
Targeting Oncometabolites in Peritoneal Cancers: Preclinical Insights and Therapeutic Strategies

 , , ,
, , ,  ,
,  and
and
Abstract
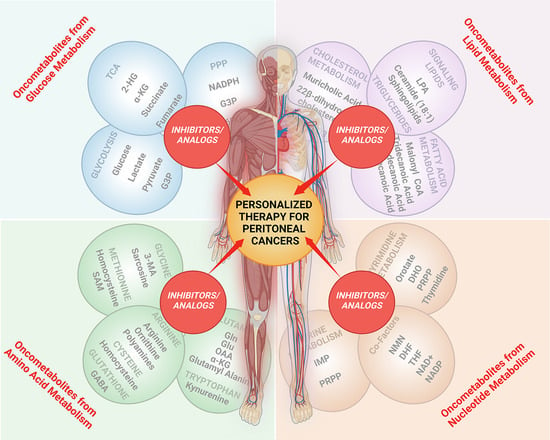
:
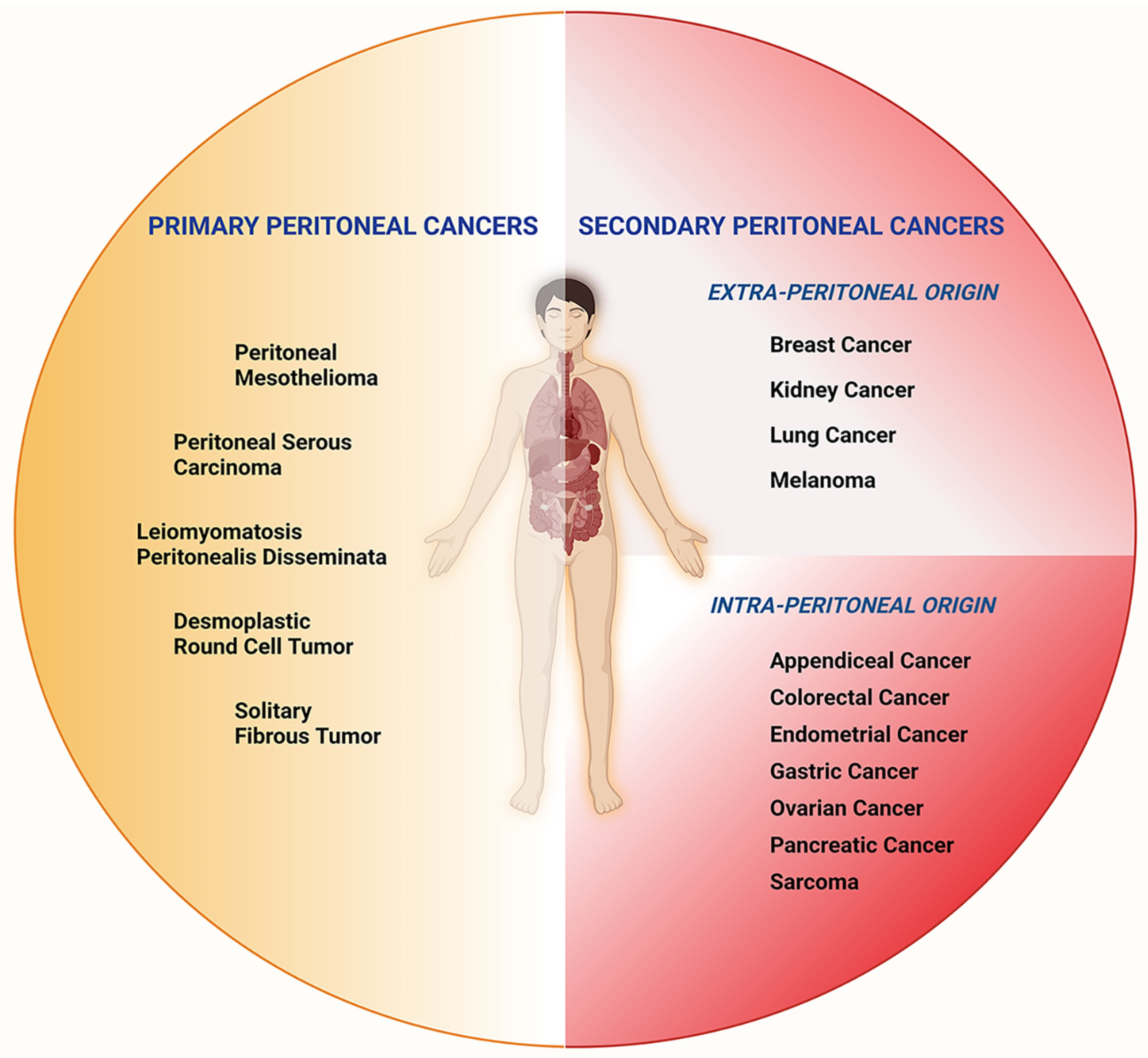
1. Introduction
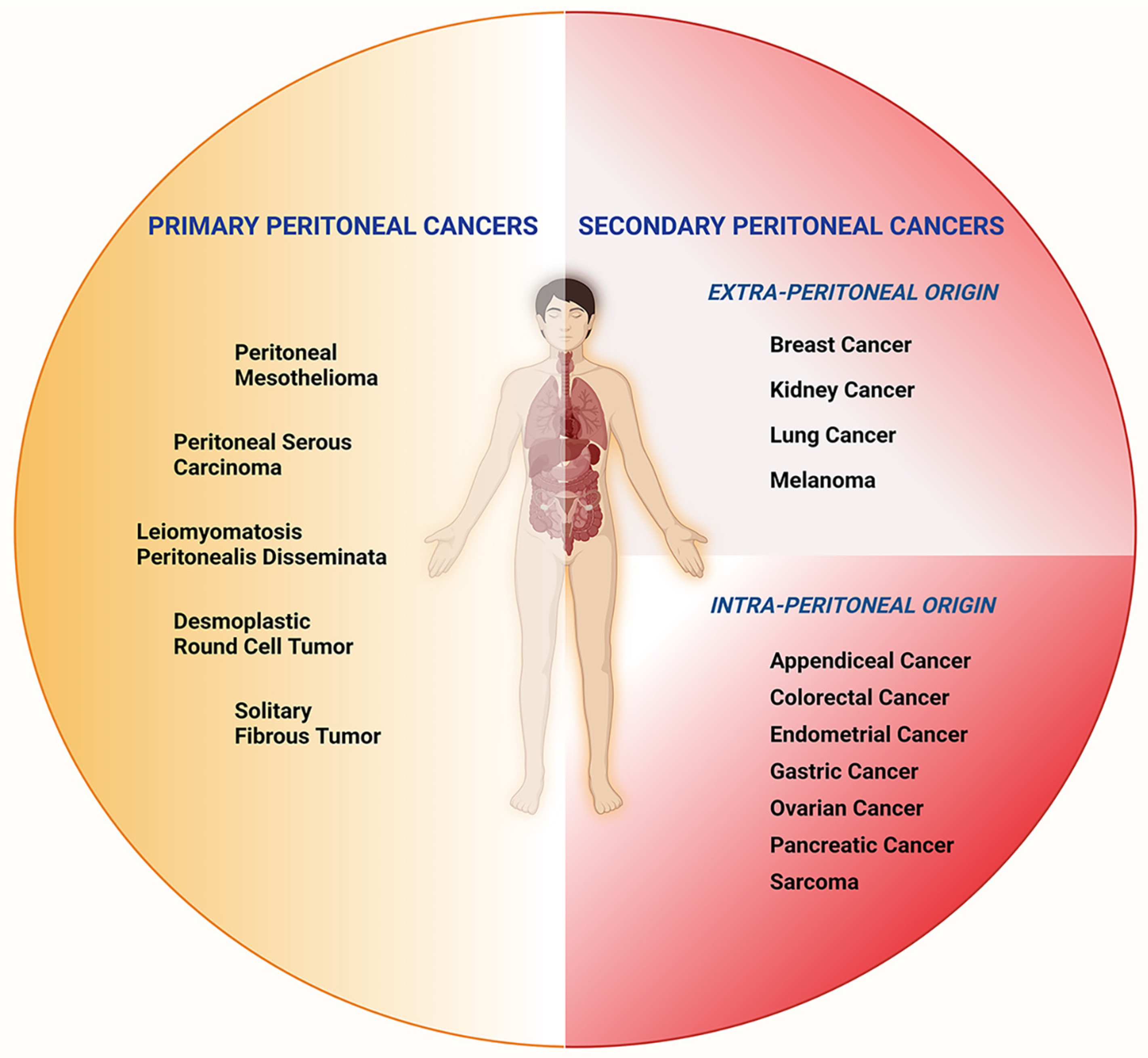
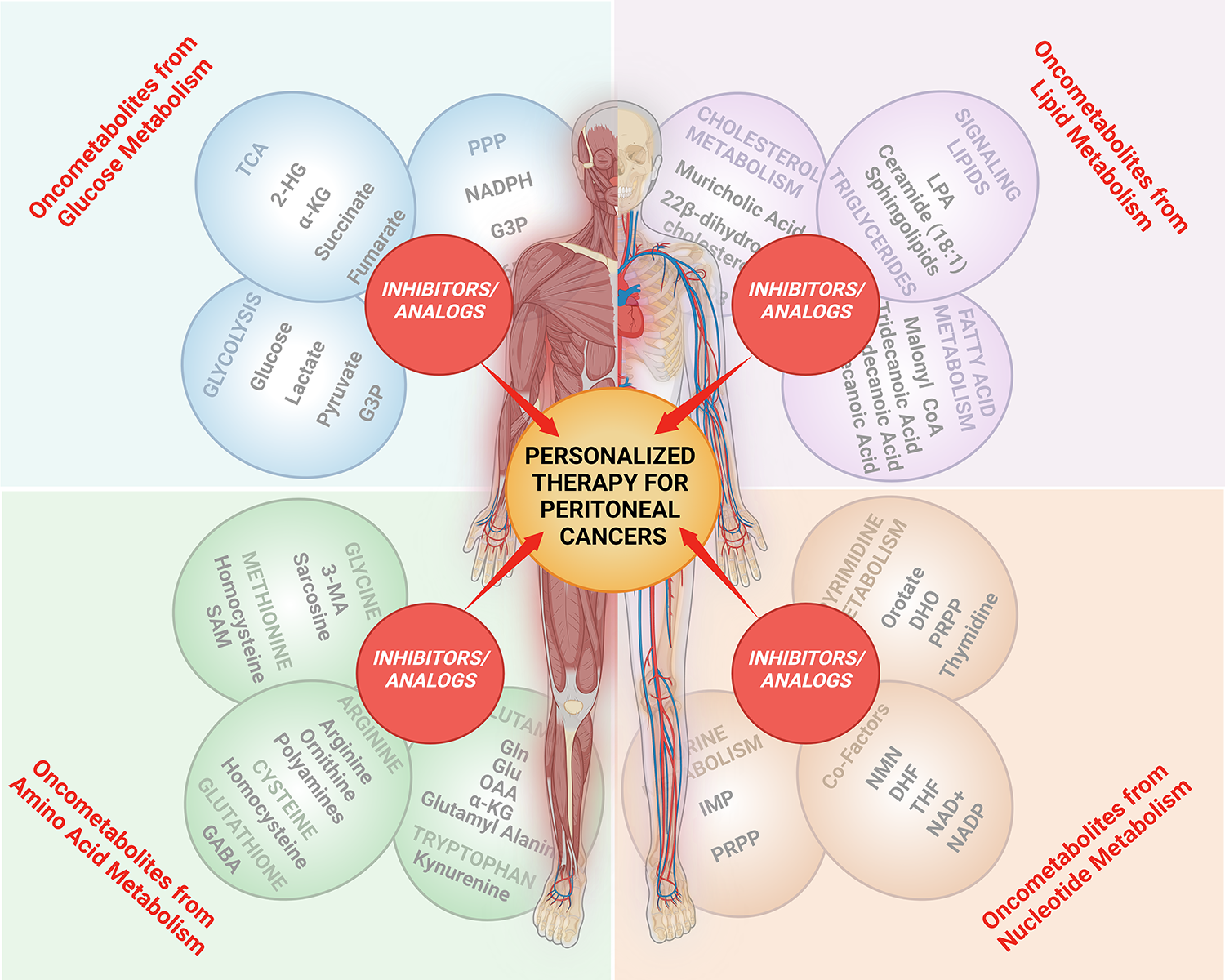
2. Dysregulated Metabolism, Oncometabolites, and Peritoneal Cancer Pathogenesis
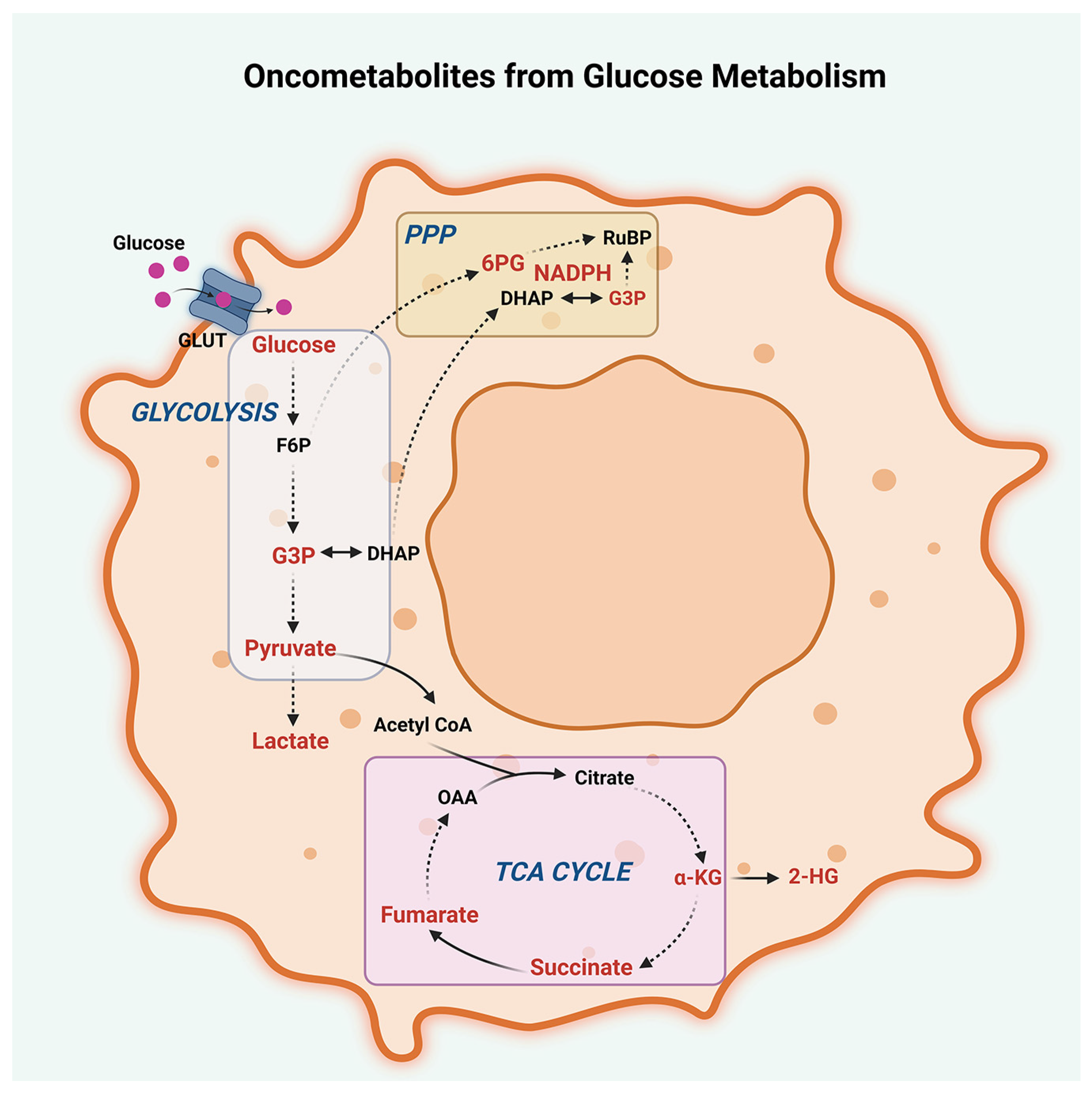
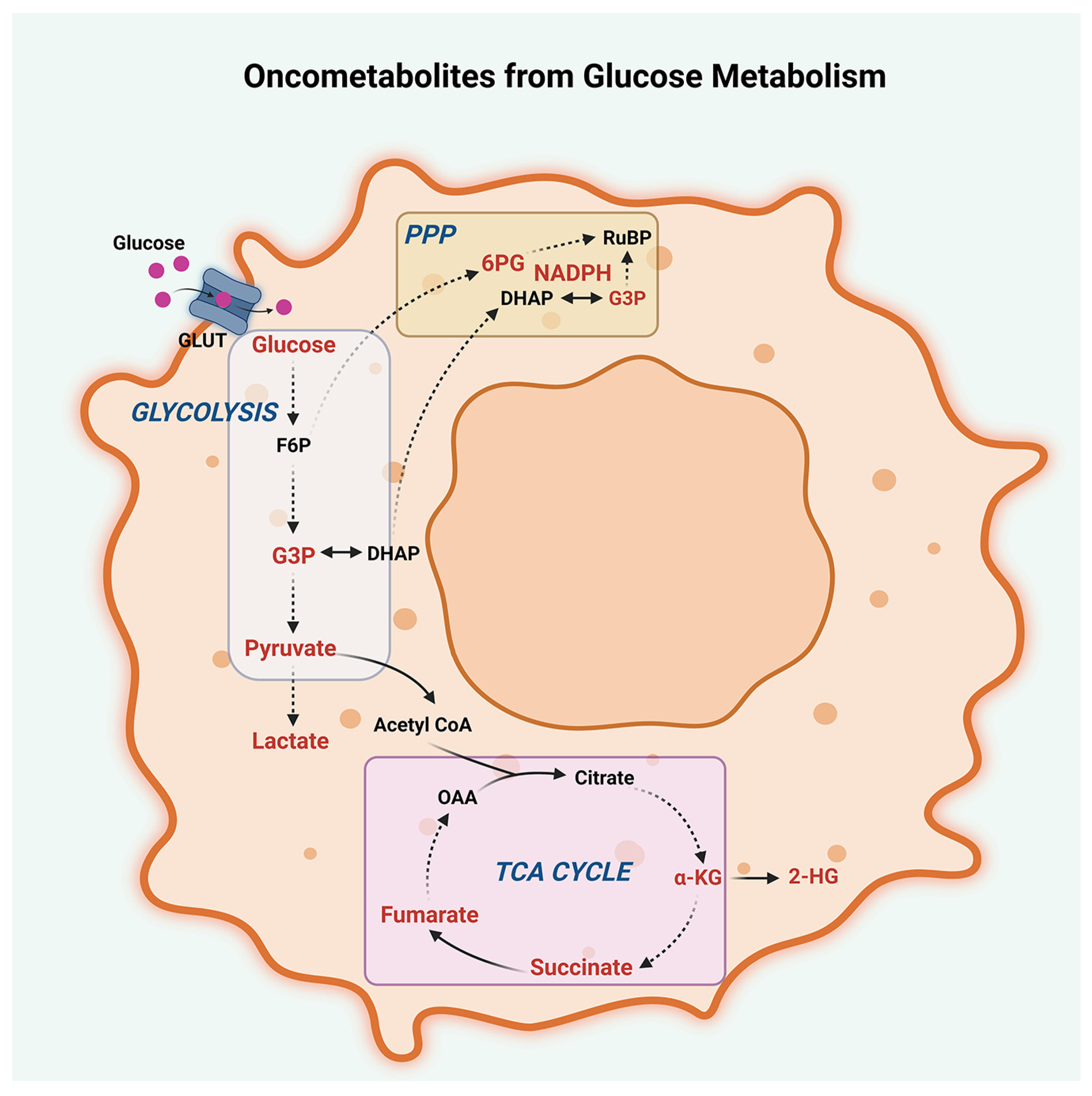
2.1. Glucose Metabolism
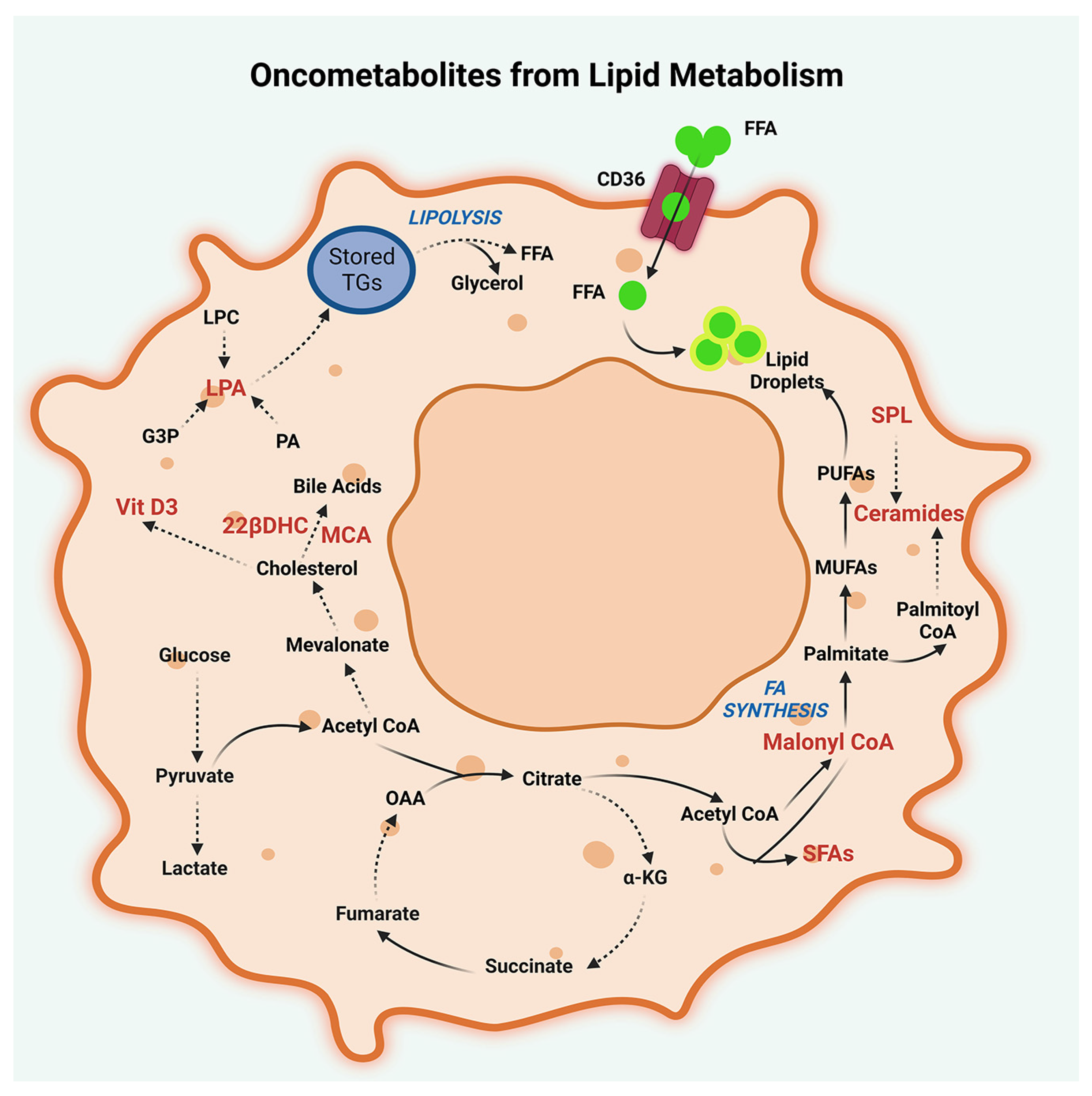
2.2. Lipid Metabolism
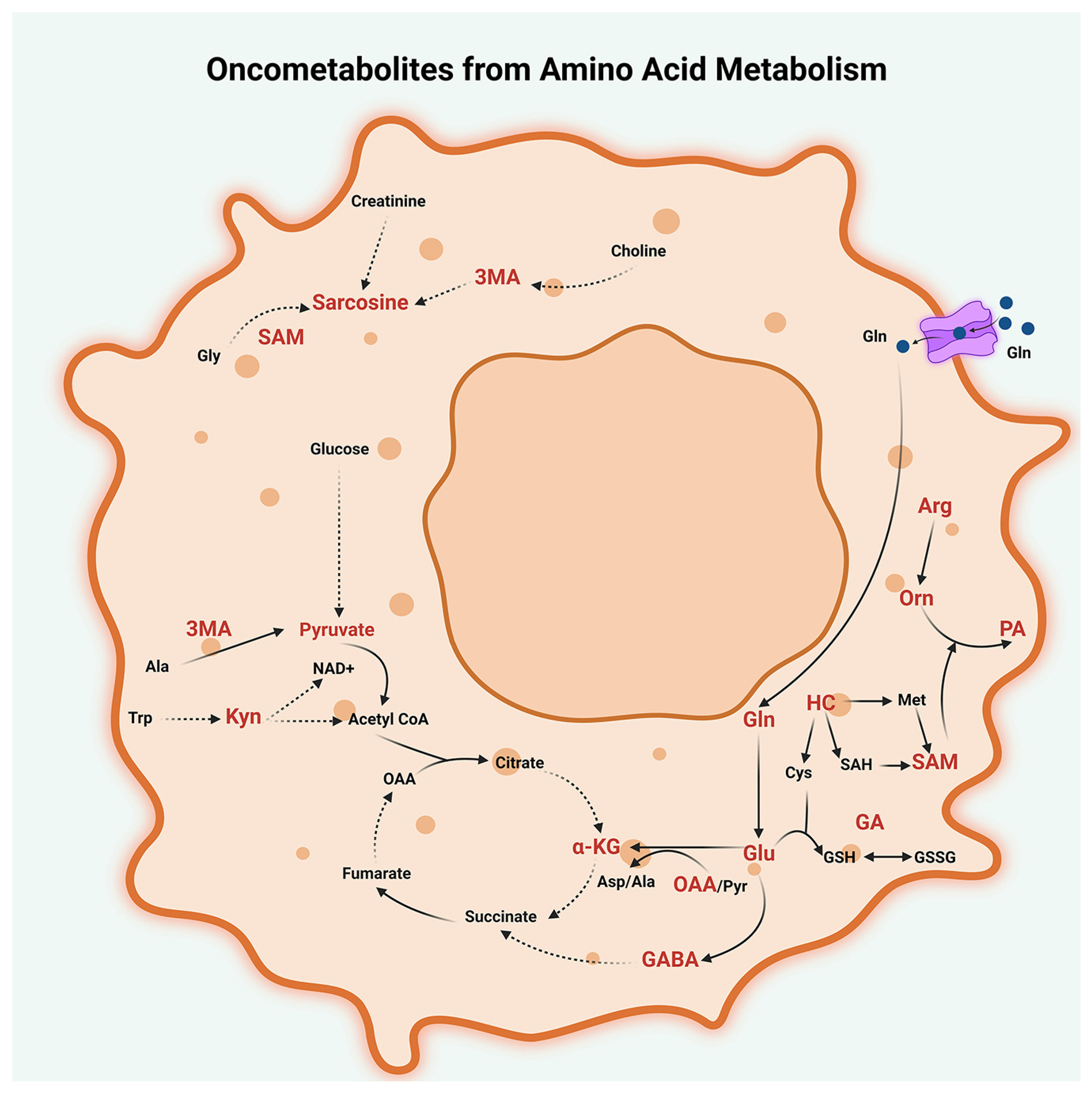
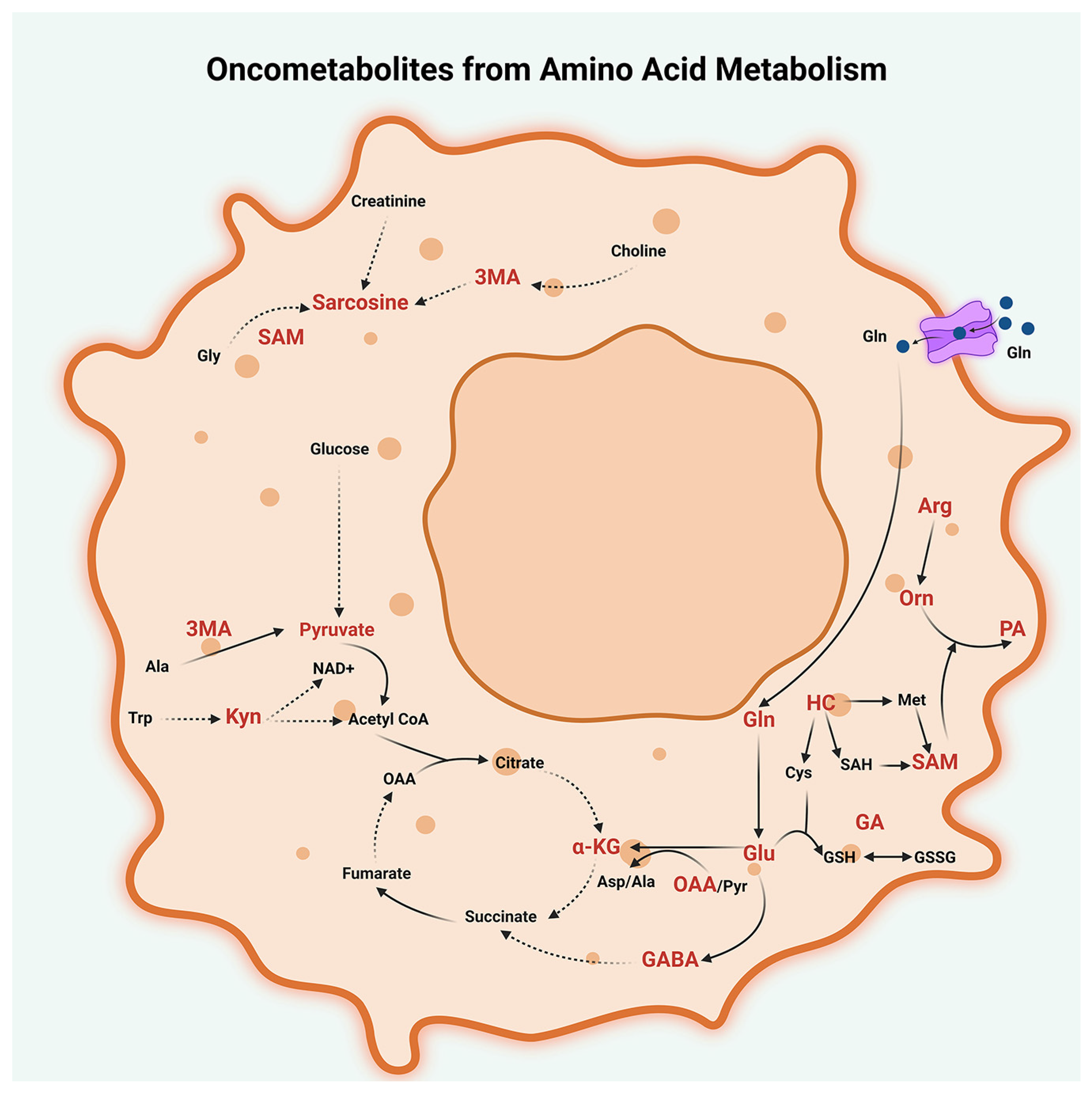
2.3. Amino Acid Metabolism
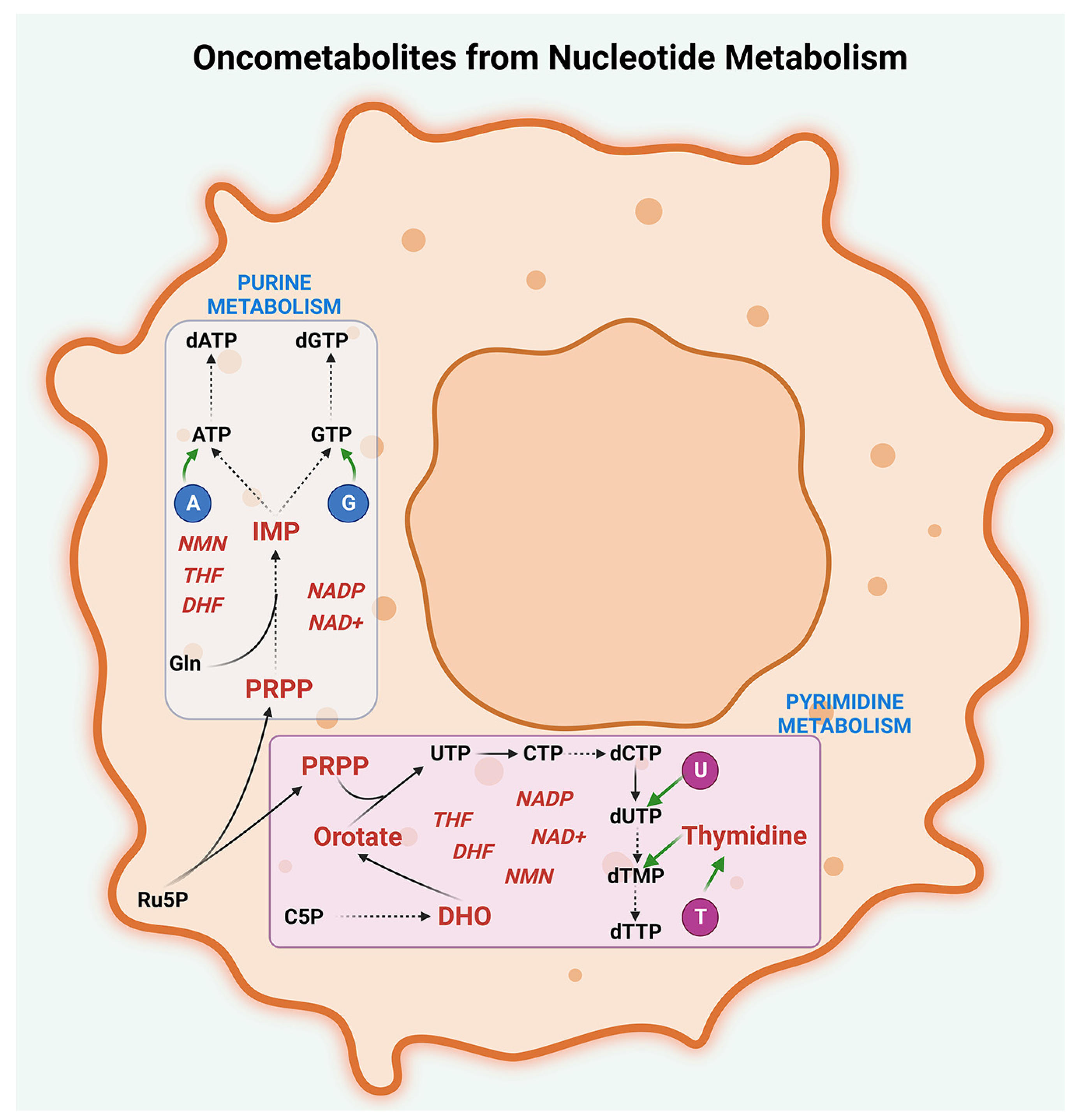
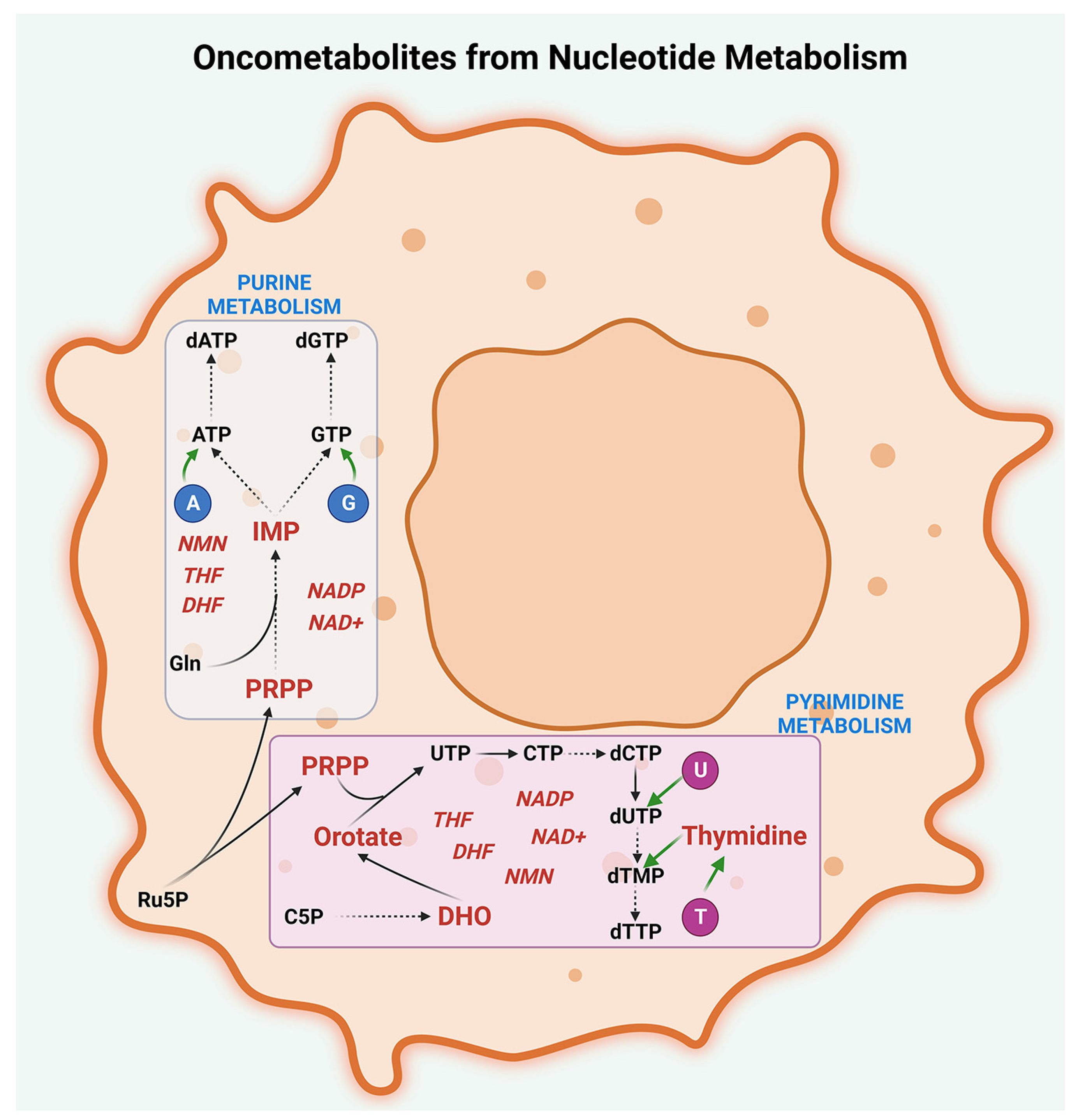
2.4. Nucleotide Metabolism
3. Oncometabolites and Precision Cancer Medicine
3.1. Targeting Glucose Metabolism
3.2. Targeting Lipid Metabolism
3.3. Targeting Amino Acid Metabolism
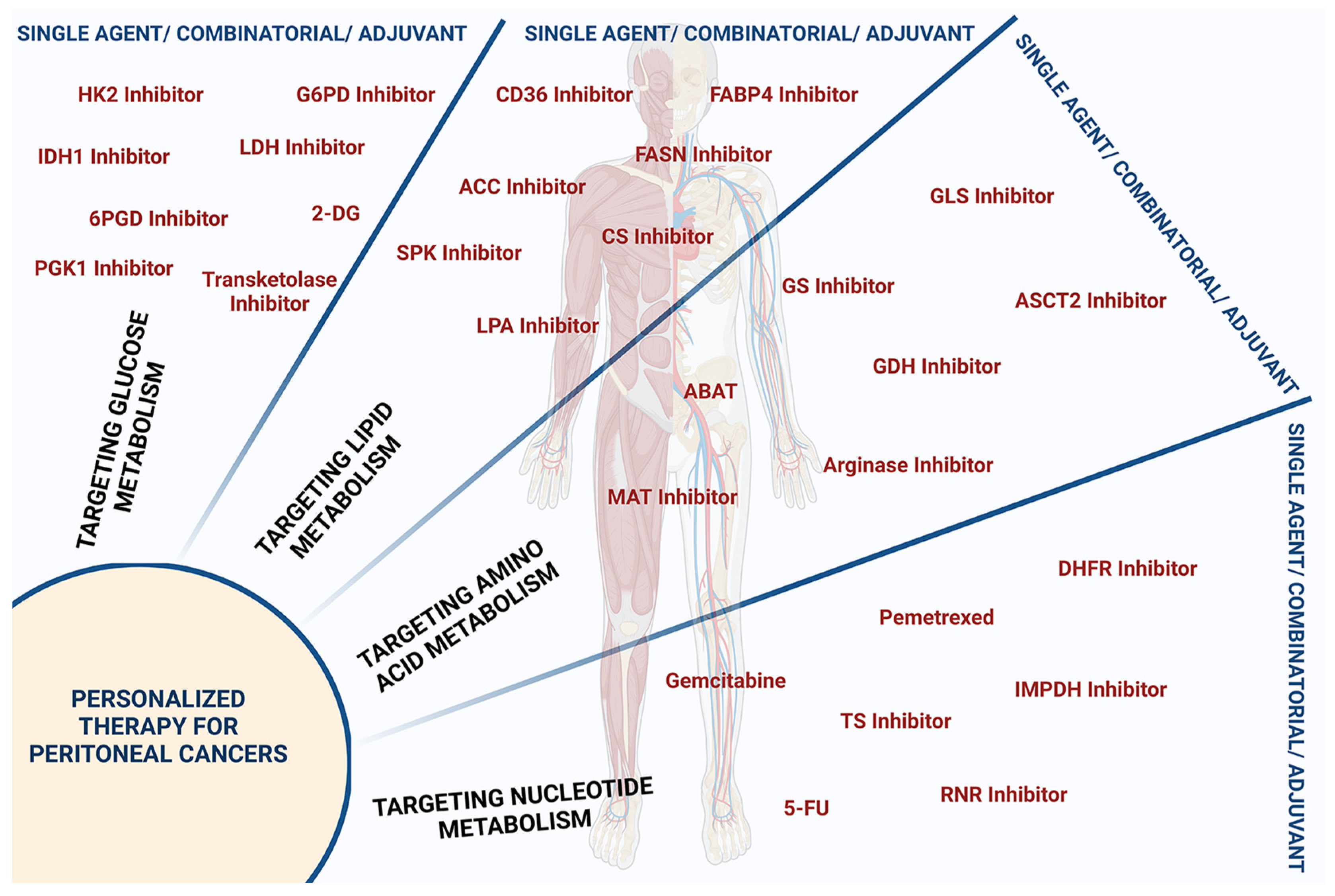
3.4. Targeting Nucleotide Metabolism
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cortés-Guiral, D.; Hübner, M.; Alyami, M.; Bhatt, A.; Ceelen, W.; Glehen, O.; Lordick, F.; Ramsay, R.; Sgarbura, O.; Van Der Speeten, K.; et al. Primary and metastatic peritoneal surface malignancies. Nat. Rev. Dis. Prim. 2021, 7, 91. [Google Scholar] [CrossRef]
- Patel, C.M.; Sahdev, A.; Reznek, R.H. CT, MRI and PET imaging in peritoneal malignancy. Cancer Imaging 2011, 11, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.; Russo, L.; Ulugoel, S.; dos Santos, R.F.; Breuer, E.; Gupta, A.; Lehmann, K. Peritoneal Metastasis: Current Status and Treatment Options. Cancers 2021, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Xie, X.; Min, T.; Sun, T.; Wang, H.; Zhang, Y.; Dang, C.; Zhang, H. Development of the Peritoneal Metastasis: A Review of Back-Grounds, Mechanisms, Treatments and Prospects. J. Clin. Med. 2022, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Laplane, L.; Duluc, D.; Bikfalvi, A.; Larmonier, N.; Pradeu, T. Beyond the tumour microenvironment. Int. J. Cancer 2019, 145, 2611–2618. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- van Baal, J.O.; Van de Vijver, K.K.; Nieuwland, R.; van Noorden, C.J.; van Driel, W.J.; Sturk, A.; Kenter, G.G.; Rikkert, L.G.; Lok, C.A. The histophysiology and pathophysiology of the peritoneum. Tissue Cell 2017, 49, 95–105. [Google Scholar] [CrossRef]
- Mei, S.; Chen, X.; Wang, K.; Chen, Y. Tumor microenvironment in ovarian cancer peritoneal metastasis. Cancer Cell Int. 2023, 23, 11. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, C. Oncometabolites in Cancer: Current Understanding and Challenges. Cancer Res. 2021, 81, 2820–2823. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Idle, J.R. Metabolic Rewiring and the Characterization of Oncometabolites. Cancers 2021, 13, 2900. [Google Scholar] [CrossRef]
- Corrado, M.; Scorrano, L.; Campello, S. Changing perspective on oncometabolites: From metabolic signature of cancer to tumorigenic and immunosuppressive agents. Oncotarget 2016, 7, 46692–46706. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F.; Aghamir, S.M.K.; Tavangar, S.M. Oncometabolites: A new insight for oncology. Mol. Genet. Genom. Med. 2019, 7, e873. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Ha, J.H.; Jayaraman, M.; Nadhan, R.; Kashyap, S.; Mukherjee, P.; Isidoro, C.; Song, Y.S.; Dhanasekaran, D.N. Unraveling Autocrine Signaling Pathways through Metabolic Fingerprinting in Serous Ovarian Cancer Cells. Biomedicines 2021, 9, 1927. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Gori, S.S.; Tsukamoto, T.; Rais, R.; Slusher, B.S. Clinical development of metabolic inhibitors for oncology. J. Clin. Investig. 2022, 132, e148550. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Sun, L.; Suo, C.; Li, S.T.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef]
- Anwar, A.; Kasi, A. Peritoneal Cancer. In StatPearls; Treasure Island: Pinellas, FL, USA, 2023. [Google Scholar]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Sainero-Alcolado, L.; Liano-Pons, J.; Ruiz-Perez, M.V.; Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef]
- Godel, M.; Ortone, G.; Anobile, D.P.; Pasino, M.; Randazzo, G.; Riganti, C.; Kopecka, J. Targeting Mitochondrial Oncometabolites: A New Approach to Overcome Drug Resistance in Cancer. Pharmaceutics 2021, 13, 762. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef]
- Alam, N.A.; Olpin, S.; Leigh, I.M. Fumarate hydratase mutations and predisposition to cutaneous leiomyomas, uterine leiomyomas and renal cancer. Br. J. Dermatol. 2005, 153, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Sajnani, K.; Islam, F.; Smith, R.A.; Gopalan, V.; Lam, A.K.-Y. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie 2017, 135, 164–172. [Google Scholar] [CrossRef]
- Nazar, E.; Khatami, F.; Saffar, H.; Tavangar, S.M. The Emerging Role of Succinate Dehyrogenase Genes (SDHx) in Tumorigenesis. Int. J. Hematol. Oncol. Stem. Cell Res. 2019, 13, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Eniafe, J.; Jiang, S. The functional roles of TCA cycle metabolites in cancer. Oncogene 2021, 40, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218, Erratum in Trends Biochem. Sci. 2016, 41, 211. [Google Scholar] [CrossRef]
- Hardie, D.G. 100 years of the Warburg effect: A historical perspective. Endocr. Relat. Cancer 2022, 29, T1–T13. [Google Scholar] [CrossRef]
- Lagana, S.M.; Taub, R.N.; Borczuk, A.C. Utility of glucose transporter 1 in the distinction of benign and malignant thoracic and abdominal mesothelial lesions. Arch. Pathol. Lab. Med. 2012, 136, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Hommell-Fontaine, J.; Isaac, S.; Passot, G.; Decullier, E.; Traverse-Glehen, A.; Cotte, E.; You, B.; Mohamed, F.; Gilly, F.N.; Glehen, O.; et al. Malignant peritoneal mesothelioma treated by cytoreductive surgery and hyperthermic intraperitoneal chemotherapy: Is GLUT1 expression a major prognostic factor? A preliminary study. Ann. Surg. Oncol. 2013, 20, 3892–3898. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, G.; Yang, D.; Guo, X.; Tian, L.; Song, H.; Liang, Y. Osteopontin, GLUT1 and Ki-67 expression in malignant peritoneal mesothelioma: Prognostic implications. Intern. Med. J. 2021, 51, 896–904. [Google Scholar] [CrossRef]
- Kucukgoz Gulec, U.; Paydas, S.; Guzel, A.B.; Buyukkurt, S.; Seydaoglu, G.; Vardar, M.A. Comparative analysis of CA 125, ferritin, beta-2 microglobulin, lactic dehydrogenase levels in serum and peritoneal fluid in patients with ovarian neoplasia. Med. Oncol. 2012, 29, 2937–2943. [Google Scholar] [CrossRef] [PubMed]
- Su, S.S.; Zheng, G.Q.; Yin, W.J.; Liang, Y.F.; Liu, Y.Y.; Song, H.; Sun, N.N.; Yang, Y.X. Prognostic Significance of Blood, Serum, and Ascites Parameters in Patients with Malignant Peritoneal Mesothelioma or Peritoneal Carcinomatosis. Gastroenterol. Res. Pract. 2018, 2018, 2619526. [Google Scholar] [CrossRef]
- Archid, R.; Solass, W.; Tempfer, C.; Konigsrainer, A.; Adolph, M.; Reymond, M.A.; Wilson, R.B. Cachexia Anorexia Syndrome and Associated Metabolic Dysfunction in Peritoneal Metastasis. Int. J. Mol. Sci. 2019, 20, 5444. [Google Scholar] [CrossRef]
- Deng, G.C.; Yan, H.; Guo, Z.P.; Dai, G. Correlation Between Baseline Serum Tumor Markers and Clinical Characteristic Factors in Patients with Advanced Pancreatic Cancer. Onco. Targets Ther. 2020, 13, 11151–11163. [Google Scholar] [CrossRef]
- Hu, J.; Yang, S.; Wang, J.; Zhang, Q.; Zhao, L.; Zhang, D.; Yu, D.; Jin, M.; Ma, H.; Liu, H.; et al. Blood alkaline phosphatase predicts prognosis of patients with advanced HER2-negative gastric cancer receiving immunotherapy. Ann. Transl. Med. 2021, 9, 1316. [Google Scholar] [CrossRef] [PubMed]
- Hervas, M.S.; Jativa-Porcar, R.; Robles-Hernandez, D.; Rubert, A.S.; Segarra, B.; Oliva, C.; Escrig, J.; Llueca, J.A. Evaluation of the relationship between lactacidemia and postoperative complications after surgery for peritoneal carcinomatosis. Korean J. Anesthesiol. 2021, 74, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ma, Y.; Suo, J.; Li, W.; Zhang, Y.; Qin, S.; Jiao, Y.; Zhang, S.; Li, S.; Kong, Y.; et al. Discovering Biomarkers in Peritoneal Metastasis of Gastric Cancer by Metabolomics. OncoTargets Ther. 2020, 13, 7199–7211. [Google Scholar] [CrossRef]
- Shender, V.O.; Pavlyukov, M.S.; Ziganshin, R.H.; Arapidi, G.P.; Kovalchuk, S.I.; Anikanov, N.A.; Altukhov, I.A.; Alexeev, D.G.; Butenko, I.O.; Shavarda, A.L.; et al. Proteome-metabolome profiling of ovarian cancer ascites reveals novel components involved in intercellular communication. Mol. Cell Proteom. 2014, 13, 3558–3571. [Google Scholar] [CrossRef]
- Yi, H.; Zheng, X.; Song, J.; Shen, R.; Su, Y.; Lin, D. Exosomes mediated pentose phosphate pathway in ovarian cancer metastasis: A proteomics analysis. Int. J. Clin. Exp. Pathol. 2015, 8, 15719–15728. [Google Scholar]
- Bose, S.; Huang, Q.; Ma, Y.; Wang, L.; Rivera, G.O.; Ouyang, Y.; Whitaker, R.; Gibson, R.A.; Kontos, C.D.; Berchuck, A.; et al. G6PD inhibition sensitizes ovarian cancer cells to oxidative stress in the metastatic omental microenvironment. Cell Rep. 2022, 39, 111012. [Google Scholar] [CrossRef]
- Zeng, X.; Guo, H.; Liu, Z.; Qin, Z.; Cong, Y.; Ren, N.; Zhang, Y.; Zhang, N. S100A11 activates the pentose phosphate pathway to induce malignant biological behaviour of pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 568. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, X.; Yong, H.; Xu, J.; Qu, P.; Qiao, S.; Hou, P.; Li, Z.; Chu, S.; Zheng, J.; et al. Transketolase promotes colorectal cancer metastasis through regulating AKT phosphorylation. Cell Death Dis. 2022, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Creaney, J.; Dick, I.M.; Leon, J.S.; Robinson, B.W. A Proteomic Analysis of the Malignant Mesothelioma Secretome Using iTRAQ. Cancer Genom. Proteom. 2017, 14, 103–117. [Google Scholar] [CrossRef]
- Tsoi, T.T.; Chiu, K.W.H.; Chu, M.Y.; Ngan, H.Y.S.; Lee, E.Y.P. Metabolic active peritoneal sites affect tumor debulking in ovarian and peritoneal cancers. J. Ovarian Res. 2020, 13, 61. [Google Scholar] [CrossRef]
- Kuribayashi, K.; Kitajima, K.; Minami, T.; Ikeda, M.; Yamakado, K.; Kijima, T. Malignant Peritoneal Mesothelioma Features Shown by FDG-PET/CT. Cancer Diagn. Progn. 2022, 2, 654–660. [Google Scholar] [CrossRef]
- Liu, M.; Liu, Y.; Feng, H.; Jing, Y.; Zhao, S.; Yang, S.; Zhang, N.; Jin, S.; Li, Y.; Weng, M.; et al. Clinical Significance of Screening Differential Metabolites in Ovarian Cancer Tissue and Ascites by LC/MS. Front. Pharmacol. 2021, 12, 701487. [Google Scholar] [CrossRef]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef]
- Fong, M.Y.; McDunn, J.; Kakar, S.S. Identification of metabolites in the normal ovary and their transformation in primary and metastatic ovarian cancer. PLoS ONE 2011, 6, e19963. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Cai, G.; Zhou, B.; Li, D.; Zhao, A.; Xie, G.; Li, H.; Cai, S.; Xie, D.; Huang, C.; et al. A distinct metabolic signature of human colorectal cancer with prognostic potential. Clin. Cancer Res. 2014, 20, 2136–2146. [Google Scholar] [CrossRef]
- Ozkan, Y.; Yardim-Akaydin, S.; Firat, H.; Caliskan-Can, E.; Ardic, S.; Simsek, B. Usefulness of homocysteine as a cancer marker: Total thiol compounds and folate levels in untreated lung cancer patients. Anticancer Res 2007, 27, 1185–1189. [Google Scholar]
- Lin, J.; Lee, I.M.; Song, Y.; Cook, N.R.; Selhub, J.; Manson, J.E.; Buring, J.E.; Zhang, S.M. Plasma homocysteine and cysteine and risk of breast cancer in women. Cancer Res. 2010, 70, 2397–2405. [Google Scholar] [CrossRef]
- Xu, W.; Cheng, Y.; Zhu, H. Evaluation of an Association of Blood Homocysteine Levels with Gastric Cancer Risk from 27 Case-Control Studies. Medicine 2016, 95, e3700. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of alpha-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; Du, A.; Li, Y.; Shi, Q.; Chen, Y.; Zhao, Y.; Wang, B.; Pan, F. Adipocytic Glutamine Synthetase Upregulation via Altered Histone Methylation Promotes 5FU Chemoresistance in Peritoneal Carcinomatosis of Colorectal Cancer. Front. Oncol. 2021, 11, 748730. [Google Scholar] [CrossRef]
- Perez-Castro, L.; Garcia, R.; Venkateswaran, N.; Barnes, S.; Conacci-Sorrell, M. Tryptophan and its metabolites in normal physiology and cancer etiology. FEBS J. 2023, 290, 7–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tao, F.; Jiang, J.; Chen, L.; Du, J.; Cheng, X.; He, Q.; Zhong, S.; Chen, W.; Wu, X.; et al. Tryptophan 2, 3-dioxygenase promotes proliferation, migration and invasion of ovarian cancer cells. Mol. Med. Rep. 2021, 23, 12084. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toth, F.; Polyak, H.; Szabo, A.; Mandi, Y.; Vecsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, N.; Conacci-Sorrell, M. Kynurenine: An oncometabolite in colon cancer. Cell Stress 2020, 4, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, H.; Saga, Y.; Fujiwara, H.; Akimoto, H.; Yamada, A.; Kagawa, S.; Takei, Y.; Machida, S.; Takikawa, O.; Suzuki, M. Indoleamine 2,3-dioxygenase promotes peritoneal dissemination of ovarian cancer through inhibition of natural killercell function and angiogenesis promotion. Int. J. Oncol. 2011, 38, 113–120. [Google Scholar]
- Wu, D.; Zhu, Y. Role of kynurenine in promoting the generation of exhausted CD8(+) T cells in colorectal cancer. Am. J. Transl. Res. 2021, 13, 1535–1547. [Google Scholar]
- Gouasmi, R.; Ferraro-Peyret, C.; Nancey, S.; Coste, I.; Renno, T.; Chaveroux, C.; Aznar, N.; Ansieau, S. The Kynurenine Pathway and Cancer: Why Keep It Simple When You Can Make It Complicated. Cancers 2022, 14, 2793. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hong, S.; Yang, J.; Zhang, X.; Wang, Y.; Wang, H.; Peng, J.; Hong, L. Targeting purine metabolism in ovarian cancer. J. Ovarian Res. 2022, 15, 93. [Google Scholar] [CrossRef]
- De Vitto, H.; Arachchige, D.B.; Richardson, B.C.; French, J.B. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 2021, 10, 2603. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Zhou, Y.; Liu, C.; Sang, N.; Liu, H.; et al. DHODH and cancer: Promising prospects to be explored. Cancer Metab. 2021, 9, 22. [Google Scholar] [CrossRef]
- Ferrari, S.; Severi, L.; Pozzi, C.; Quotadamo, A.; Ponterini, G.; Losi, L.; Marverti, G.; Costi, M.P. Human Thymidylate Synthase Inhibitors Halting Ovarian Cancer Growth. Vitam. Horm. 2018, 107, 473–513. [Google Scholar] [CrossRef] [PubMed]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD(+) metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Kato, T.; Nakazato, H.; Ito, K.; Mizuno, I.; Kanemitsu, T.; Matsumoto, K.; Yamaguchi, A.; Nakai, K.; Inada, K.; et al. Retrospective study on thymidylate synthase as a predictor of outcome and sensitivity to adjuvant chemotherapy in patients with curatively resected colorectal cancer. Anticancer Drugs 2002, 13, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tang, S. Metabolic reprogramming and cancer precision medicine: A narrative review. Precis. Cancer Med. 2021, 4, 35. [Google Scholar] [CrossRef]
- Yonemura, Y.; Endo, Y.; Obata, T.; Sasaki, T. Recent advances in the treatment of peritoneal dissemination of gastrointestinal cancers by nucleoside antimetabolites. Cancer Sci. 2007, 98, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.R.; Yung, M.M.H.; Xuan, Y.; Zhan, S.; Leung, L.L.; Liang, R.R.; Leung, T.H.Y.; Yang, H.; Xu, D.; Sharma, R.; et al. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun. Biol. 2019, 2, 281. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Yung, M.M.H.; Siu, M.K.Y.; Jiao, P.; Ngan, H.Y.S.; Chan, D.W.; Chan, K.K.L. New Insights into Ferroptosis Initiating Therapies (FIT) by Targeting the Rewired Lipid Metabolism in Ovarian Cancer Peritoneal Metastases. Int. J. Mol. Sci. 2022, 23, 15263. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, Y.; Zhang, H.; Sun, H. Targeting cancer cell metabolism: The combination of metformin and 2-Deoxyglucose regulates apoptosis in ovarian cancer cells via p38 MAPK/JNK signaling pathway. Am. J. Transl. Res. 2016, 8, 4812–4821. [Google Scholar] [PubMed]
- Icard, P.; Zhang, X.D.; Lemoisson, E.; Louis, M.H.; Allouche, S.; Lincet, H.; Poulain, L. Experimental results using 3-bromopyruvate in mesothelioma: In vitro and in vivo studies. J. Bioenerg. Biomembr. 2012, 44, 81–90. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Archid, R.; Zieker, D.; Weinreich, F.J.; Hönes, F.; Königsrainer, A.; Quintanilla-Martínez, L.; Reymond, M.A.; Solass, W. shRNA-mediated inhibition of PhosphoGlycerate Kinase 1 (PGK1) enhances cytotoxicity of intraperitoneal chemotherapy in peritoneal metastasis of gastric origin. Eur. J. Surg. Oncol. 2020, 46, 613–619. [Google Scholar] [CrossRef]
- Dahl, E.S.; Buj, R.; Leon, K.E.; Newell, J.M.; Imamura, Y.; Bitler, B.G.; Snyder, N.W.; Aird, K.M. Targeting IDH1 as a Prosenescent Therapy in High-grade Serous Ovarian Cancer. Mol. Cancer Res. 2019, 17, 1710–1720. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, F.; Zhang, Y.; Lin, Z.; Yang, J.; Han, X.; Feng, Y.; Pei, X.; Li, F.; Liu, Q.; et al. Targeting glucose-6-phosphate dehydrogenase by 6-AN induces ROS-mediated autophagic cell death in breast cancer. FEBS J. 2023, 290, 763–779. [Google Scholar] [CrossRef]
- Zheng, W.; Feng, Q.; Liu, J.; Guo, Y.; Gao, L.; Li, R.; Xu, M.; Yan, G.; Yin, Z.; Zhang, S.; et al. Inhibition of 6-phosphogluconate Dehydrogenase Reverses Cisplatin Resistance in Ovarian and Lung Cancer. Front. Pharmacol. 2017, 8, 421. [Google Scholar] [CrossRef]
- Lemberger, L.; Wagner, R.; Heller, G.; Pils, D.; Grunt, T.W. Pharmacological Inhibition of Lipid Import and Transport Proteins in Ovarian Cancer. Cancers 2022, 14, 6004. [Google Scholar] [CrossRef]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef]
- Huang, P.L.; Zhu, S.N.; Lu, S.L.; Dai, Z.S.; Jin, Y.L. Inhibitor of fatty acid synthase induced apoptosis in human colonic cancer cells. World J. Gastroenterol. 2000, 6, 295–297. [Google Scholar] [PubMed]
- Janneh, A.H.; Ogretmen, B. Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment. Cancers 2022, 14, 2183. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Taiyab, A.; Hussain, A.; Alajmi, M.F.; Islam, A.; Hassan, M.I. Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy. Cancers 2021, 13, 1898. [Google Scholar] [CrossRef]
- Wang, W.; Pan, H.; Ren, F.; Chen, H.; Ren, P. Targeting ASCT2-mediated glutamine metabolism inhibits proliferation and promotes apoptosis of pancreatic cancer cells. Biosci. Rep. 2022, 42, BSR20212171. [Google Scholar] [CrossRef]
- Wang, J.J.; Siu, M.K.; Jiang, Y.X.; Leung, T.H.; Chan, D.W.; Wang, H.G.; Ngan, H.Y.; Chan, K.K. A Combination of Glutaminase Inhibitor 968 and PD-L1 Blockade Boosts the Immune Response against Ovarian Cancer. Biomolecules 2021, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. Glutaminase GLS1 Inhibitors as Potential Cancer Treatment. ACS Med. Chem. Lett. 2016, 7, 207–208. [Google Scholar] [CrossRef]
- Wanders, D.; Hobson, K.; Ji, X. Methionine Restriction and Cancer Biology. Nutrients 2020, 12, 684. [Google Scholar] [CrossRef]
- Niu, F.; Yu, Y.; Li, Z.; Ren, Y.; Li, Z.; Ye, Q.; Liu, P.; Ji, C.; Qian, L.; Xiong, Y. Arginase: An emerging and promising therapeutic target for cancer treatment. Biomed. Pharm. 2022, 149, 112840. [Google Scholar] [CrossRef]
- Paz, E.A.; LaFleur, B.; Gerner, E.W. Polyamines are oncometabolites that regulate the LIN28/let-7 pathway in colorectal cancer cells. Mol. Carcinog. 2014, 53 (Suppl. S1), E96–E106. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.P.; Rajendiran, T.M.; Ateeq, B.; Asangani, I.A.; Athanikar, J.N.; Yocum, A.K.; Mehra, R.; Siddiqui, J.; Palapattu, G.; Wei, J.T.; et al. The role of sarcosine metabolism in prostate cancer progression. Neoplasia 2013, 15, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, G.; Li, N.; Luo, Z.; Wang, X.; Gu, J. Role of 4-aminobutyrate aminotransferase (ABAT) and the lncRNA co-expression network in the development of myelodysplastic syndrome. Oncol. Rep. 2019, 42, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Ciszewski, W.M.; Chmielewska-Kassassir, M.; Wozniak, L.A.; Sobierajska, K. Thymidylate Synthase Overexpression Drives the Invasive Phenotype in Colon Cancer Cells. Biomedicines 2022, 10, 1267. [Google Scholar] [CrossRef]
- Ikushima, H.; Sakatani, T.; Ohara, S.; Takeshima, H.; Horiuchi, H.; Morikawa, T.; Usui, K. Cisplatin plus pemetrexed therapy and subsequent immune checkpoint inhibitor administration for malignant peritoneal mesothelioma without pleural lesions: Case report. Medicine 2020, 99, e19956. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.; Parisi, L.; Li, L.; Knehans, A.; Phaeton, R.; Kesterson, J.P. The role of pemetrexed in recurrent epithelial ovarian cancer: A scoping review. Oncol. Rev. 2018, 12, 346. [Google Scholar] [CrossRef]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef]
- Naffouje, R.; Grover, P.; Yu, H.; Sendilnathan, A.; Wolfe, K.; Majd, N.; Smith, E.P.; Takeuchi, K.; Senda, T.; Kofuji, S.; et al. Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story. Cancers 2019, 11, 1346. [Google Scholar] [CrossRef]
- Huff, S.E.; Winter, J.M.; Dealwis, C.G. Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present. Biomolecules 2022, 12, 815. [Google Scholar] [CrossRef]
- Suprasert, P.; Cheewakriangkrai, C.; Manopunya, M. Outcome of single agent generic gemcitabine in platinum-resistant ovarian cancer, fallopian tube cancer and primary peritoneal adenocarcinoma. Asian Pac. J. Cancer Prev. 2012, 13, 517–520. [Google Scholar] [CrossRef]
- Tate, S.; Nishikimi, K.; Matsuoka, A.; Otsuka, S.; Kato, K.; Takahashi, Y.; Shozu, M. Tailored-dose chemotherapy with gemcitabine and irinotecan in patients with platinum-refractory/resistant ovarian or primary peritoneal cancer: A phase II trial. J. Gynecol. Oncol. 2021, 32, e8. [Google Scholar] [CrossRef] [PubMed]
- Sauermann, G. The Effect of 2-Deoxy-D-Glucose on Substrate Oxidation of Ascites Tumor Cells. Arch. Biochem. Biophys. 1964, 104, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, J.; Giammarile, F.; Rousset, P.; Rubello, D.; Bakrin, N.; Passot, G.; Isaac, S.; Glehen, O.; Skanjeti, A. The role of 18F-FDG-PET/ceCT in peritoneal mesothelioma. Nucl. Med. Commun. 2017, 38, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nakamoto, Y.; Ishimori, T.; Saga, T.; Kido, A.; Hamanishi, J.; Hamanaka, Y.; Togashi, K. Prognostic utility of FDG PET/CT in advanced ovarian, fallopian and primary peritoneal high-grade serous cancer patients before and after neoadjuvant chemotherapy. Ann. Nucl. Med. 2020, 34, 128–135. [Google Scholar] [CrossRef]
- Moindjie, H.; Rodrigues-Ferreira, S.; Nahmias, C. Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy. Cancers 2021, 13, 3311. [Google Scholar] [CrossRef]
- Rakheja, D.; Medeiros, L.; Bevan, S.; Chen, W. The Emerging Role of D-2-Hydroxyglutarate as an Oncometabolite in Hematolymphoid and Central Nervous System Neoplasms. Front. Oncol. 2013, 3, 169. [Google Scholar] [CrossRef]
- Borger, D.R.; Goyal, L.; Yau, T.; Poon, R.T.; Ancukiewicz, M.; Deshpande, V.; Christiani, D.C.; Liebman, H.M.; Yang, H.; Kim, H.; et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2014, 20, 1884–1890. [Google Scholar] [CrossRef]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef]
- Gupta, V.K.; Sharma, N.S.; Durden, B.; Garrido, V.T.; Kesh, K.; Edwards, D.; Wang, D.; Myer, C.; Mateo-Victoriano, B.; Kollala, S.S.; et al. Hypoxia-Driven Oncometabolite L-2HG Maintains Stemness-Differentiation Balance and Facilitates Immune Evasion in Pancreatic Cancer. Cancer Res. 2021, 81, 4001–4013. [Google Scholar] [CrossRef]
- Sonego, M.; Baldassarre, G. A new role for IDH1 in the control of ovarian cancer cells metabolism and senescence. Ann. Transl. Med. 2020, 8, 780. [Google Scholar] [CrossRef]
- Salati, M.; Caputo, F.; Baldessari, C.; Galassi, B.; Grossi, F.; Dominici, M.; Ghidini, M. IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting. Cancers 2020, 12, 3310. [Google Scholar] [CrossRef]
- Zhang, S.; Li, W.; Liang, F. Clinical value of fluorine-18 2-fluoro-2-deoxy-D-glucose positron emission tomography/computed tomography in penile cancer. Oncotarget 2016, 7, 48600–48606. [Google Scholar] [CrossRef]
- Garcia, S.N.; Guedes, R.C.; Marques, M.M. Unlocking the Potential of HK2 in Cancer Metabolism and Therapeutics. Curr. Med. Chem. 2019, 26, 7285–7322. [Google Scholar] [CrossRef]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695. [Google Scholar] [CrossRef]
- Xiang, J.; Zhou, L.; He, Y.; Wu, S. LDH-A inhibitors as remedies to enhance the anticancer effects of PARP inhibitors in ovarian cancer cells. Aging 2021, 13, 25920–25930. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Sun, H.; Zhang, S.; Shan, C. The Multiple Roles of Glucose-6-Phosphate Dehydrogenase in Tumorigenesis and Cancer Chemoresistance. Life 2022, 12, 271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, Y.; Lu, W.; Li, J.; Yu, S.; Brown, E.J.; Stanger, B.Z.; Rabinowitz, J.D.; Yang, X. G6PD-mediated increase in de novo NADP(+) biosynthesis promotes antioxidant defense and tumor metastasis. Sci. Adv. 2022, 8, eabo0404. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, D.; Bao, L.; Yin, T.; Lei, D.; Yu, J.; Tong, X. 6PGD inhibition sensitizes hepatocellular carcinoma to chemotherapy via AMPK activation and metabolic reprogramming. Biomed. Pharmacother. 2019, 111, 1353–1358. [Google Scholar] [CrossRef]
- Poff, A.M.; Ari, C.; Seyfried, T.N.; D’Agostino, D.P. The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS ONE 2013, 8, e65522. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.W.; Wang, J.; Guo, H.; Zhao, Y.Y.; Sun, H.H.; Li, Y.F.; Lai, X.Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765. [Google Scholar] [CrossRef]
- Mukherjee, A.; Chiang, C.Y.; Daifotis, H.A.; Nieman, K.M.; Fahrmann, J.F.; Lastra, R.R.; Romero, I.L.; Fiehn, O.; Lengyel, E. Adipocyte-Induced FABP4 Expression in Ovarian Cancer Cells Promotes Metastasis and Mediates Carboplatin Resistance. Cancer Res. 2020, 80, 1748–1761. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, J.; Yeo, J.H.; Kwon, D.; Min, B.S.; Cha, Y.J.; Koo, J.S.; Jeong, J.; Lee, J.; Choi, J. Interaction between CD36 and FABP4 modulates adipocyte-induced fatty acid import and metabolism in breast cancer. NPJ Breast Cancer 2021, 7, 129. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef]
- Tomacha, J.; Dokduang, H.; Padthaisong, S.; Namwat, N.; Klanrit, P.; Phetcharaburanin, J.; Wangwiwatsin, A.; Khampitak, T.; Koonmee, S.; Titapun, A.; et al. Targeting Fatty Acid Synthase Modulates Metabolic Pathways and Inhibits Cholangiocarcinoma Cell Progression. Front. Pharm. 2021, 12, 696961. [Google Scholar] [CrossRef]
- Wu, X.; Qin, L.; Fako, V.; Zhang, J.T. Molecular mechanisms of fatty acid synthase (FASN)-mediated resistance to anti-cancer treatments. Adv. Biol. Regul. 2014, 54, 214–221. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Lewis, C.S.; Voelkel-Johnson, C.; Smith, C.D. Targeting Sphingosine Kinases for the Treatment of Cancer. Adv. Cancer Res. 2018, 140, 295–325. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.H.; Radhakrishnan, R.; Jayaraman, M.; Yan, M.; Ward, J.D.; Fung, K.M.; Moxley, K.; Sood, A.K.; Isidoro, C.; Mukherjee, P.; et al. LPA Induces Metabolic Reprogramming in Ovarian Cancer via a Pseudohypoxic Response. Cancer Res. 2018, 78, 1923–1934. [Google Scholar] [CrossRef]
- Benesch, M.G.; Tang, X.; Venkatraman, G.; Bekele, R.T.; Brindley, D.N. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J. Biomed. Res. 2016, 30, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, L.; Qiu, Z.; Deng, W.; Wang, W. Key Molecules of Fatty Acid Metabolism in Gastric Cancer. Biomolecules 2022, 12, 706. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef]
- Shen, Y.A.; Chen, C.L.; Huang, Y.H.; Evans, E.E.; Cheng, C.C.; Chuang, Y.J.; Zhang, C.; Le, A. Inhibition of glutaminolysis in combination with other therapies to improve cancer treatment. Curr. Opin. Chem. Biol. 2021, 62, 64–81. [Google Scholar] [CrossRef]
- Yang, W.H.; Qiu, Y.; Stamatatos, O.; Janowitz, T.; Lukey, M.J. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends Cancer 2021, 7, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Suhre, K. Advancing Cancer Treatment by Targeting Glutamine Metabolism—A Roadmap. Cancers 2022, 14, 553. [Google Scholar] [CrossRef]
- Fasoulakis, Z.; Koutras, A.; Ntounis, T.; Prokopakis, I.; Perros, P.; Chionis, A.; Sapantzoglou, I.; Katrachouras, A.; Konis, K.; Samara, A.A.; et al. Ovarian Cancer and Glutamine Metabolism. Int. J. Mol. Sci. 2023, 24, 5041. [Google Scholar] [CrossRef]
- Hudson, C.D.; Savadelis, A.; Nagaraj, A.B.; Joseph, P.; Avril, S.; DiFeo, A.; Avril, N. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget 2016, 7, 41637–41649. [Google Scholar] [CrossRef]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. eLife 2020, 9, e56749. [Google Scholar] [CrossRef]
- Chen, R.; Lai, L.A.; Sullivan, Y.; Wong, M.; Wang, L.; Riddell, J.; Jung, L.; Pillarisetty, V.G.; Brentnall, T.A.; Pan, S. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci. Rep. 2017, 7, 7950. [Google Scholar] [CrossRef]
- Mazumder, M.E.; Beale, P.; Chan, C.; Yu, J.Q.; Huq, F. Epigallocatechin gallate acts synergistically in combination with cisplatin and designed trans-palladiums in ovarian cancer cells. Anticancer Res. 2012, 32, 4851–4860. [Google Scholar] [PubMed]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P. Methionine Dependence of Cancer. Biomolecules 2020, 10, 568. [Google Scholar] [CrossRef]
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Tajan, M.; Vousden, K.H. Dietary Approaches to Cancer Therapy. Cancer Cell 2020, 37, 767–785. [Google Scholar] [CrossRef]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P., Jr.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Albaugh, V.L.; Pinzon-Guzman, C.; Barbul, A. Arginine-Dual roles as an onconutrient and immunonutrient. J. Surg. Oncol. 2017, 115, 273–280. [Google Scholar] [CrossRef]
- Fultang, L.; Vardon, A.; De Santo, C.; Mussai, F. Molecular basis and current strategies of therapeutic arginine depletion for cancer. Int. J. Cancer 2016, 139, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.K.; Rial, N.S.; Kachel, K.L.; Gerner, E.W. Activated K-RAS increases polyamine uptake in human colon cancer cells through modulation of caveolar endocytosis. Mol. Carcinog. 2008, 47, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Holbert, C.E.; Cullen, M.T.; Casero, R.A., Jr.; Stewart, T.M. Polyamines in cancer: Integrating organismal metabolism and antitumour immunity. Nat. Rev. Cancer 2022, 22, 467–480. [Google Scholar] [CrossRef]
- Geeraerts, S.L.; Heylen, E.; De Keersmaecker, K.; Kampen, K.R. The ins and outs of serine and glycine metabolism in cancer. Nat. Metab. 2021, 3, 131–141. [Google Scholar] [CrossRef]
- Pan, S.; Fan, M.; Liu, Z.; Li, X.; Wang, H. Serine, glycine and one-carbon metabolism in cancer (Review). Int. J. Oncol. 2021, 58, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.C.; Chen, W.C.; Teo, X.Q.; Radda, G.K.; Lee, P.T. Downregulating serine hydroxymethyltransferase 2 (SHMT2) suppresses tumorigenesis in human hepatocellular carcinoma. Oncotarget 2016, 7, 53005–53017. [Google Scholar] [CrossRef]
- Tajan, M.; Hennequart, M.; Cheung, E.C.; Zani, F.; Hock, A.K.; Legrave, N.; Maddocks, O.D.K.; Ridgway, R.A.; Athineos, D.; Suarez-Bonnet, A.; et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat. Commun. 2021, 12, 366. [Google Scholar] [CrossRef]
- Geeraerts, S.L.; Kampen, K.R.; Rinaldi, G.; Gupta, P.; Planque, M.; Louros, N.; Heylen, E.; De Cremer, K.; De Brucker, K.; Vereecke, S.; et al. Repurposing the Antidepressant Sertraline as SHMT Inhibitor to Suppress Serine/Glycine Synthesis-Addicted Breast Tumor Growth. Mol. Cancer Ther. 2021, 20, 50–63. [Google Scholar] [CrossRef]
- Van Nyen, T.; Planque, M.; van Wagensveld, L.; Duarte, J.A.G.; Zaal, E.A.; Talebi, A.; Rossi, M.; Korner, P.R.; Rizzotto, L.; Moens, S.; et al. Serine metabolism remodeling after platinum-based chemotherapy identifies vulnerabilities in a subgroup of resistant ovarian cancers. Nat. Commun. 2022, 13, 4578. [Google Scholar] [CrossRef]
- Pundir, C.S.; Deswal, R.; Kumar, P. Quantitative analysis of sarcosine with special emphasis on biosensors: A review. Biomarkers 2019, 24, 415–422. [Google Scholar] [CrossRef]
- Atalay, E.B.; Kayali, H.A. The elevated D-2-hydroxyglutarate level found as a characteristic metabolic change of colon cancer in both in vitro and in vivo models. Biochem. Biophys. Res. Commun. 2022, 627, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, X.; Sun, S.; Ni, P.; Li, C.; Ren, A.; Wang, W.; Zhu, L. Homocysteine and Digestive Tract Cancer Risk: A Dose-Response Meta-Analysis. J. Oncol. 2018, 2018, 3720684. [Google Scholar] [CrossRef]
- Rose, M.G.; Farrell, M.P.; Schmitz, J.C. Thymidylate synthase: A critical target for cancer chemotherapy. Clin. Color. Cancer 2002, 1, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, E.; Kijima, T.; Kuribayashi, K.; Negi, Y.; Kanemura, S.; Mikami, K.; Doi, H.; Kitajima, K.; Nakano, T. First-line chemotherapy with pemetrexed plus cisplatin for malignant peritoneal mesothelioma. Expert Rev. Anticancer 2017, 17, 865–872. [Google Scholar] [CrossRef]
- Lee, Y.K.; Jun, H.J.; Nahm, J.H.; Lim, T.S.; Park, J.S.; Ahn, J.B.; Rha, S.Y.; Chung, H.C.; Oh, H.E.; Song, J.S.; et al. Therapeutic strategies for well-differentiated papillary mesothelioma of the peritoneum. Jpn. J. Clin. Oncol. 2013, 43, 996–1003. [Google Scholar] [CrossRef]
- Minami, K.; Shinsato, Y.; Yamamoto, M.; Takahashi, H.; Zhang, S.; Nishizawa, Y.; Tabata, S.; Ikeda, R.; Kawahara, K.; Tsujikawa, K.; et al. Ribonucleotide reductase is an effective target to overcome gemcitabine resistance in gemcitabine-resistant pancreatic cancer cells with dual resistant factors. J. Pharmacol. Sci. 2015, 127, 319–325. [Google Scholar] [CrossRef]
- Rose, P.G. Gemcitabine reverses platinum resistance in platinum-resistant ovarian and peritoneal carcinoma. Int. J. Gynecol. Cancer 2005, 15 (Suppl. S1), 18–22. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bhagwandin, S.; Labow, D.M. Malignant peritoneal mesothelioma: A review. Ann. Transl. Med. 2017, 5, 236. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
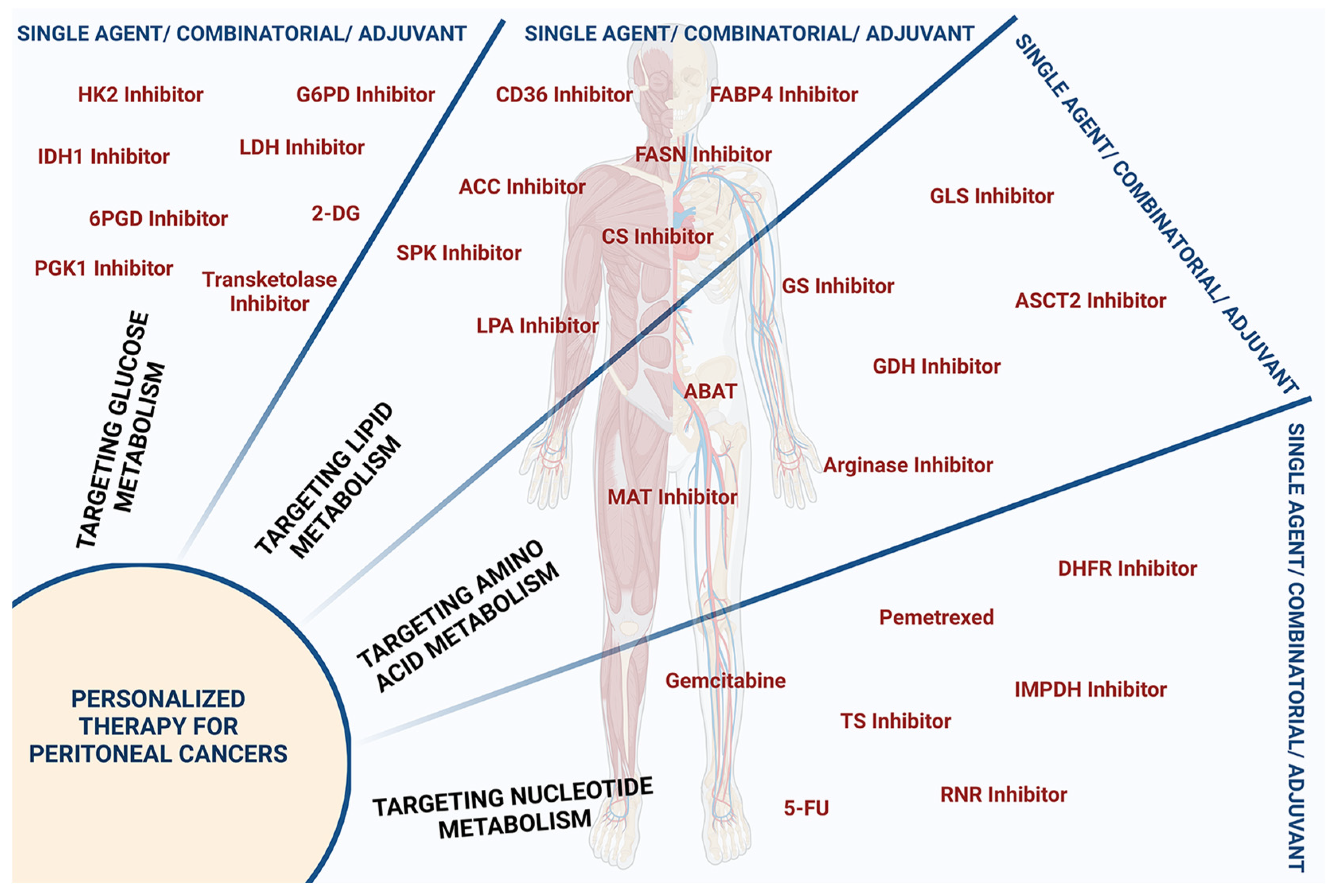{kind=link}
| S. No. | Target Molecule | Targeted Pathway | Drug/Inhibitor | Cancer Type | Reference |
|---|---|---|---|---|---|
| Targeting Glucose Metabolism | |||||
| 1. | Glu | Glycolysis | 2-deoxyglucose | PPC; OvCa | [95] |
| 2. | HK2 | Glycolysis | 3-bromopyruvate | PPC | [96] |
| 3. | LDH | Glycolysis | LDH Inhibitor | PPC | [97] |
| 4. | PGK1 | Glycolysis | 5-FU + shRNA-PGK1 | PPC; GC | [98] |
| 5. | 2HG | TCA Cycle | IDH1 Inhibitor | PPC; OvCa | [99] |
| 6. | G6PD | PPP | G6PD Inhibitor | BrCa; OvCA; PaCa | [100] |
| 7. | 6PGD | PPP | 6PGD Inhibitor | OvCa; HCC | [101] |
| Targeting Lipid Metabolism | |||||
| 8. | CD36 | FA uptake | CD36 Inhibition | OvCa | [102] |
| 9. | FABP4 | FA uptake | FABP4 Inhibitor | OvCa | [102] |
| 10. | ACC | FA synthesis | ACC Inhibitor | BrCa; CCC | [103] |
| 11. | FASN | FA synthesis | Orlistat, Cerulenin | PrCa; CC | [104,105] |
| 12. | CS | Sphingolipid synthesis | CS Inhibitor | OvCa; BrCa; CC | [106] |
| 13. | SPK | Sphingolipid synthesis | SPK Inhibitor | BrCa; PrCa; CC | [107] |
| Targeting Amino Acid Metabolism | |||||
| 14. | ASCT2 | Glutamine uptake | ASCT2 Inhibitor | PaCa; OvCa | [108] |
| 15. | Glutaminase | Glutaminolysis | 968 + Anti-PD-L1, Glutaminase Inhibitor + Doxorubicin | OvCa, PaCa | [109,110] |
| 17. | Met | Met-metabolism | Met-restriction | BrCa; CC; PrCa | [111] |
| 18. | Arginase | Arg-metabolism | Arginase Inhibitor | CRC; BrCa; LC; OvCa; HCC | [112] |
| 19. | Polycationic polyamines | Arg-metabolism | Polyamine Inhibitor | CC; CRC | [113] |
| 20. | Sarcosine | Ser-Gly metabolism | Sarcosine Inhibitor | PrCa; GC; CRC | [114] |
| 21. | α-Amino butyric acid | Met/Ser/Thr-metabolism | ABAT | CRC | [115] |
| Targeting Nucleotide Metabolism | |||||
| 22. | TYMS | Purine & Pyrimidine | ZD9331, AG337 | CRC; CC | [116] |
| 23. | DHFR | Purine & Pyrimidine | Pemetrexed, Raltitrexed | PPC; Ovary; CRC | [117,118,119] |
| 25. | IMPDH | Purine | Tiazofurin | PaCa; OvCa; CC; BrCa | [120] |
| 26. | RNR | Purine & Pyrimidine | Gemcitabine | OvCa; FTC; PPC | [121,122,123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadhan, R.; Kashyap, S.; Ha, J.H.; Jayaraman, M.; Song, Y.S.; Isidoro, C.; Dhanasekaran, D.N. Targeting Oncometabolites in Peritoneal Cancers: Preclinical Insights and Therapeutic Strategies. Metabolites 2023, 13, 618. https://doi.org/10.3390/metabo13050618
Nadhan R, Kashyap S, Ha JH, Jayaraman M, Song YS, Isidoro C, Dhanasekaran DN. Targeting Oncometabolites in Peritoneal Cancers: Preclinical Insights and Therapeutic Strategies. Metabolites. 2023; 13(5):618. https://doi.org/10.3390/metabo13050618
Chicago/Turabian StyleNadhan, Revathy, Srishti Kashyap, Ji Hee Ha, Muralidharan Jayaraman, Yong Sang Song, Ciro Isidoro, and Danny N. Dhanasekaran. 2023. "Targeting Oncometabolites in Peritoneal Cancers: Preclinical Insights and Therapeutic Strategies" Metabolites 13, no. 5: 618. https://doi.org/10.3390/metabo13050618
APA StyleNadhan, R., Kashyap, S., Ha, J. H., Jayaraman, M., Song, Y. S., Isidoro, C., & Dhanasekaran, D. N. (2023). Targeting Oncometabolites in Peritoneal Cancers: Preclinical Insights and Therapeutic Strategies. Metabolites, 13(5), 618. https://doi.org/10.3390/metabo13050618