

Understanding Transcription Factors and How They Affect Processes in Cucumber Sex Determination


Abstract
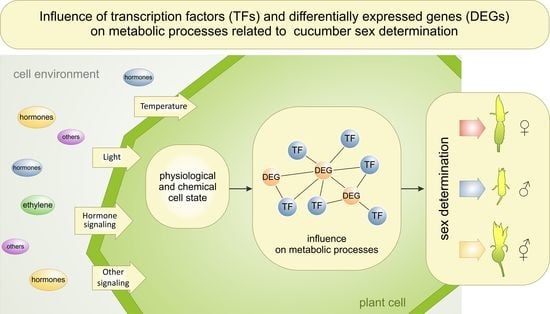
:
1. Introduction
2. Materials and Methods
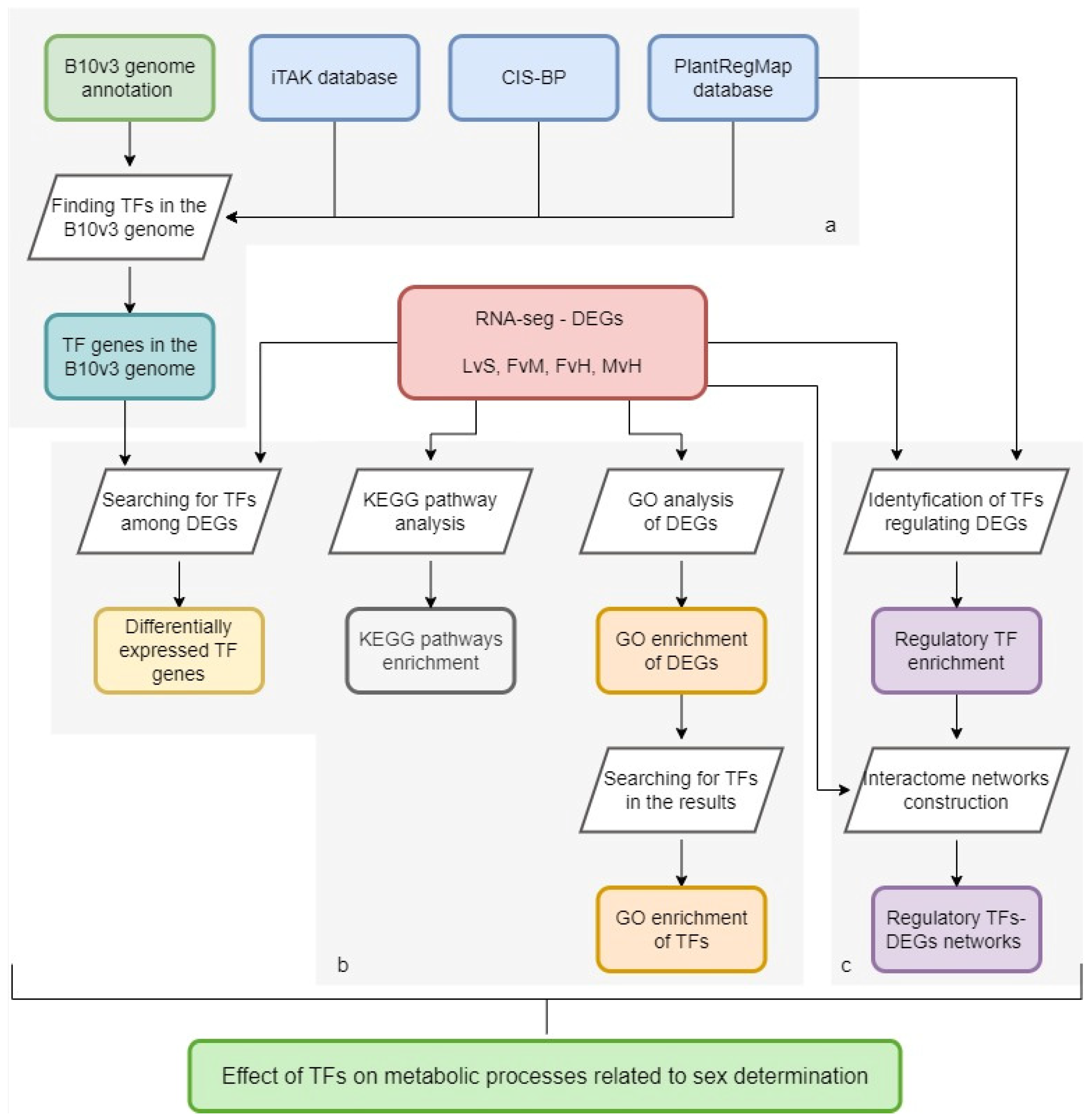
2.1. Finding the TFs in the B10v3 Genome
2.2. TFs among the Results of RNA-Seq Experiments
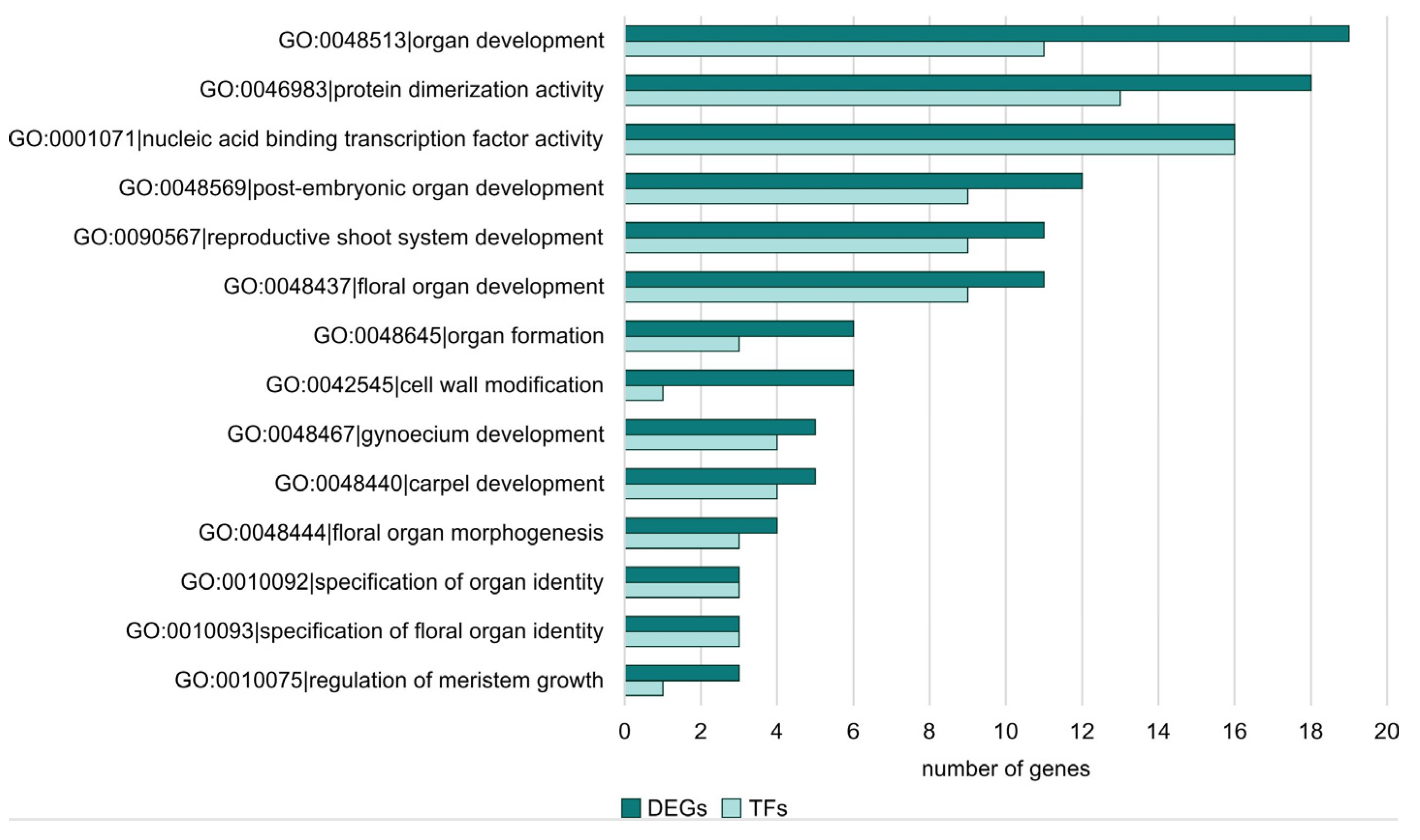
2.3. Gene Ontology Analysis among DEGs
2.4. KEGG Pathway Enrichment Analysis of DEGs
2.5. DEG’s Regulatory TFs Enrichment
3. Results and Discussions
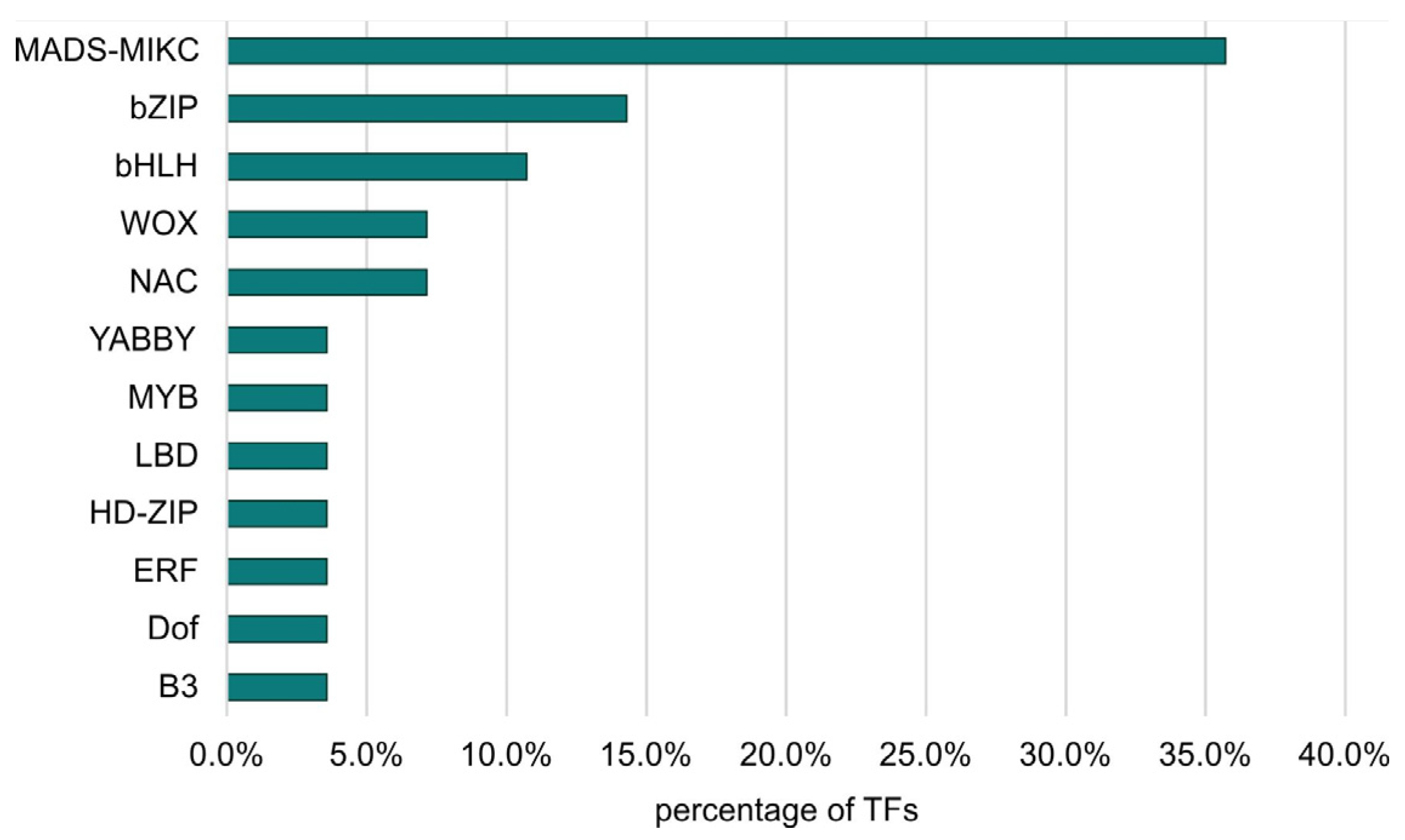
3.1. TF Search Results in the B10v3 Genome
3.2. TFs among DEGs
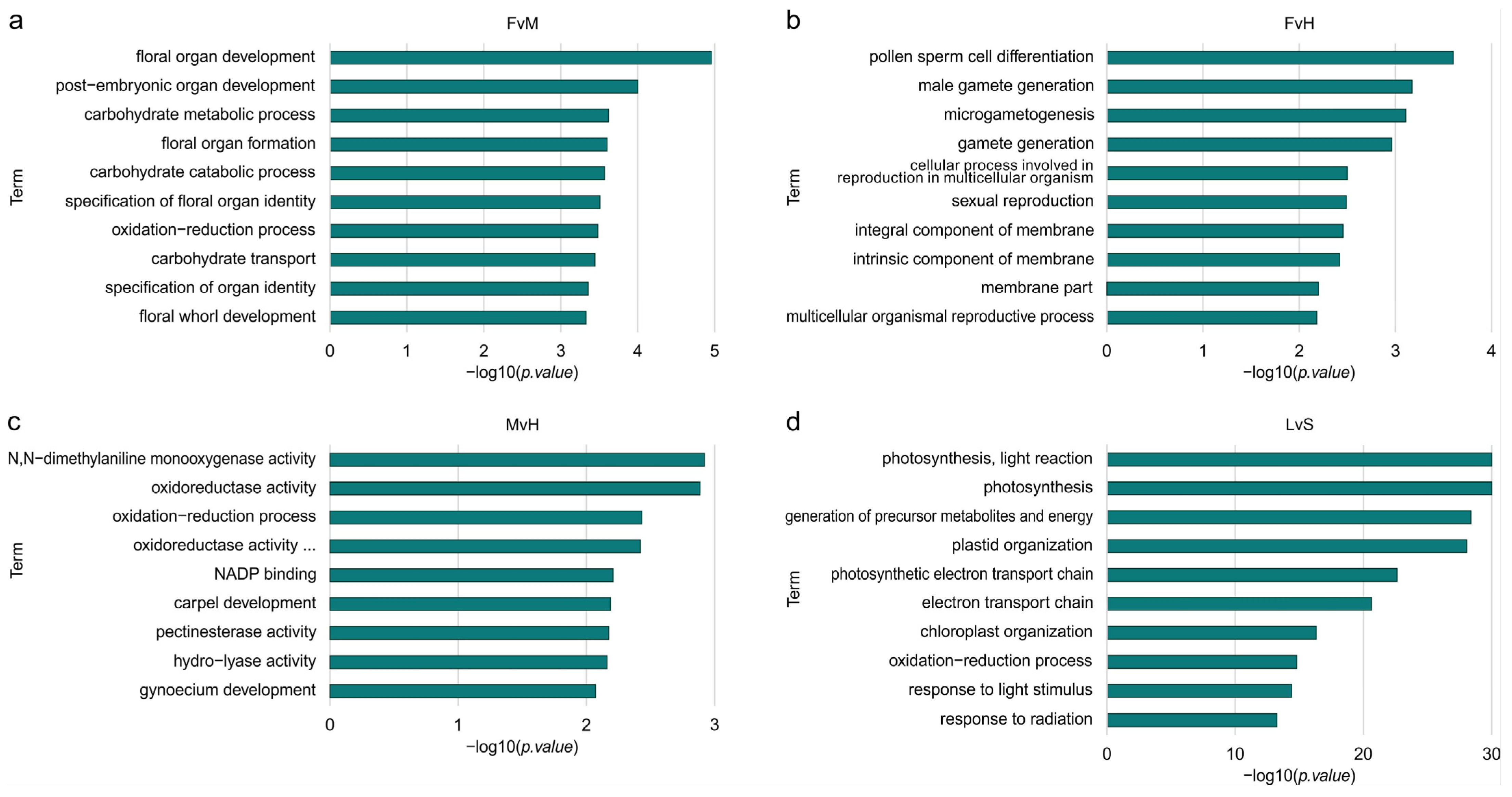
3.3. Ontology Analysis among DEGs
3.4. KEGG Enrichment Results in DEGs
3.5. DEGs’ Regulatory TFs
3.5.1. DEG’s Regulatory TFs’ Enrichment Results
3.5.2. Interaction Networks between Regulatory TFs and DEGs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The Genome of the Cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Koo, D.H.; Li, Y.; Zhang, X.; Luan, F.; Havey, M.J.; Jiang, J.; Weng, Y. Chromosome Rearrangements during Domestication of Cucumber as Revealed by High-Density Genetic Mapping and Draft Genome Assembly. Plant J. 2012, 71, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Osipowski, P.; Pawełkowicz, M.; Wojcieszek, M.; Skarzyńska, A.; Przybecki, Z.; Pląder, W. A High-Quality Cucumber Genome Assembly Enhances Computational Comparative Genomics. Mol. Genet. Genom. 2020, 295, 177–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turek, S.; Pląder, W.; Hoshi, Y.; Skarzyńska, A.; Pawełkowicz, M. Insight into the Organization of the B10v3 Cucumber Genome by Integration of Biological and Bioinformatic Data. Int. J. Mol. Sci. 2023, 24, 4011. [Google Scholar] [CrossRef]
- Biłas, R.; Szafran, K.; Hnatuszko-Konka, K.; Kononowicz, A.K. Cis-Regulatory Elements Used to Control Gene Expression in Plants. Plant Cell Tissue Organ Cult. (PCTOC) 2016, 127, 269–287. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. ITAK: A Program for Genome-Wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, M. Genome-Scale Characterization of Transcription Factors. Nat. Genet. 2023, 55, 357. [Google Scholar] [CrossRef]
- Dai, X.; Sinharoy, S.; Udvardi, M.; Zhao, P.X. PlantTFcat: An Online Plant Transcription Factor and Transcriptional Regulator Categorization and Analysis Tool. BMC Bioinform. 2013, 14, 321. [Google Scholar] [CrossRef] [Green Version]
- Franco-Zorrilla, J.M.; López-Vidriero, I.; Carrasco, J.L.; Godoy, M.; Vera, P.; Solano, R. DNA-Binding Specificities of Plant Transcription Factors and Their Potential to Define Target Genes. Proc. Natl. Acad. Sci. USA 2014, 111, 2367–2372. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.; Gao, G. PlantRegMap: Charting Functional Regulatory Maps in Plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and Inference of Eukaryotic Transcription Factor Sequence Specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Pawełkowicz, M.; Pryszcz, L.; Skarzyńska, A.; Wóycicki, R.K.; Posyniak, K.; Rymuszka, J.; Przybecki, Z.; Pląder, W. Comparative Transcriptome Analysis Reveals New Molecular Pathways for Cucumber Genes Related to Sex Determination. Plant Reprod. 2019, 32, 193–216. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- GitHub—Christophergandrud/NetworkD3: D3 JavaScript Network Graphs from R. Available online: https://github.com/christophergandrud/networkD3 (accessed on 25 April 2023).
- Meraj, T.A.; Fu, J.; Raza, M.A.; Zhu, C.; Shen, Q.; Xu, D.; Wang, Q. Transcriptional Factors Regulate Plant Stress Responses Through Mediating Secondary Metabolism. Genes 2020, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Lai, X.; Daher, H.; Galien, A.; Hugouvieux, V.; Zubieta, C. Structural Basis for Plant MADS Transcription Factor Oligomerization. Comput. Struct. Biotechnol. J. 2019, 17, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Theißen, G.; Gramzow, L. Structure and Evolution of Plant MADS Domain Transcription Factors. In Plant Transcription Factors: Evolutionary, Structural and Functional Aspects; Academic Press: Cambridge, MA, USA, 2016; pp. 127–138. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, L.; Song, J.; Jiang, L.; Liu, S. Isolation and Characterization of a MADS-Box Gene in Cucumber (Cucumis sativus L.) That Affects Flowering Time and Leaf Morphology in Transgenic Arabidopsis. Biotechnol. Biotechnol. Equip. 2019, 33, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Theißen, G.; Melzer, R.; Ruümpler, F. MADS-Domain Transcription Factors and the Floral Quartet Model of Flower Development: Linking Plant Development and Evolution. Development 2016, 143, 3259–3271. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhang, L.; Chen, P.; Wu, C.; Zhang, H.; Yuan, J.; Zhou, J.; Li, X. Genome-Wide Identification of APETALA2/ETHYLENE RESPONSIVE FACTOR Transcription Factors in Cucurbita moschata and Their Involvement in Ethylene Response. Front. Plant Sci. 2022, 13, 847754. [Google Scholar] [CrossRef] [PubMed]
- Pawełkowicz, M.E.; Skarzyńska, A.; Pląder, W.; Przybecki, Z. Genetic and Molecular Bases of Cucumber (Cucumis sativus L.) Sex Determination. Mol. Breed. 2019, 39, 50. [Google Scholar] [CrossRef] [Green Version]
- Boualem, A.; Troadec, C.; Camps, C.; Lemhemdi, A.; Morin, H.; Sari, M.A.; Fraenkel-Zagouri, R.; Kovalski, I.; Dogimont, C.; Perl-Treves, R.; et al. A Cucurbit Androecy Gene Reveals How Unisexual Flowers Develop and Dioecy Emerges. Science 2015, 350, 688–691. [Google Scholar] [CrossRef]
- Boualem, A.; Troadec, C.; Kovalski, I.; Sari, M.A.; Perl-Treves, R.; Bendahmane, A. A Conserved Ethylene Biosynthesis Enzyme Leads to Andromonoecy in Two Cucumis Species. PLoS ONE 2009, 4, e6144. [Google Scholar] [CrossRef] [PubMed]
- Trebitsh, T.; Staub, J.E.; O’Neill, S.D. Identification of a 1-Aminocyclopropane-1-Carboxylic Acid Synthase Gene Linked to the Female (F) Locus That Enhances Female Sex Expression in Cucumber. Plant Physiol. 1997, 113, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, S.; Fujii, N.; Takahashi, H. Characterization of Ethylene Effects on Sex Determination in Cucumber Plants. Sex. Plant Reprod. 2003, 16, 103–111. [Google Scholar] [CrossRef]
- Bhowmick, B.K.; Jha, S. Dynamics of Sex Expression and Chromosome Diversity in Cucurbitaceae: A Story in the Making. J. Genet. 2015, 94, 793–808. [Google Scholar] [CrossRef]
- Zhang, H.; Pan, X.; Liu, S.; Lin, W.; Li, Y.; Zhang, X. Genome-Wide Analysis of AP2/ERF Transcription Factors in Pineapple Reveals Functional Divergence during Flowering Induction Mediated by Ethylene and Floral Organ Development. Genomics 2021, 113, 474–489. [Google Scholar] [CrossRef]
- Shan, H.; Chen, S.; Jiang, J.; Chen, F.; Chen, Y.; Gu, C.; Li, P.; Song, A.; Zhu, X.; Gao, H.; et al. Heterologous Expression of the Chrysanthemum R2R3-MYB Transcription Factor CmMYB2 Enhances Drought and Salinity Tolerance, Increases Hypersensitivity to ABA and Delays Flowering in Arabidopsis thaliana. Mol. Biotechnol. 2012, 51, 160–173. [Google Scholar] [CrossRef]
- Pei, H.; Ma, N.; Tian, J.; Luo, J.; Chen, J.; Li, J.; Zheng, Y.; Chen, X.; Fei, Z.; Gao, J. An NAC Transcription Factor Controls Ethylene-Regulated Cell Expansion in Flower Petals. Plant Physiol. 2013, 163, 775. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, L.; Yu, Y.; Quan, R.; Zhang, Z.; Zhang, H.; Huang, R. The Ethylene Response Factor AtERF11 That Is Transcriptionally Modulated by the BZIP Transcription Factor HY5 Is a Crucial Repressor for Ethylene Biosynthesis in Arabidopsis. Plant J. 2011, 68, 88–99. [Google Scholar] [CrossRef]
- Alessio, V.M.; Cavaçana, N.; Dantas, L.L.D.B.; Lee, N.; Hotta, C.T.; Imaizumi, T.; Menossi, M. The FBH Family of BHLH Transcription Factors Controls ACC Synthase Expression in Sugarcane. J. Exp. Bot. 2018, 69, 2511–2525. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, X.; Wang, Y.; Sun, M.; Zhao, P.; Wang, Q.; Yang, B.; Li, J.; Jiang, Y.Q. WRKY29 Transcription Factor Regulates Ethylene Biosynthesis and Response in Arabidopsis. Plant Physiol. Biochem. 2023, 194, 134–145. [Google Scholar] [CrossRef] [PubMed]
- van Es, S.W.; Silveira, S.R.; Rocha, D.I.; Bimbo, A.; Martinelli, A.P.; Dornelas, M.C.; Angenent, G.C.; Immink, R.G.H. Novel Functions of the Arabidopsis Transcription Factor TCP5 in Petal Development and Ethylene Biosynthesis. Plant J. 2018, 94, 867–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, M.; Matsui, K.; Hiratsu, K.; Shinshi, H.; Ohme-Takagi, M. Repression Domains of Class II ERF Transcriptional Repressors Share an Essential Motif for Active Repression. Plant Cell 2001, 13, 1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamant, O.; Nogué, F.; Belles-Boix, E.; Jublot, D.; Grandjean, O.; Traas, J.; Pautot, V. The KNAT2 Homeodomain Protein Interacts with Ethylene and Cytokinin Signaling. Plant Physiol. 2002, 130, 657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brian, L.; Warren, B.; McAtee, P.; Rodrigues, J.; Nieuwenhuizen, N.; Pasha, A.; David, K.M.; Richardson, A.; Provart, N.J.; Allan, A.C.; et al. A Gene Expression Atlas for Kiwifruit (Actinidia Chinensis) and Network Analysis of Transcription Factors. BMC Plant Biol. 2021, 21, 121. [Google Scholar] [CrossRef] [PubMed]
- Trebitsh, T.; Rudich, J.; Riov, J. Auxin, Biosynthesis of Ethylene and Sex Expression in Cucumber (Cucumis sativus). Plant Growth Regul. 1987, 5, 105–113. [Google Scholar] [CrossRef]
- Shannon, S.; De La Guardia, M.D. Sex Expression and the Production of Ethylene Induced by Auxin in the Cucumber (Cucumis Sativum L.). Nature 1969, 223, 186. [Google Scholar] [CrossRef]
- He, X.J.; Mu, R.L.; Cao, W.H.; Zhang, Z.G.; Zhang, J.S.; Chen, S.Y. AtNAC2, a Transcription Factor Downstream of Ethylene and Auxin Signaling Pathways, Is Involved in Salt Stress Response and Lateral Root Development. Plant J. 2005, 44, 903–916. [Google Scholar] [CrossRef]
- Reyes-Olalde, J.I.; Zúñiga-Mayo, V.M.; Serwatowska, J.; Chavez Montes, R.A.; Lozano-Sotomayor, P.; Herrera-Ubaldo, H.; Gonzalez-Aguilera, K.L.; Ballester, P.; Ripoll, J.J.; Ezquer, I.; et al. The BHLH Transcription Factor SPATULA Enables Cytokinin Signaling, and Both Activate Auxin Biosynthesis and Transport Genes at the Medial Domain of the Gynoecium. PLoS Genet. 2017, 13, e1006726. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N.; Wu, M.F.; Winter, C.M.; Wagner, D. LEAFY and Polar Auxin Transport Coordinately Regulate Arabidopsis Flower Development. Plants 2014, 3, 251. [Google Scholar] [CrossRef] [Green Version]
- Shanks, C.M.; Hecker, A.; Cheng, C.; Brand, L.; Collani, S.; Schmid, M.; Schaller, G.E.; Wanke, D.; Harter, K.; Kieber, J.J. Role of BASIC PENTACYSTEINE Transcription Factors in a Subset of Cytokinin Signaling Responses. Plant J. 2018, 95, 458–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theune, M.L.; Bloss, U.; Brand, L.H.; Ladwig, F.; Wanke, D. Phylogenetic Analyses and GAGA-Motif Binding Studies of BBR/BPC Proteins Lend to Clues in GAGA-MOTIF Recognition and a Regulatory Role in Brassinosteroid Signaling. Front. Plant Sci. 2019, 10, 466. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Quinn, J.A. Tests of a Mechanistic Model of One Hormone Regulating Both Sexes in Cucumis sativus (Cucurbitaceae). Am. J. Bot. 1995, 82, 1537–1546. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, G.; Li, Y.; Mo, N.; Zhang, J.; Liang, Y. Transcriptomic Analysis Implies That GA Regulates Sex Expression via Ethylene-Dependent and Ethylene-Independent Pathways in Cucumber (Cucumis sativus L.). Front. Plant Sci. 2017, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, N.; Winter, C.M.; Wu, M.F.; Kanno, Y.; Yamaguchi, A.; Seo, M.; Wagner, D. Gibberellin Acts Positively Then Negatively to Control Onset of Flower Formation in Arabidopsis. Science 2014, 344, 638–641. [Google Scholar] [CrossRef]
- Yang, C.; Ma, Y.; Li, J. The Rice YABBY4 Gene Regulates Plant Growth and Development through Modulating the Gibberellin Pathway. J. Exp. Bot. 2016, 67, 5545–5556. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Zhao, Y.; Ma, Q.; Hu, Y.; Hedden, P.; Zhang, Q.; Zhou, D.X. The Rice YABBY1 Gene Is Involved in the Feedback Regulation of Gibberellin Metabolism. Plant Physiol. 2007, 144, 121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, X.; Liu, B.; Wang, W.; Liu, X.; Chen, C.; Liu, X.; Yang, S.; Ren, H. A GAMYB Homologue CsGAMYB1 Regulates Sex Expression of Cucumber via an Ethylene-Independent Pathway. J. Exp. Bot. 2014, 65, 3201. [Google Scholar] [CrossRef] [Green Version]
- Jia, P.; Xing, L.; Zhang, C.; Chen, H.; Li, Y.; Zhang, D.; Ma, J.; Zhao, C.; Han, M.; Ren, X.; et al. MdKNOX15, a Class I Knotted-like Transcription Factor of Apple, Controls Flowering and Plant Height by Regulating GA Levels through Promoting the MdGA2ox7 Transcription. Environ. Exp. Bot. 2021, 185, 104411. [Google Scholar] [CrossRef]
- Lin, R.C.; Park, H.J.; Wang, H.Y. Role of Arabidopsis RAP2.4 in Regulating Light- and Ethylene-Mediated Developmental Processes and Drought Stress Tolerance. Mol. Plant 2008, 1, 42–57. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Zhao, L.; Chong, K.; Wang, T. Overexpression of OsERF1, a Novel Rice ERF Gene, up-Regulates Ethylene-Responsive Genes Expression besides Affects Growth and Development in Arabidopsis. J. Plant Physiol. 2008, 165, 1717–1725. [Google Scholar] [CrossRef]
- Tao, Q.; Niu, H.; Wang, Z.; Zhang, W.; Wang, H.; Wang, S.; Zhang, X.; Li, Z. Ethylene Responsive Factor ERF110 Mediates Ethylene-Regulated Transcription of a Sex Determination-Related Orthologous Gene in Two Cucumis Species. J. Exp. Bot. 2018, 69, 2953–2965. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Xu, L.; Jia, Z.; Xu, Y.; Yang, Q.; Fei, Z.; Lu, X.; Chen, H.; Huang, S. Genetic Association of ETHYLENE-INSENSITIVE3-like Sequence with the Sex-Determining M Locus in Cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2008, 117, 927–933. [Google Scholar] [CrossRef]
- Zhao, S.; Luo, Y.; Zhang, Z.; Xu, M.; Wang, W.; Zhao, Y.; Zhang, L.; Fan, Y.; Wang, L. ZmSOC1, an MADS-Box Transcription Factor from Zea Mays, Promotes Flowering in Arabidopsis. Int. J. Mol. Sci. 2014, 15, 19987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Zhuo, S.; Liu, X.; Che, G.; Wang, Z.; Gu, R.; Shen, J.; Song, W.; Zhou, Z.; Han, D.; et al. The MADS-Box Gene CsSHP Participates in Fruit Maturation and Floral Organ Development in Cucumber. Front. Plant Sci. 2020, 10, 1781. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Xin, R.; Kim, D.H.; Sung, S.; Lange, T.; Huq, E. NO FLOWERING IN SHORT DAY (NFL) Is a BHLH Transcription Factor That Promotes Flowering Specifically under Short-Day Conditions in Arabidopsis. Development 2016, 143, 682–690. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Xin, H.; Gu, X.; Ma, J.; Li, L. Genome-Wide Identification and Functional Analysis of the Basic Helix-Loop-Helix (Bhlh) Transcription Family Reveals Candidate Ptfbh Genes Involved in the Flowering Process of Populus trichocarpa. Forests 2021, 12, 1439. [Google Scholar] [CrossRef]
- Li, X.; Tian, X.; He, M.; Liu, X.; Li, Z.; Tang, J.; Mei, E.; Xu, M.; Liu, Y.; Wang, Z.; et al. BZIP71 Delays Flowering by Suppressing Ehd1 Expression in Rice. J. Integr. Plant Biol. 2022, 64, 1352–1363. [Google Scholar] [CrossRef]
- Hendelman, A.; Stav, R.; Zemach, H.; Arazi, T. The Tomato NAC Transcription Factor SlNAM2 Is Involved in Flower-Boundary Morphogenesis. J. Exp. Bot. 2013, 64, 5497–5507. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Dai, S.; Singh, P.K.; Wang, H.; Wang, Y.; Tan, J.L.H.; Wee, W.; Ito, T. A Membrane-Bound NAC-like Transcription Factor OsNTL5 Represses the Flowering in Oryza sativa. Front. Plant Sci. 2018, 9, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liu, G.; Jia, J.; Zhao, G.; Xia, C.; Zhang, L.; Li, F.; Zhang, Q.; Dong, C.; Gao, S.; et al. The Wheat MYB-Related Transcription Factor TaMYB72 Promotes Flowering in Rice. J. Integr. Plant Biol. 2016, 58, 701–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, M.; Kobayashi, Y.; Yamamoto, S.; Daimon, Y.; Yamaguchi, A.; Ikeda, Y.; Ichinoki, H.; Notaguchi, M.; Goto, K.; Araki, T. FD, a BZIP Protein Mediating Signals from the Floral Pathway Integrator FT at the Shoot Apex. Science 2005, 309, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Z.; Wang, L.; Kim, S.G.; Seo, P.J.; Qiao, M.; Wang, N.; Li, S.; Cao, X.; Park, C.M.; et al. WRKY71 Accelerates Flowering via the Direct Activation of FLOWERING LOCUS T and LEAFY in Arabidopsis thaliana. Plant J. 2016, 85, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waseem, M.; Li, N.; Su, D.; Chen, J.; Li, Z. Overexpression of a Basic Helix-Loop-Helix Transcription Factor Gene, SlbHLH22, Promotes Early Flowering and Accelerates Fruit Ripening in Tomato (Solanum Lycopersicum L.). Planta 2019, 250, 173–185. [Google Scholar] [CrossRef]
- Zhang, Y.; Verhoeff, N.I.; Chen, Z.; Chen, S.; Wang, M.; Zhu, Z.; Ouwerkerk, P.B.F. Functions of OsDof25 in Regulation of OsC4PPDK. Plant Mol. Biol. 2015, 89, 229. [Google Scholar] [CrossRef] [Green Version]
- Noguero, M.; Atif, R.M.; Ochatt, S.; Thompson, R.D. The Role of the DNA-Binding One Zinc Finger (DOF) Transcription Factor Family in Plants. Plant Sci. 2013, 209, 32–45. [Google Scholar] [CrossRef]
- Lorrai, R.; Gandolfi, F.; Boccaccini, A.; Ruta, V.; Possenti, M.; Tramontano, A.; Costantino, P.; Lepore, R.; Vittorioso, P. Genome-Wide RNA-Seq Analysis Indicates That the DAG1 Transcription Factor Promotes Hypocotyl Elongation Acting on ABA, Ethylene and Auxin Signaling. Sci. Rep. 2018, 8, 15895. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Gracia, P.; Roque, E.; Medina, M.; López-Martín, M.J.; Cañas, L.A.; Beltrán, J.P.; Gómez-Mena, C. The DOF Transcription Factor Sldof10 Regulates Vascular Tissue Formation during Ovary Development in Tomato. Front. Plant Sci. 2019, 10, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Choi, K.; Park, C.; Hwang, H.J.; Lee, I. SUPPRESSOR OF FRIGIDA4, Encoding a C2H2-Type Zinc Finger Protein, Represses Flowering by Transcriptional Activation of Arabidopsis FLOWERING LOCUS C. Plant Cell 2006, 18, 2985–2998. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; You, C.; Li, C.; Long, T.; Chen, G.; Byrne, M.E.; Zhang, Q. RID1, Encoding a Cys2/His2-Type Zinc Finger Transcription Factor, Acts as a Master Switch from Vegetative to Floral Development in Rice. Proc. Natl. Acad. Sci. USA 2008, 105, 12915–12920. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-c.; Fu, C.-c.; Kuang, J.-f.; Chen, J.-y.; Lu, W.-j. Two Banana Fruit Ripening-Related C2H2 Zinc Finger Proteins Are Transcriptional Repressors of Ethylene Biosynthetic Genes. Postharvest Biol. Technol. 2016, 116, 8–15. [Google Scholar] [CrossRef]
- Huang, R.; Liu, D.; Huang, M.; Ma, J.; Li, Z.; Li, M.; Sui, S. CpWRKY71, a WRKY Transcription Factor Gene of Wintersweet (Chimonanthus praecox), Promotes Flowering and Leaf Senescence in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 5325. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ning, K.; Che, G.; Yan, S.; Han, L.; Gu, R.; Li, Z.; Weng, Y.; Zhang, X. CsSPL Functions as an Adaptor between HD-ZIP III and CsWUS Transcription Factors Regulating Anther and Ovule Development in Cucumis sativus (Cucumber). Plant J. 2018, 94, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Unte, U.S.; Sorensen, A.M.; Pesaresi, P.; Gandikota, M.; Leister, D.; Saedler, H.; Huijser, P. SPL8, an SBP-Box Gene That Affects Pollen Sac Development in Arabidopsis. Plant Cell 2003, 15, 1009–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.K.; Skinner, D.J.; Gallagher, T.L.; Gasser, C.S. Integument Development in Arabidopsis Depends on Interaction of YABBY Protein INNER NO OUTER with Coactivators and Corepressors. Genetics 2017, 207, 1489–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.; Hur, J.; Kim, S.J.; Han, M.J.; Kim, S.R.; An, G. Ectopic Expression of OsYAB1 Causes Extra Stamens and Carpels in Rice. Plant Mol. Biol. 2004, 56, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.; Broholm, S.; Becker, A. CRABS CLAW Acts as a Bifunctional Transcription Factor in Flower Development. Front. Plant Sci. 2018, 9, 835. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N.; Jeong, C.W.; Nole-Wilson, S.; Krizek, B.A.; Wagner, D. AINTEGUMENTA and AINTEGUMENTA-LIKE6/PLETHORA3 Induce LEAFY Expression in Response to Auxin to Promote the Onset of Flower Formation in Arabidopsis. Plant Physiol. 2016, 170, 283–293. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Number of DEGs | Number of TFs Found for the Comparison | % of all DEGs |
|---|---|---|---|
| LvS | 2852 | 248 | 8.70% |
| FvM | 260 | 28 | 10.77% |
| FvH | 36 | 2 | 5.88% |
| MvH | 55 | 6 | 10.91% |
| TF Family | Count for FvH | Count for FvM | Count for MvH | Count for LvS |
|---|---|---|---|---|
| bHLH | 12 | 3 | 3 | 0 |
| NAC | 9 | 0 | 14 | 2 |
| bZIP | 6 | 0 | 0 | 0 |
| E2F/DP | 3 | 0 | 0 | 0 |
| BBR-BPC | 3 | 0 | 0 | 3 |
| FAR1 | 2 | 0 | 0 | 0 |
| MYB | 2 | 5 | 2 | 10 |
| ERF | 2 | 0 | 0 | 0 |
| ARR-B | 1 | 1 | 1 | 0 |
| MADS-MIKC | 1 | 0 | 0 | 1 |
| BES1 | 1 | 0 | 0 | 0 |
| AP2 | 1 | 0 | 1 | 1 |
| GATA | 1 | 0 | 0 | 0 |
| C2H2 | 1 | 0 | 4 | 5 |
| CPP | 1 | 0 | 0 | 0 |
| Dof | 1 | 1 | 3 | 4 |
| WOX | 1 | 0 | 1 | 0 |
| HD-ZIP | 0 | 2 | 6 | 2 |
| G2-like | 0 | 1 | 0 | 1 |
| SBP | 0 | 2 | 0 | 0 |
| TCP | 0 | 1 | 0 | 7 |
| ARF | 0 | 1 | 0 | 0 |
| Trihelix | 0 | 1 | 0 | 0 |
| C3H | 0 | 0 | 2 | 0 |
| WRKY | 0 | 0 | 1 | 6 |
| MYB-related | 0 | 0 | 0 | 0 |
| GRAS | 0 | 0 | 0 | 1 |
| TALE | 0 | 0 | 0 | 1 |
| EIL | 0 | 0 | 0 | 4 |
| FvM | FvH | MvH | LvS | |
|---|---|---|---|---|
| DEGs | 186 | 31 | 35 | 244 |
| TFs | 282 | 147 | 156 | 2785 |
| Comparison | Transcription Factor | No. of Edges | ||
|---|---|---|---|---|
| Gene ID | TF Family | Description | ||
| FvM | Cucsa.362960 | MADS-MIKC | MADS box transcription factor | 116 |
| Cucsa.277740 | AP2 | AP2-like ethylene-responsive transcription factor | 109 | |
| Cucsa.026600 | BBR-BPC | GAGA-binding transcriptional activator | 93 | |
| Cucsa.307870 | C2H2 | Transcription factor IIIA | 82 | |
| Cucsa.102120 | Dof | Dof zinc finger protein | 81 | |
| Cucsa.280310 | GRAS | DELLA protein GAI | 81 | |
| Cucsa.213830 | Dof | Dof zinc finger protein | 76 | |
| Cucsa.159750 | BBR-BPC | GAGA-binding transcriptional activator | 69 | |
| Cucsa.341290 | Dof | Dof zinc finger protein | 60 | |
| Cucsa.098430 | Dof | Dof zinc finger protein | 48 | |
| FvH | Cucsa.362960 | MADS-MIKC | MADS box transcription factor | 23 |
| Cucsa.026600 | BBR-BPC | GAGA-binding transcriptional activator | 21 | |
| Cucsa.277740 | AP2 | AP2-like ethylene-responsive transcription factor | 19 | |
| Cucsa.307870 | C2H2 | Transcription factor IIIA | 17 | |
| Cucsa.159750 | BBR-BPC | GAGA-binding transcriptional activator | 14 | |
| Cucsa.280310 | GRAS | DELLA protein GAI | 13 | |
| Cucsa.353140 | TALE | Homeobox protein knotted-1-like 1 | 13 | |
| Cucsa.102120 | Dof | Dof zinc finger protein | 12 | |
| Cucsa.213830 | Dof | Dof zinc finger protein | 12 | |
| Cucsa.098430 | Dof | Dof zinc finger protein | 9 | |
| MvH | Cucsa.362960 | MADS-MIKC | MADS box transcription factor | 21 |
| Cucsa.102120 | Dof | Dof zinc finger protein | 17 | |
| Cucsa.277740 | AP2 | AP2-like ethylene-responsive transcription factor | 16 | |
| Cucsa.213830 | Dof | Dof zinc finger protein | 12 | |
| Cucsa.307870 | C2H2 | Transcription factor IIIA | 12 | |
| Cucsa.341290 | Dof | Dof zinc finger protein | 12 | |
| Cucsa.026600 | BBR-BPC | GAGA-binding transcriptional activator | 11 | |
| Cucsa.159750 | BBR-BPC | GAGA-binding transcriptional activator | 11 | |
| Cucsa.136780 | ERF | Dehydration responsive element binding transcription factor | 9 | |
| Cucsa.237150 | ERF | Ethylene-responsive transcription factor ERF021 | 9 | |
| LvS | Cucsa.362960 | MADS-MIKC | MADS box transcription factor | 1579 |
| Cucsa.277740 | AP2 | AP2-like ethylene-responsive transcription factor | 1485 | |
| Cucsa.026600 | BBR-BPC | GAGA-binding transcriptional activator | 1324 | |
| Cucsa.307870 | C2H2 | Transcription factor IIIA | 1205 | |
| Cucsa.280310 | GRAS | DELLA protein GAI | 1090 | |
| Cucsa.102120 | Dof | Dof zinc finger protein | 1076 | |
| Cucsa.213830 | Dof | Dof zinc finger protein | 1031 | |
| Cucsa.159750 | BBR-BPC | GAGA-binding transcriptional activator | 1000 | |
| Cucsa.353140 | TALE | Homeobox protein knotted-1-like 1 | 920 | |
| Cucsa.341290 | Dof | Dof zinc finger protein | 823 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turek, S.; Aparna; Skarzyńska, A.; Pląder, W.; Pawełkowicz, M. Understanding Transcription Factors and How They Affect Processes in Cucumber Sex Determination. Metabolites 2023, 13, 740. https://doi.org/10.3390/metabo13060740
Turek S, Aparna, Skarzyńska A, Pląder W, Pawełkowicz M. Understanding Transcription Factors and How They Affect Processes in Cucumber Sex Determination. Metabolites. 2023; 13(6):740. https://doi.org/10.3390/metabo13060740
Chicago/Turabian StyleTurek, Szymon, Aparna, Agnieszka Skarzyńska, Wojciech Pląder, and Magdalena Pawełkowicz. 2023. "Understanding Transcription Factors and How They Affect Processes in Cucumber Sex Determination" Metabolites 13, no. 6: 740. https://doi.org/10.3390/metabo13060740
APA StyleTurek, S., Aparna, Skarzyńska, A., Pląder, W., & Pawełkowicz, M. (2023). Understanding Transcription Factors and How They Affect Processes in Cucumber Sex Determination. Metabolites, 13(6), 740. https://doi.org/10.3390/metabo13060740