carbon in the aromatic ring adjacent to the aromatic nitrogen with exactly one hydrogen |  |
| B | [$([#6D2R;H][*][*][#7X2R])] | The carbon in the aromatic ring conjugated addition with γ-position nitrogen with exactly one hydrogen |  |
| Step | Examples: Reactants >> Products |
|---|---|
| Reaction |  |
| Reaction SMILES | CC1 = NC2 = C(C = CC = C2)C = N1>>CC3 = NC(O) = C4C = CC = CC4 = N3 |
| Atom-mapping Reaction SMILES | [N:1]1 = [CH:2][C:3] = 2[CH:4] = [CH:5][CH:6] = [CH:7][C:8]2[N:9] = [C:10]1[CH3:11] > > [CH3:11][C:10] = 1[N:9] = [C:8]2[CH:7] = [CH:6][CH:5] = [CH:4][C:3]2 = [C:2]([OH:12])[N:1]1 |
| Step | Reactant | Product |
|---|---|---|
| SMILES | CC(CO)NC1 = NC = C2C(N(CC(O)C)C(C(OC3 = CC = C(F)C = C3F) = C2) = O) = N1 | CC(NC1 = NC(O) = C2C(N(C(C(OC3 = CC = C(C = C3F)F) = C2) = O)CC(C)O) = N1)CO |
| Tokenization | C C (C O) N C 1 = N C = C 2 C (N (C C (O) C) C (C (O C 3 = C C = C (F) C = C 3 F) = C 2) = O) = N 1 | C C (N C 1 = N C (O) = C 2 C (N (C (C (O C 3 = C C = C (C = C 3 F) F) = C 2) = O) C C (C) O) = N 1) C O |
| Model | SMILES Validity | Top 1 Accuracy | Top 2 Accuracy | Top 3 Accuracy |
|---|---|---|---|---|
| Transformer–baseline model | 0.69 | 0.08 | 0.08 | 0.10 |
| Transformer–transfer learning model | 0.93 | 0.57 | 0.63 | 0.67 |
| Model | SE | SP | ACC | F1 |
|---|---|---|---|---|
| Meta-hAOX | 0.77 | 0.93 | 0.91 | 0.77 |
| DTAOX | 0.50 | 0.87 | 0.81 | 0.47 |
| DTNAOX | 0.79 | 0.91 | 0.89 | 0.71 |
| NMR shielding | 0.71 | 0.77 | 0.76 | 0.50 |
| ESP charge | 0.79 | 0.83 | 0.82 | 0.59 |
| Chemical shift | 0.71 | 0.77 | 0.76 | 0.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Zhu, K.; Wang, Y.; Lou, C.; Sun, H.; Li, W.; Tang, Y.; Liu, G. In Silico Prediction of Metabolic Reaction Catalyzed by Human Aldehyde Oxidase. Metabolites 2023, 13, 449. https://doi.org/10.3390/metabo13030449
Huang M, Zhu K, Wang Y, Lou C, Sun H, Li W, Tang Y, Liu G. In Silico Prediction of Metabolic Reaction Catalyzed by Human Aldehyde Oxidase. Metabolites. 2023; 13(3):449. https://doi.org/10.3390/metabo13030449
Chicago/Turabian StyleHuang, Mengting, Keyun Zhu, Yimeng Wang, Chaofeng Lou, Huimin Sun, Weihua Li, Yun Tang, and Guixia Liu. 2023. "In Silico Prediction of Metabolic Reaction Catalyzed by Human Aldehyde Oxidase" Metabolites 13, no. 3: 449. https://doi.org/10.3390/metabo13030449
APA StyleHuang, M., Zhu, K., Wang, Y., Lou, C., Sun, H., Li, W., Tang, Y., & Liu, G. (2023). In Silico Prediction of Metabolic Reaction Catalyzed by Human Aldehyde Oxidase. Metabolites, 13(3), 449. https://doi.org/10.3390/metabo13030449