

Design of Novel Letrozole Analogues Targeting Aromatase for Breast Cancer: Molecular Docking, Molecular Dynamics, and Theoretical Studies on Gold Nanoparticles
 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Protein Preparation
2.2. Preparation of the Ligand
2.3. Quantum-Polarized Ligand Docking (QPLD) and Design of New Aromatase Inhibitors
2.4. Glide Docking
2.5. ADME Prediction
2.6. Molecular Dynamics (MD) Simulation and Post-MD Binding Free Energy Calculation
2.7. Preparation of Gold Nanoparticles and DFT Calculations
3. Results
3.1. Molecular Docking and ADME Studies
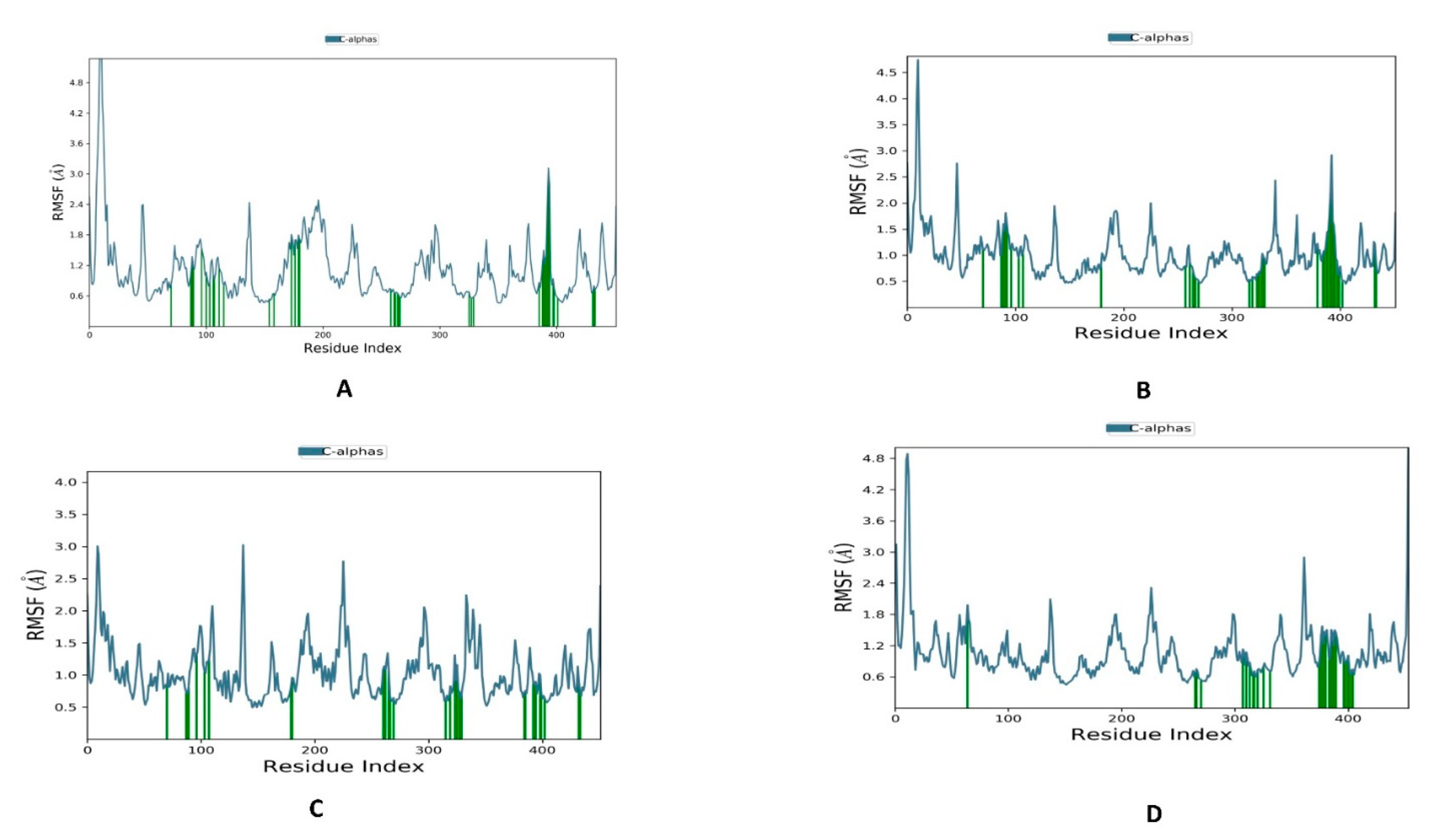
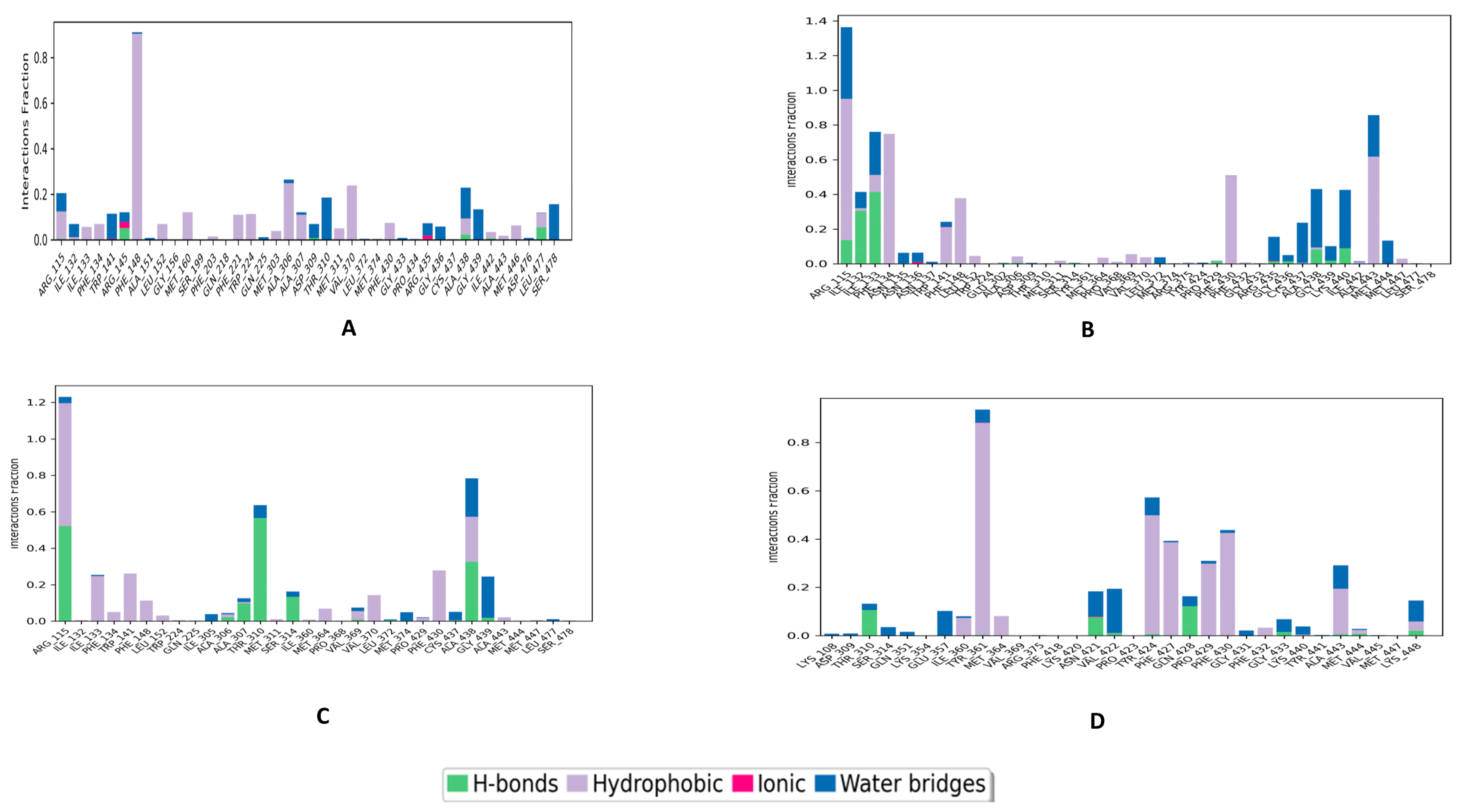
3.2. Molecular Dynamics and Post-MD MM-GBSA Calculations
3.3. DFT Calculations
3.4. Molecular Docking with Human Off-Target Desmolase
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naghavi, M.; Wang, H.; Lozano, R.; Davis, A.; Liang, X.; Zhou, M.; Vollset, S.E.; Abbasoglu Ozgoren, A.; Abdalla, S.; Abd-Allah, F.; et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- van Landeghem, A.A.J.; Poortman, J.; Nabuurs, M.; Thijssen, J.H.H. Endogenous Concentration and Subcellular Distribution of Estrogens in Normal and Malignant Human Breast Tissue. Cancer Res. 1985, 45, 2900–2906. [Google Scholar] [PubMed]
- Bulun, S.E.; Takayama, K.; Suzuki, T.; Sasano, H.; Yilmaz, B.; Sebastian, S. Organization of the Human Aromatase P450 (CYP19) Gene. Semin. Reprod. Med. 2004, 22, 5–9. [Google Scholar] [CrossRef]
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. J. Steroid Biochem. Mol. Biol. 2011, 125, 13–22. [Google Scholar] [CrossRef]
- Riemsma, R.; Forbes, C.A.; Kessels, A.; Lykopoulos, K.; Amonkar, M.M.; Rea, D.W.; Kleijnen, J. Systematic review of aromatase inhibitors in the first-line treatment for hormone sensitive advanced or metastatic breast cancer. Breast Cancer Res. Treat. 2010, 123, 9–24. [Google Scholar] [CrossRef]
- Sledge, G.W.; Mamounas, E.P.; Hortobagyi, G.N.; Burstein, H.J.; Goodwin, P.J.; Wolff, A.C. Past, present, and future challenges in breast cancer treatment. J. Clin. Oncol. 2014, 32, 1979–1986. [Google Scholar] [CrossRef]
- Haynes, B.P.; Dowsett, M.; Miller, W.R.; Dixon, J.M.; Bhatnagar, A.S. The pharmacology of letrozole. J. Steroid Biochem. Mol. Biol. 2003, 87, 35–45. [Google Scholar] [CrossRef]
- Buzdar, A.U.; Robertson, J.F.R.; Eiermann, W.; Nabholtz, J.M. An overview of the pharmacology and pharmacokinetics of the newer generation aromatase inhibitors anastrozole, letrozole, and exemestane. Cancer 2002, 95, 2006–2016. [Google Scholar] [CrossRef]
- Mumford, S.L.; Schisterman, E.F.; Siega-Riz, A.M.; Browne, R.W.; Gaskins, A.J.; Trevisan, M.; Steiner, A.Z.; Daniels, J.L.; Zhang, C.; Perkins, N.J.; et al. A longitudinal study of serum lipoproteins in relation to endogenous reproductive hormones during the menstrual cycle: Findings from the BioCycle study. J. Clin. Endocrinol. Metab. 2010, 95, E80–E85. [Google Scholar] [CrossRef]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem.-Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Ortiz, R.; Fernández-De Gortari, E. Overview of Computer-Aided Drug Design for Epigenetic Targets; Elsevier Inc.: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Liu, J.; Wang, R. Classification of Current Scoring Functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2012, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Gao, T.; Hong, H.; Sun, J. Applications of gold nanoparticles in cancer nanotechnology. Nanotechnol. Sci. Appl. 2008, 1, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; Armenia, E.; Aurilio, C.; Rosso, F.; Clemente, O.; Sena, G.D.E.; Barbarisi, M.; Barbarisi, A. New Treatment of Medullary and Papillary Human Thyroid Cancer: Biological Effects of Hyaluronic Acid Hydrogel Loaded with Quercetin Alone or in Combination to an Inhibitor of Aurora Kinase. J. Cell. Physiol. 2015, 231, 1784–1795. [Google Scholar] [CrossRef]
- Maestro; Schrödinger, LLC: New York, NY, USA, 2021.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Schrödinger LLC. Schrödinger Release 2018-3; Glide, Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- De Luca, M.; Occhiuzzi, M.A.; Rizzuti, B.; Ioele, G.; Ragno, G.; Garofalo, A.; Grande, F. Interaction of letrozole and its degradation products with aromatase: Chemometric assessment of kinetics and structure-based binding validation. J. Enzym. Inhib. Med. Chem. 2022, 37, 1600–1609. [Google Scholar] [CrossRef]
- Schrödinger. Schrödinger Release 2018-1; LigPrep|Schrödinger: New York, NY, USA, 2021. [Google Scholar]
- Alzain, A.A.; Ismail, A.; Fadlelmola, M.; Mohamed, M.A.; Mahjoub, M.; Makki, A.A.; Elsaman, T. De novo design of novel spike glycoprotein inhibitors using e-pharmacophore modeling, molecular hybridization, ADMET, quantum mechanics and molecular dynamics studies for COVID-19. Pak. J. Pharm. Sci. 2022, 35, 313–321. [Google Scholar]
- Cho, A.E.; Guallar, V.; Berne, B.J.; Friesner, R. Importance of accurate charges in molecular docking: Quantum Mechanical/Molecular Mechanical (QM/MM) approach. J. Comput. Chem. 2005, 26, 915–931. [Google Scholar] [CrossRef]
- Sirous, H.; Chemi, G.; Gemma, S.; Butini, S.; Debyser, Z.; Christ, F.; Saghaie, L.; Brogi, S.; Fassihi, A.; Campiani, G.; et al. Identification of Novel 3-Hydroxy-pyran-4-One Derivatives as Potent HIV-1 Integrase Inhibitors Using in silico Structure-Based Combinatorial Library Design Approach. Front. Chem. 2019, 7, 574. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]
- Alzain, A.A.; Elbadwi, F.A. Identification of novel TMPRSS2 inhibitors for COVID-19 using e-pharmacophore modelling, molecular docking, molecular dynamics and quantum mechanics studies. Inform. Med. Unlocked 2021, 26, 100758. [Google Scholar] [CrossRef]
- Elbadwi, F.A.; Khairy, E.A.; Alsamani, F.O.; Mahadi, M.A.; Abdalrahman, S.E.; Alsharf, Z.; Ahmed, M.; Elsayed, I.; Ibraheem, W.; Alzain, A.A. Identification of novel transmembrane Protease Serine Type 2 drug candidates for COVID-19 using computational studies. Inform. Med. Unlocked 2021, 26, 100725. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. GaussView 5.0.; E.U.A.: Wallingford, UK, 2016. [Google Scholar]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- De Abreu, F.; Schwartz, G.; Wells, W.; Tsongalis, G. Personalized therapy for breast cancer. Clin. Genet. 2014, 86, 62–67. [Google Scholar] [CrossRef]
- Caciolla, J.; Spinello, A.; Martini, S.; Bisi, A.; Zaffaroni, N.; Gobbi, S.; Magistrato, A. Targeting Orthosteric and Allosteric Pockets of Aromatase via Dual-Mode Novel Azole Inhibitors. ACS Med. Chem. Lett. 2020, 11, 732–739. [Google Scholar] [CrossRef]
- Suvannang, N.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Molecular docking of aromatase inhibitors. Molecules 2011, 16, 3597–3617. [Google Scholar] [CrossRef]
- Chamduang, C.; Pingaew, R.; Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Novel triazole-tetrahydroisoquinoline hybrids as human aromatase inhibitors. Bioorg. Chem. 2019, 93, 103327. [Google Scholar] [CrossRef]
- Duncan, B.; Kim, C.; Rotello, V.M. Gold nanoparticle platforms as drug and biomacromolecule delivery systems. J. Control. Release 2010, 148, 122–127. [Google Scholar] [CrossRef]
- Vecchione, R.; Quagliariello, V.; Giustetto, P.; Calabria, D.; Sathya, A.; Marotta, R.; Profeta, M.; Nitti, S.; Silvestri, N.; Pellegrino, T.; et al. Oil/water nano-emulsion loaded with cobalt ferrite oxide nanocubes for photo-acoustic and magnetic resonance dual imaging in cancer: In vitro and preclinical studies. Nanomed. Nanotechnol. Biol. Med. 2016, 13, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Sitia, L.; Sevieri, M.; Signati, L.; Bonizzi, A.; Chesi, A.; Mainini, F.; Corsi, F.; Mazzucchelli, S. HER-2-Targeted Nanoparticles for Breast Cancer Diagnosis and Treatment. Cancers 2022, 14, 2424. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
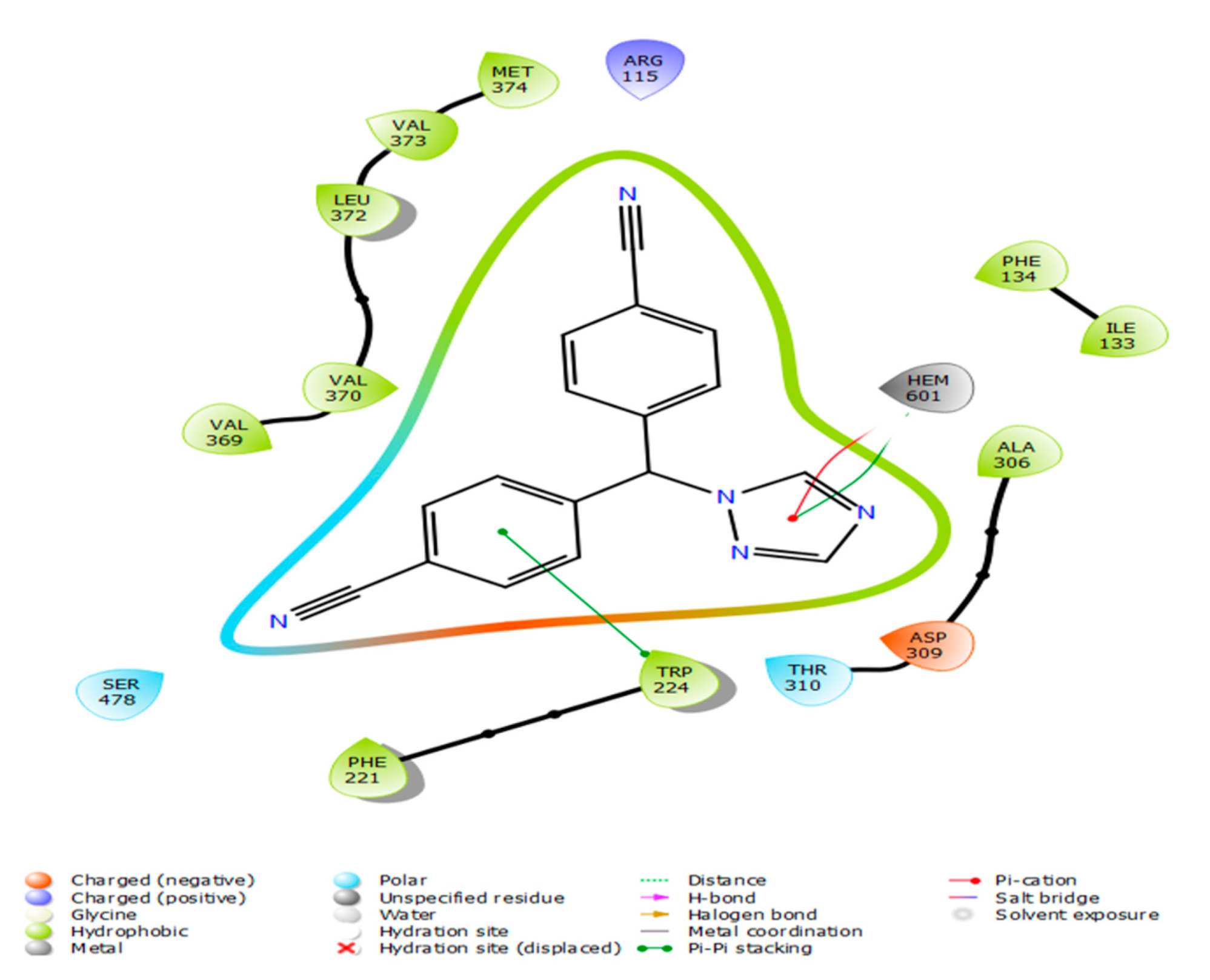
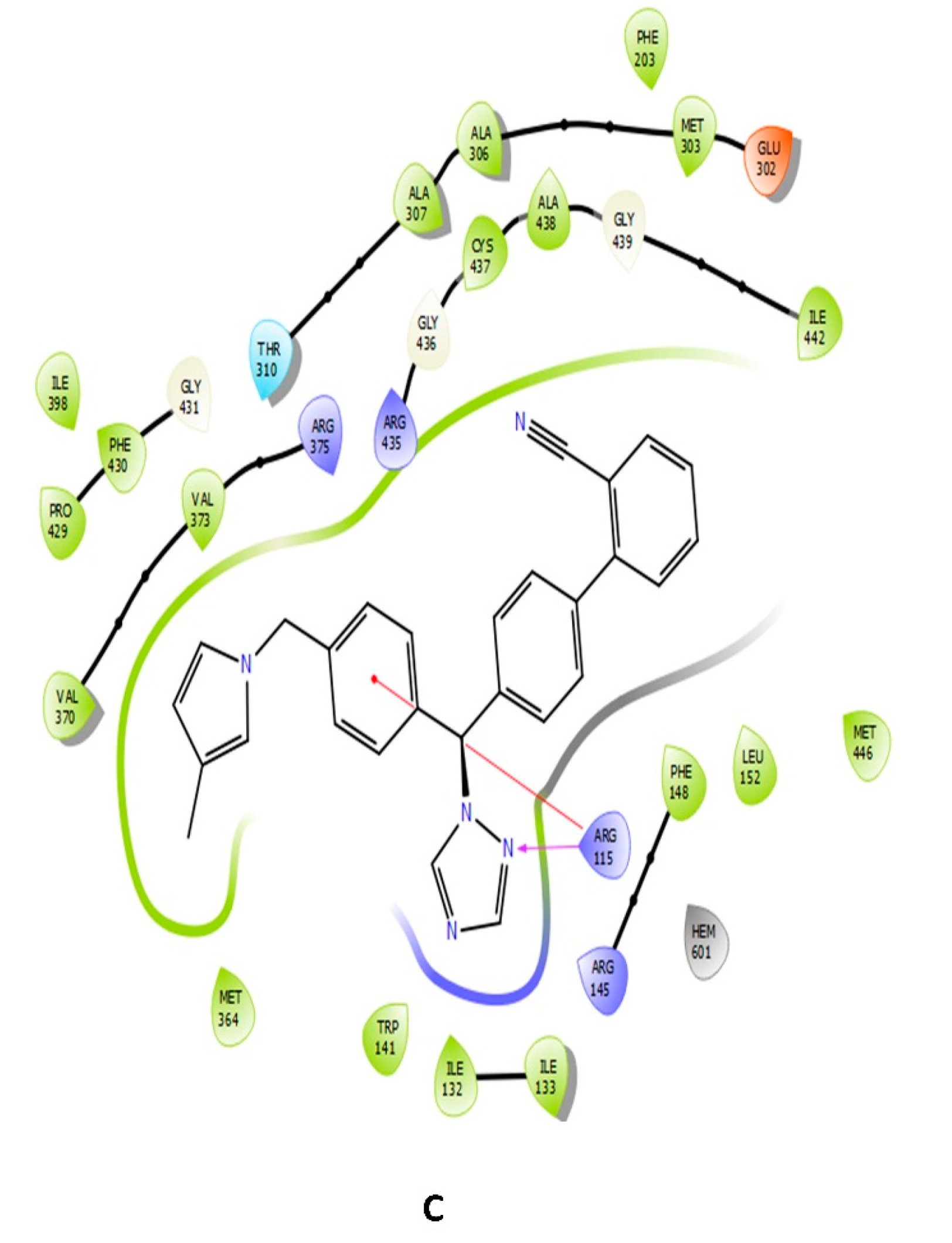
| Name | Docking Scores (Kcal/mol) | Pi-Cation | Hydrogen Bond | Hydrophobic Interaction |
|---|---|---|---|---|
| Combine1 | −8.117 | ARG115 | ARG115 | ALA306/ALA307/MET303/VAL370/VAL373/ARG375/THR310/GLU302 |
| Combine2 | −8.053 | ARG115 | ARG115 | PRO429/PHE430/ALA307/ALA306/ALA443 |
| Combine3 | −8.83 | ARG115 | ARG115 | ARG435/MET364/ARG375/ARG435/VAL370/VAL373 |
| Combine4 | −7.737 | ARG115 | GLN248 | ALA443/MET446/MET311/ILE442 |
| Combine5 | −7.704 | ARG115 | ARG115/PHE430 | GLU302/TRP141/MET303 |
| Combine6 | −7.411 | ARG115 | ARG115 | VAL373/GLU431 |
| Combine7 | −7.342 | HEM601 | ARG115/ARG375/ARG345 | ILE398/PRO429/PHE430/PHE432 |
| Combine8 | −7.324 | HEM601 | PHA430/ARG375 | PRO429/PHE430/ALA438/CYS437 |
| Combine9 | −7.304 | ARG115 | MET303/ALA306/ALA307/VAL373/PHE430 | |
| Combine10 | −7.287 | ARG145/PHE430 | ALA306/ALA307/ALA438/MET303/ILE398 | |
| Combine11 | −7.119 | ARG115 | GLN428 | ALA438/GLU439/VAL370/VAL373 |
| Combine12 | −7.09 | TRP141/ARG115 | PRO429/GLY431/ILE398 | |
| Combine13 | −7.059 | ARG115 | ARG115 | ALA307/ALA438/ALA443/CYS437/PRO429/PHE430/GLY431 |
| Combine14 | −7.041 | TRP141 | ALA306/ALA438/MET303/GLY430/GLY493/ARG375 | |
| Letrozole | −4.109 | ARG115 | MET444/TRY441/TRY424/PHE430/PRO329/PHE427 | |
| Testosterone | −5.322 | MET374 | ILE133, PHE134, PHE221, TRP224, ILE305, ALA306, VAL370, LEU372, VAL373, LEU477 |
| Compound | QPlogPo/w a | QPlogS b | QPPCaco c | QPlogBB d | QPPMDCK e | %HOA | RoF g |
|---|---|---|---|---|---|---|---|
| Combine1 | 4.680 | −6.913 | 693.833 | −1.167 | 333.246 | 100 | 0 |
| Combine2 | 4.639 | −7.150 | 463.709 | −1.395 | 215.576 | 100 | 0 |
| Combine3 | 5.797 | −8.228 | 846.562 | −1.109 | 413.198 | 100 | 1 |
| Combine4 | 4.444 | −7.404 | 497.569 | −1.291 | 232.640 | 100 | 0 |
| Combine5 | 4.530 | −6.276 | 681.210 | −1.161 | 326.698 | 100 | 0 |
| Combine6 | 4.715 | −6.897 | 349.469 | −1.527 | 158.792 | 100 | 0 |
| Combine7 | 4.421 | −7.404 | 313.812 | −1.741 | 141.355 | 100 | 0 |
| Combine8 | 4.345 | −6.140 | 602.982 | −1.321 | 286.342 | 100 | 0 |
| Combine9 | 4.566 | −6.298 | 946.340 | −0.952 | 466.079 | 100 | 0 |
| Combine10 | 4.763 | −7.227 | 444.186 | −1.340 | 205.783 | 100 | 0 |
| Combine11 | 4.410 | −7.335 | 402.658 | −1.369 | 185.069 | 100 | 0 |
| Combine12 | 4.397 | −5.987 | 817.738 | −1.009 | 398.012 | 100 | 0 |
| Combine13 | 4.630 | −7.118 | 466.185 | −1.387 | 216.821 | 100 | 0 |
| Combine14 | 4.117 | −5.968 | 437.232 | −1.383 | 202.303 | 100 | 0 |
| Letrozole | 1.459 | −3.693 | 138.864 | −1.570 | 58.559 | 73.838 | 0 |
| Compound | MM-GBSA Free Binding Energy (kcal/mol) |
|---|---|
| Combine1 | −68.9075 ± 5.304 |
| Combine2 | −63.5514 ± 4.219 |
| Combine3 | −66.2378 ± 5.1799 |
| Letrozole | −44.3891 ± 8.014567 |
| Complex | Interact Atom | E(UB3LYP) | Dipole Moment | Zero Energy |
|---|---|---|---|---|
| Complex1 | Nitrogen47 | −1932.195781 | 7.401879 | −1931.767112 |
| Complex2 | Oxygen45 | −1932.163889 | 5.534319 | −1931.736428 |
| Complex3 | Nitrogen30 | −1932.209405 | 14.785182 | −1931.780390 |
| Complex4 | Nitrogen3 | −1932.197730 | 8.697839 | −1931.769055 |
| Complex5 | Nitrogen5 | −1932.191763 | 10.960059 | −1931.763301 |
| Complex6 | Nitrogen1 | −1932.201165 | 6.402050 | −1931.772674 |
| Combine1 | - | −1390.215008 | 6.376844 | −1389.789215 |
| Gold | - | −541.930470 | 0.00 | −541.929251 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edris, A.; Abdelrahman, M.; Osman, W.; Sherif, A.E.; Ashour, A.; Garelnabi, E.A.E.; Ibrahim, S.R.M.; Bafail, R.; Samman, W.A.; Ghazawi, K.F.; et al. Design of Novel Letrozole Analogues Targeting Aromatase for Breast Cancer: Molecular Docking, Molecular Dynamics, and Theoretical Studies on Gold Nanoparticles. Metabolites 2023, 13, 583. https://doi.org/10.3390/metabo13050583
Edris A, Abdelrahman M, Osman W, Sherif AE, Ashour A, Garelnabi EAE, Ibrahim SRM, Bafail R, Samman WA, Ghazawi KF, et al. Design of Novel Letrozole Analogues Targeting Aromatase for Breast Cancer: Molecular Docking, Molecular Dynamics, and Theoretical Studies on Gold Nanoparticles. Metabolites. 2023; 13(5):583. https://doi.org/10.3390/metabo13050583
Chicago/Turabian StyleEdris, Alaa, Mohammed Abdelrahman, Wadah Osman, Asmaa E. Sherif, Ahmed Ashour, Elrashied A. E. Garelnabi, Sabrin R. M. Ibrahim, Rawan Bafail, Waad A. Samman, Kholoud F. Ghazawi, and et al. 2023. "Design of Novel Letrozole Analogues Targeting Aromatase for Breast Cancer: Molecular Docking, Molecular Dynamics, and Theoretical Studies on Gold Nanoparticles" Metabolites 13, no. 5: 583. https://doi.org/10.3390/metabo13050583
APA StyleEdris, A., Abdelrahman, M., Osman, W., Sherif, A. E., Ashour, A., Garelnabi, E. A. E., Ibrahim, S. R. M., Bafail, R., Samman, W. A., Ghazawi, K. F., Mohamed, G. A., & Alzain, A. A. (2023). Design of Novel Letrozole Analogues Targeting Aromatase for Breast Cancer: Molecular Docking, Molecular Dynamics, and Theoretical Studies on Gold Nanoparticles. Metabolites, 13(5), 583. https://doi.org/10.3390/metabo13050583