MESSES: Software for Transforming Messy Research Datasets into Clean Submissions to Metabolomics Workbench for Public Sharing


Abstract
:1. Introduction
2. Materials and Methods
2.1. Third Party Packages
2.2. Package Organization and Module Description
2.3. Tagging System
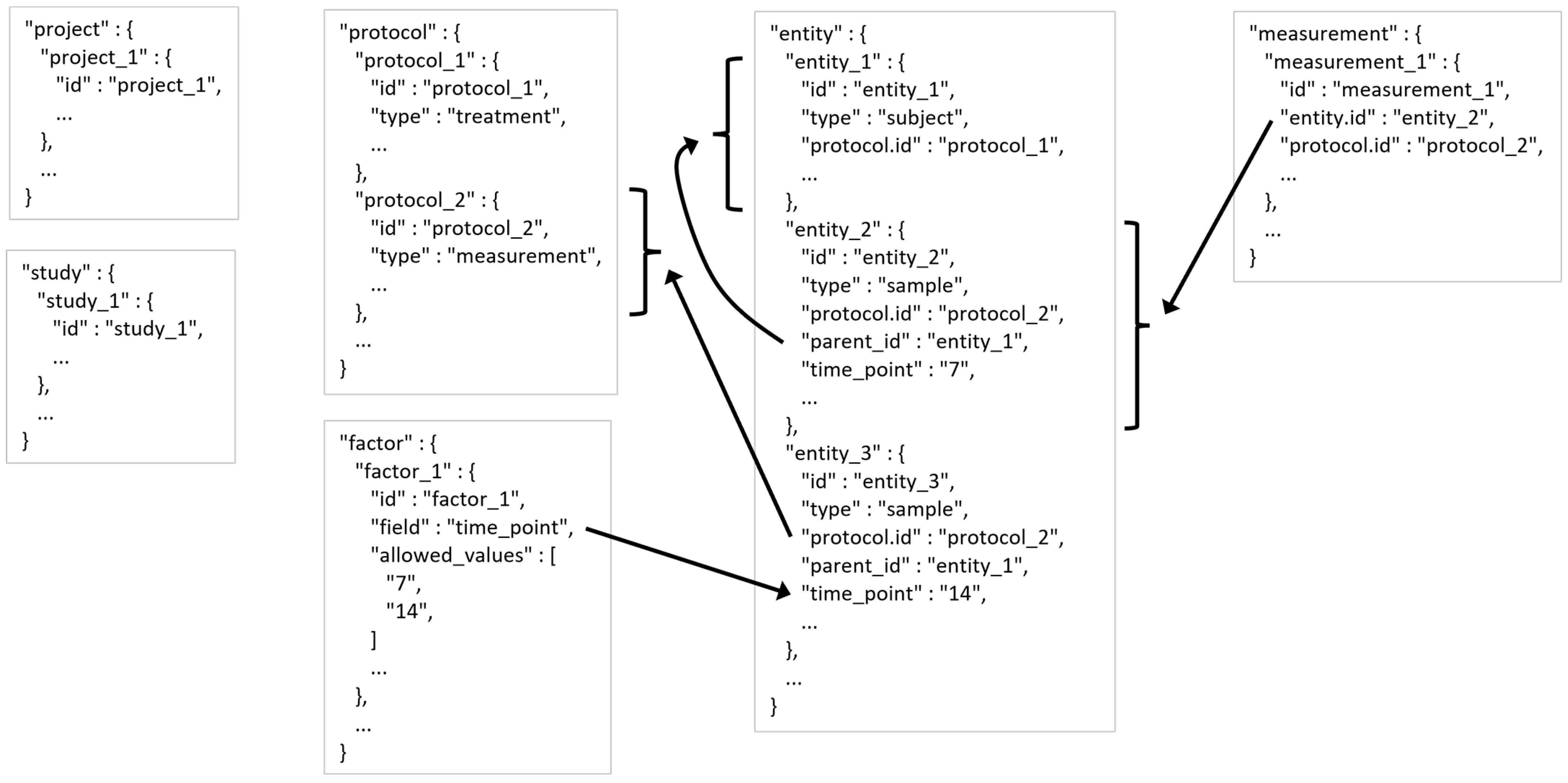
2.4. MESSES JSONized Data and Metadata Representation
- A project generally refers to a research project with multiple analytical datasets derived from one or more experimental designs.
- ○
- The project table entries would have information about the project, such as PI contact information and a description of the project.
- A study is generally one experimental design or analytical experiment inside of the project.
- ○
- The study table entries would have information about each study, such as PI contact information and a description of the study.
- A protocol describes an operation or set of operations done on a subject or sample entity.
- ○
- The protocol table entries would have information about each protocol, such as a description of the procedure and details about the equipment used.
- Entities are either subjects or samples that were collected or experimented on.
- ○
- The entity table entries would have information about each entity, such as sex and age of a subject or weight and units of weight of a sample. These latter examples demonstrate a descriptive attribute relationship between the weight field and the units of weight field typically indicated by ‘weight%unit’ used as the field name for units of weight.
- A measurement is typically the results acquired after putting a sample through an assay or analytical instrument such as a mass spectrometer or nuclear magnetic resonance spectrometer as well as any data calculation steps applied to raw measurements to generate usable processed results for downstream analysis.
- ○
- The measurement table entries would have information about each measurement, such as intensity, peak area, or compound assignment.
- A factor is a controlled independent variable of the experimental design. Experimental factors are conditions set in the experiment. Other factors may be other classifications such as male or female gender.
- ○
- The factor table entries would have information about each factor, such as the name of the factor and the allowed values of the factor.
- A treatment protocol describes the experimental factors performed on subject entities.
- ○
- For example, if a cell line is given 2 different media solutions to observe the different growth behavior between the 2, then this would be a treatment type protocol.
- A collection protocol describes how samples are collected from subject entities.
- ○
- For example, if media is taken out of a cell culture at various time points, this would be a collection protocol.
- A sample_prep protocol describes operations performed on sample entities.
- ○
- For example, once the cells in a culture are collected, they may be spun in a centrifuge or have solvents added to separate out protein, lipids, etc.
- A measurement protocol describes operations performed on samples to measure features about them.
- ○
- For example, if a sample is put through a mass spectrometer or into an NMR.
- A storage protocol describes where and/or how things (mainly samples) are stored.
- ○
- This was created mostly to help keep track of where samples were physically stored in freezers or where measurement data files were located on a share drive.
- If a sample comes from a sample, it must have a sample_prep type protocol.
- If a sample comes from a subject, it must have a collection type protocol.
- Subjects should have a treatment type protocol associated with it.
2.5. Testing
3. Results
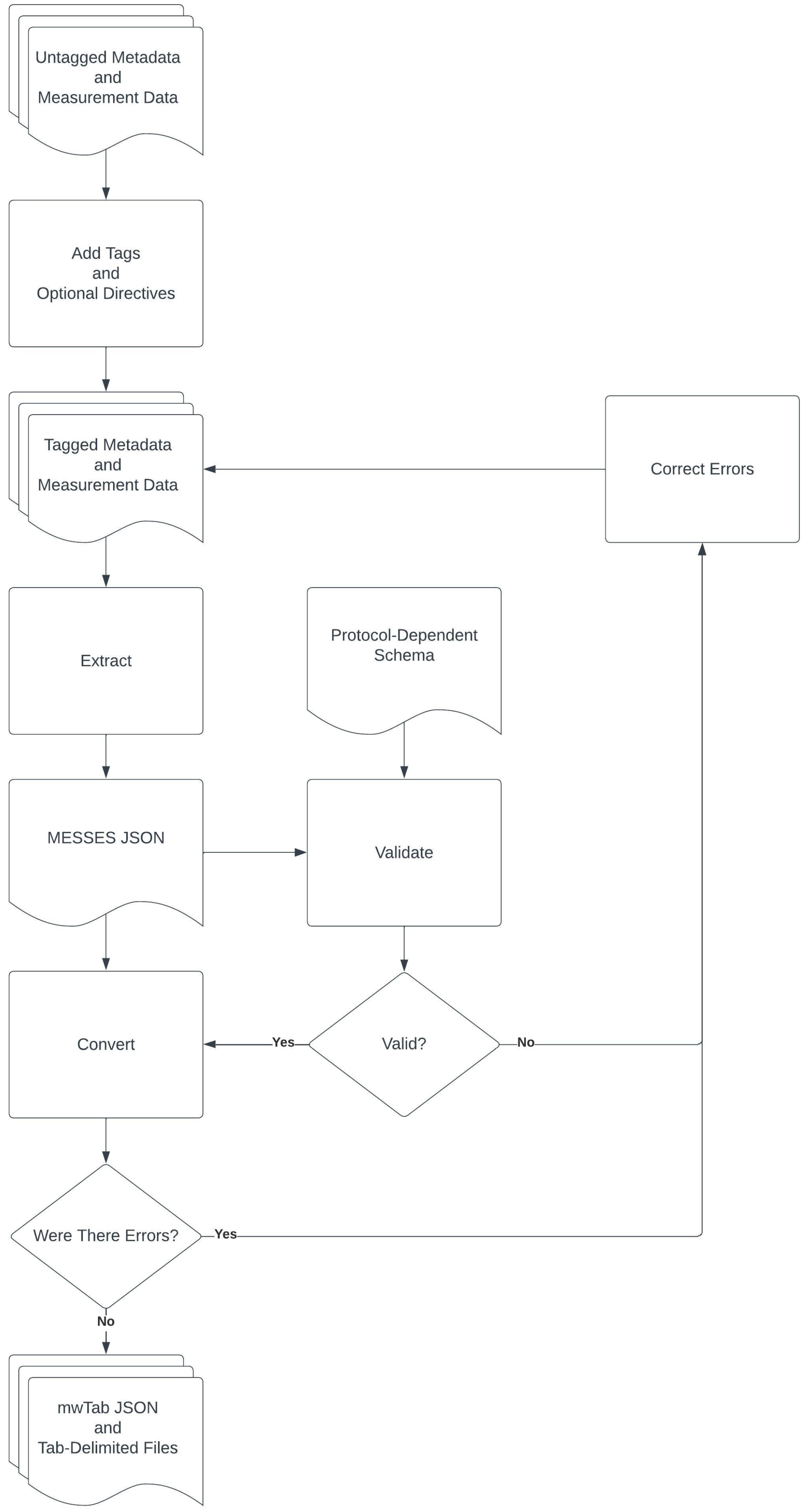
3.1. The Command Line Interface and Overall Metabolomics Workbench Deposition Workflow
3.2. Creation of an Example Mass Spectrometry Deposition
3.2.1. Extraction from Spreadsheets
3.2.2. Validation of Extracted Data and Metadata
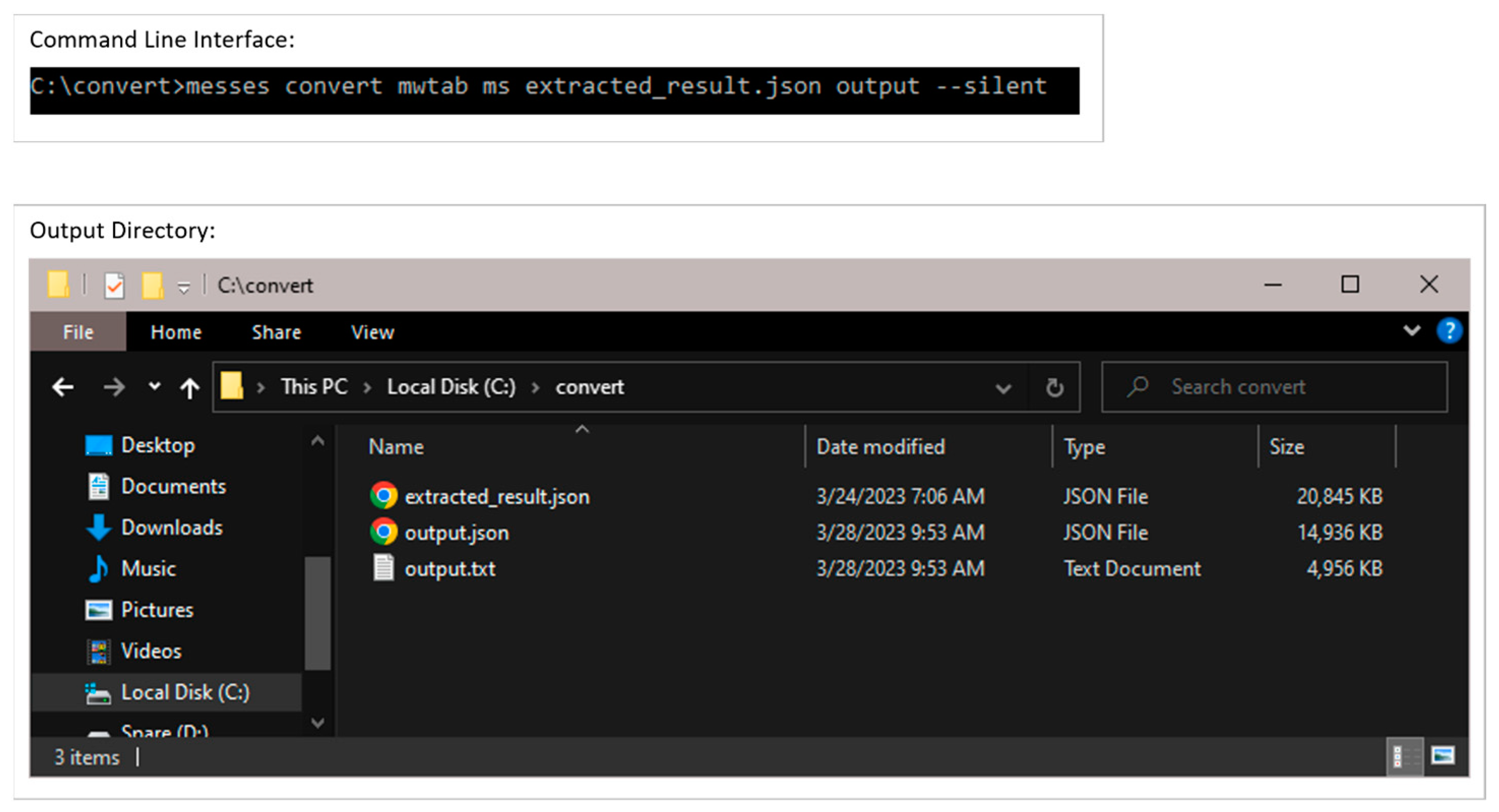
3.2.3. Conversion into mwTab Formats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FAIR | Findable, Accessible, Interoperable, Reusable. |
| NIH | National Institutes of Health |
| DMS | Data Management Sharing |
| MESSES | Metadata from Experimental SpreadSheets Extraction System |
| LIMS | Laboratory Information Management System |
| JSON | JavaScript Object Notation |
| mwTab | Metabolomics Workbench Tabular format |
| NMR | Nuclear Magnetic Resonance |
| MS | Mass Spectrometry |
| PyPi | Python Package Index |
| CLI | Command Line Interface |
| API | Application Programming Interface |
| PD schema | Protocol-Dependent schema |
| EDS | Experiment Description Schema |
| CSV | Comma Separated Values |
Appendix A
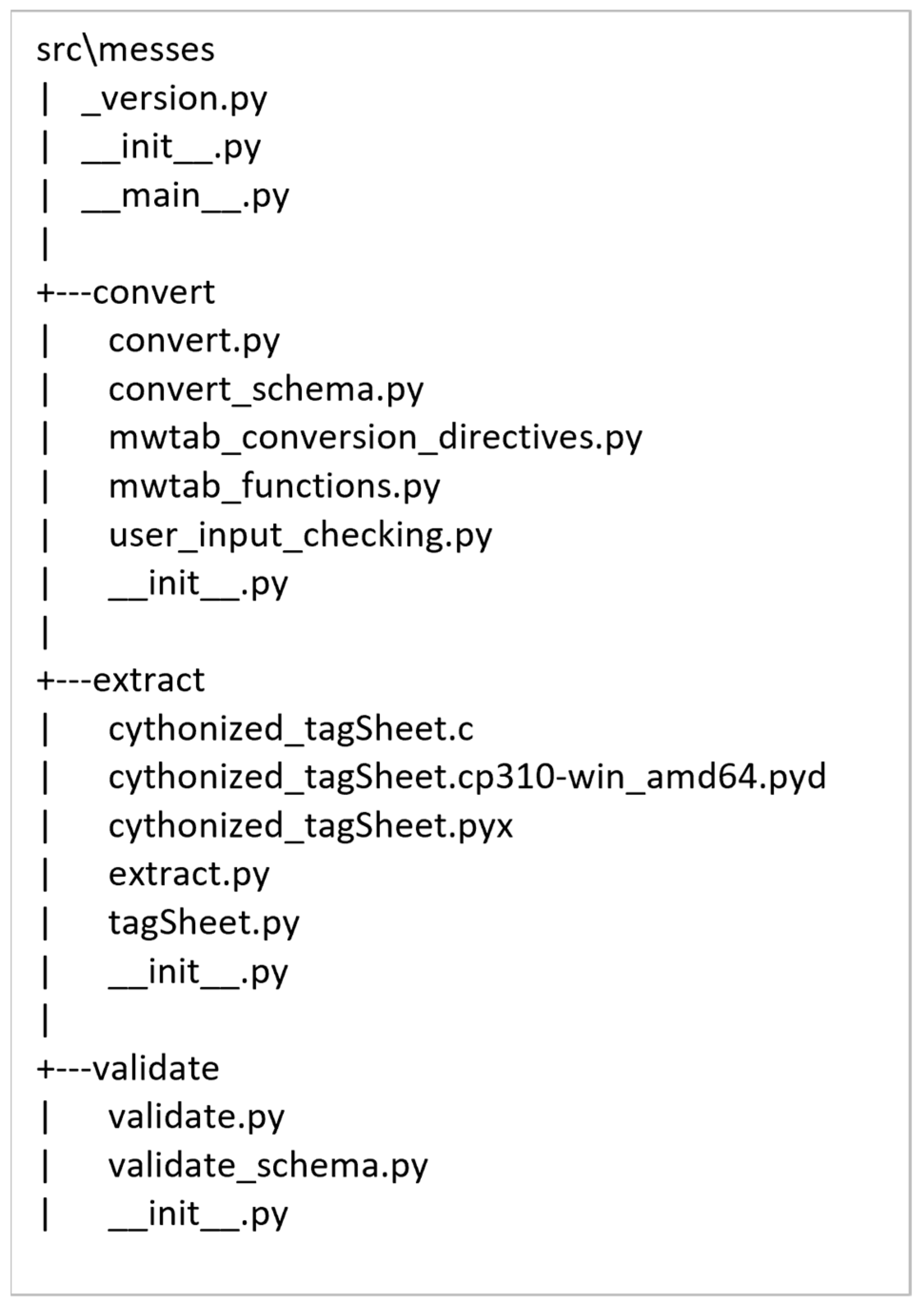






Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study ID | Title |
|---|---|
| ST000076 | A549 Cell Study |
| ST000110 | SIRM Analysis of human P493 cells under hypoxia in [U-13C/15N] labeled Glutamine medium (Both positive and ion mode FTMS) |
| ST000111 | Study of biological variation in PC9 cell culture |
| ST000113 | SIRM Analysis of human P493 cells under hypoxia in [U-13C/15N] labeled Glutamine medium (Positive ion mode FTMS) |
| ST000114 | SIRM Analysis of human P493 cells under hypoxia in [U-13C] labeled Glucose medium |
| ST000142 | H1299 13C-labeled Cell Study |
| ST000148 | A549 13C-labeled Cell Study |
| ST000367 | Distinctly perturbed metabolic networks underlie differential tumor tissue damages induced by immune modulator b-glucan in a two-case ex vivo non-small cell lung cancer study |
| ST000949 | Human NK vs. T cell metabolism using 13C-Glucose tracer (part I) |
| ST000950 | Human NK vs. T cell metabolism using 13C-Glucose tracer with/out galactose (part II) |
| ST000951 | Human NK vs. T cell metabolism using 13C-Glucose tracer with/out oligomycin (part III) |
| ST000952 | Human NK vs. T cell metabolism using 13C-Glucose tracer with/out oligomycin and galactose (part IV) |
| ST001044 | PGC1-A effect on TCA enzymes |
| ST001045 | FASN effect on HCT116 metabolism probed by 13C6-glucose tracer (part I) |
| ST001046 | FASN effect on HCT116 metabolism probed by 13C6-glucose tracer (part II) |
| ST001049 | P4HA1 knockdown in the breast cell line MDA231 (part I) |
| ST001050 | P4HA1 knockdown in the breast cell line MDA231 Gln metabolism (part II) |
| ST001129 | P4HA1 knockdown in the breast cell line MDA231 (part III) |
| ST001138 | P4HA1 knockdown in the breast cell line MDA231 Gln metabolism (part V) |
| ST001139 | P4HA1 knockdown in the breast cell line MDA231 Gln metabolism (part VI) |
| ST001445 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Colon NMR 1D |
| ST001446 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Colon NMR HSQC |
| ST001447 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Colon ICMS |
| ST001449 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Colon DI-FTMS |
| ST001453 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Liver ICMS |
| ST001455 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Liver NMR 1D |
| ST001456 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Liver NMR HSQC |
| ST001459 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Lung NMR 1D |
| ST001460 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Lung NMR HSQC |
| ST001461 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Plasma NMR 1D |
| ST001462 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Plasma NMR HSQC |
| ST001463 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Small Intenstines NMR 1D |
| ST001465 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Spleen NMR 1D |
| ST001466 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation Spleen—NMR HSQC |
| ST001469 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Lung DI-FTMS |
| ST001470 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Lung ICMS |
| ST001471 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Small Intestines DI-FTMS |
| ST001472 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Small Intestines ICMS |
| ST001473 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Spleen DI-FTMS |
| ST001474 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Spleen ICMS |
| ST001475 | Metabolomics of lung injury after allogeneic hematopoietic cell transplantation—Liver DI-FTMS |
References
- Carroll, M. National Academies of Sciences, Engineering, and Medicine, Open Science by Design: Realizing a Vision for 21st Century Research; The National Academies Press: Washington, DC, USA, 2018. [Google Scholar]
- Vicente-Saez, R.; Martinez-Fuentes, C. Open Science now: A systematic literature review for an integrated definition. J. Bus. Res. 2018, 88, 428–436. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Final NIH policy for data management and sharing. In NOT-OD-21-013. Vol NOT-OD-21-013. NIH Grants & Funding; National Institutes of Health: Bethesda, MD, USA, 2020.
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.D.; Moseley, H.N. The mwtab Python Library for RESTful Access and Enhanced Quality Control, Deposition, and Curation of the Metabolomics Workbench Data Repository. Metabolites 2021, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.D.; Moseley, H.N. The Metabolomics Workbench File Status Website: A Metadata Repository Promoting FAIR Principles of Metabolomics Data. bioRxiv 2022. [Google Scholar] [CrossRef]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P. MetaboLights—An open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2012, 41, D781–D786. [Google Scholar] [CrossRef] [PubMed]
- Salek, R.M.; Haug, K.; Conesa, P.; Hastings, J.; Williams, M.; Mahendraker, T.; Maguire, E.; Gonzalez-Beltran, A.N.; Rocca-Serra, P.; Sansone, S.-A. The MetaboLights repository: Curation challenges in metabolomics. Database 2013, 2013, bat029. [Google Scholar] [CrossRef] [PubMed]
- Docopt Python Library for Creating Command-Line Interfaces. Available online: http://docopt.readthedocs.io/en/latest/ (accessed on 1 January 2023).
- Pezoa, F.; Reutter, J.L.; Suarez, F.; Ugarte, M.; Vrgoč, D. Foundations of JSON schema. In Proceedings of the 25th International Conference on World Wide Web, Montreal, QC, Canada, 11–15 April 2016; pp. 263–273. [Google Scholar]
- Droettboom, M. Understanding JSON Schema. 2014. Available online: http://spacetelescope.github.io/understanding-jsonschema/UnderstandingJSONSchema.pdf (accessed on 1 January 2023).
- Open JS Foundation. 2019. Available online: https://openjsf.org/ (accessed on 1 January 2023).
- McKinney, W. Pandas: A foundational Python library for data analysis and statistics. Python High Perform. Sci. Comput. 2011, 14, 1–9. [Google Scholar]
- Oliphant, T.E. A Guide to NumPy; Trelgol Publishing: Spanish Fork, UT, USA, 2006; Volume 1. [Google Scholar]
- Gazoni, E.; Clark, C. openpyxl—A Python Library to Read/Write Excel 2010 xlsx/xlsm Files. 2016. Available online: http://openpyxl.readthedocs.org/en/default (accessed on 1 January 2023).
- Behnel, S.; Bradshaw, R.; Citro, C.; Dalcin, L.; Seljebotn, D.S.; Smith, K. Cython: The best of both worlds. Comput. Sci. Eng. 2011, 13, 31–39. [Google Scholar] [CrossRef]
- Smelter, A.; Moseley, H.N. A Python library for FAIRer access and deposition to the Metabolomics Workbench Data Repository. Metabolomics 2018, 14, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, G. Metabolomics of Lung Injury after Allogeneic Hematopoietic Cell Transplantation—Colon ICMS. Available online: https://www.metabolomicsworkbench.org/data/DRCCMetadata.php?Mode=Project&ProjectID=PR000993 (accessed on 1 January 2020).
- Organisation for Economic Co-Operation Development. Draft Advisory Document of the Working Group on Good Laboratory Practice on GLP Data Integrity. Available online: https://www.oecd.org/env/ehs/testing/DRAFT_OECD_Advisory_Document_on_GLP_Data_Integrity_07_August_2020.pdf (accessed on 1 January 2023).
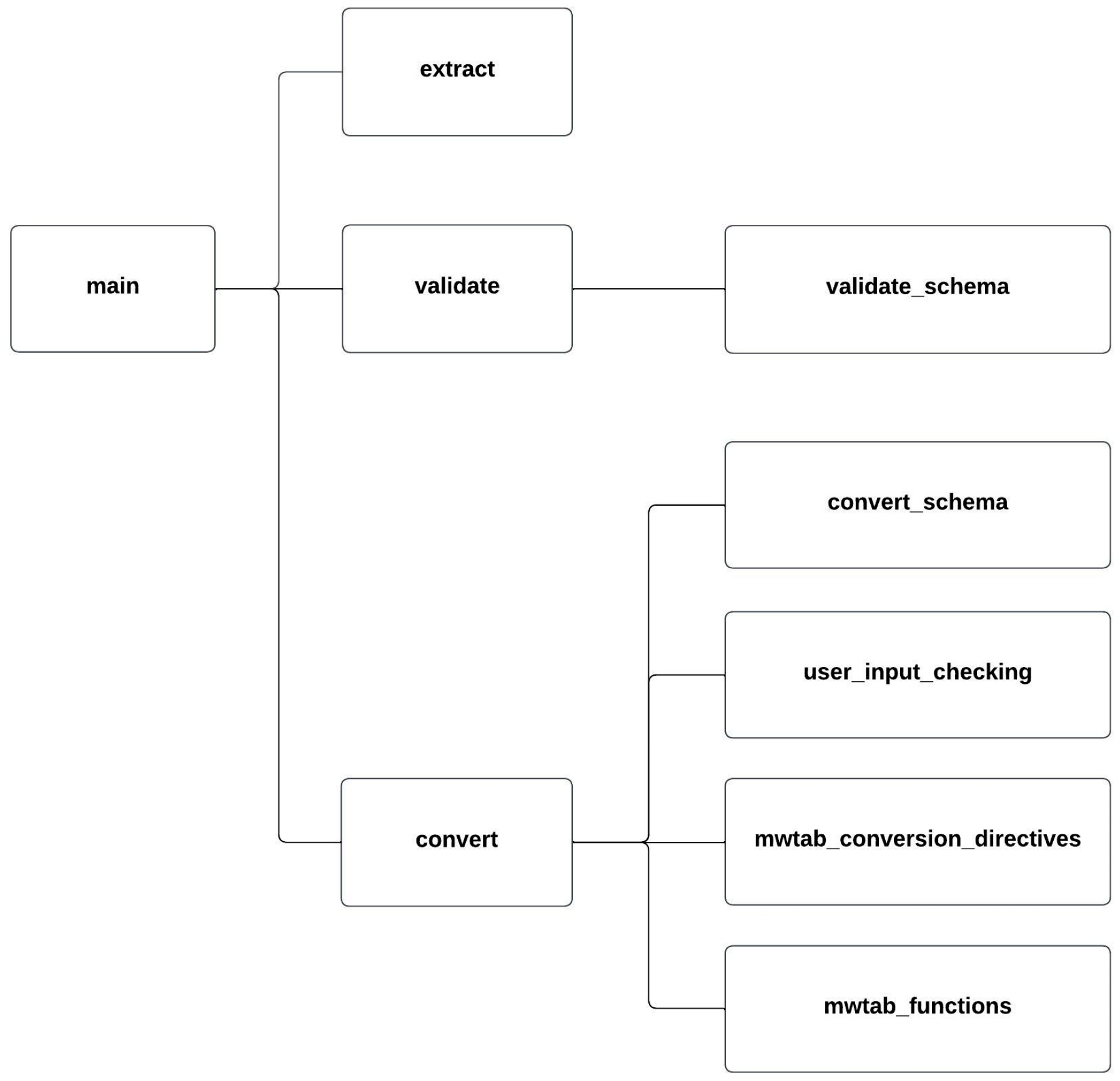

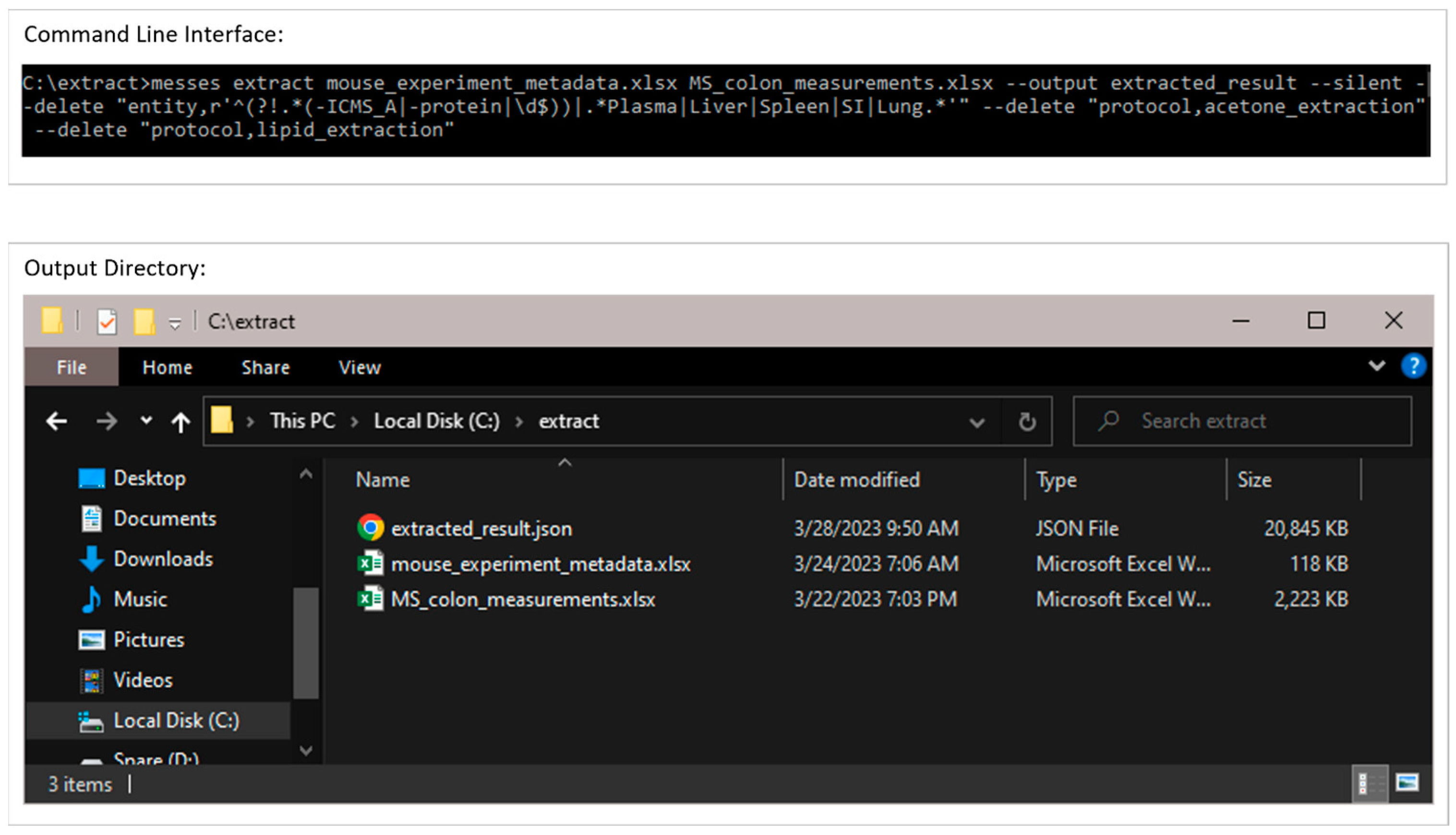

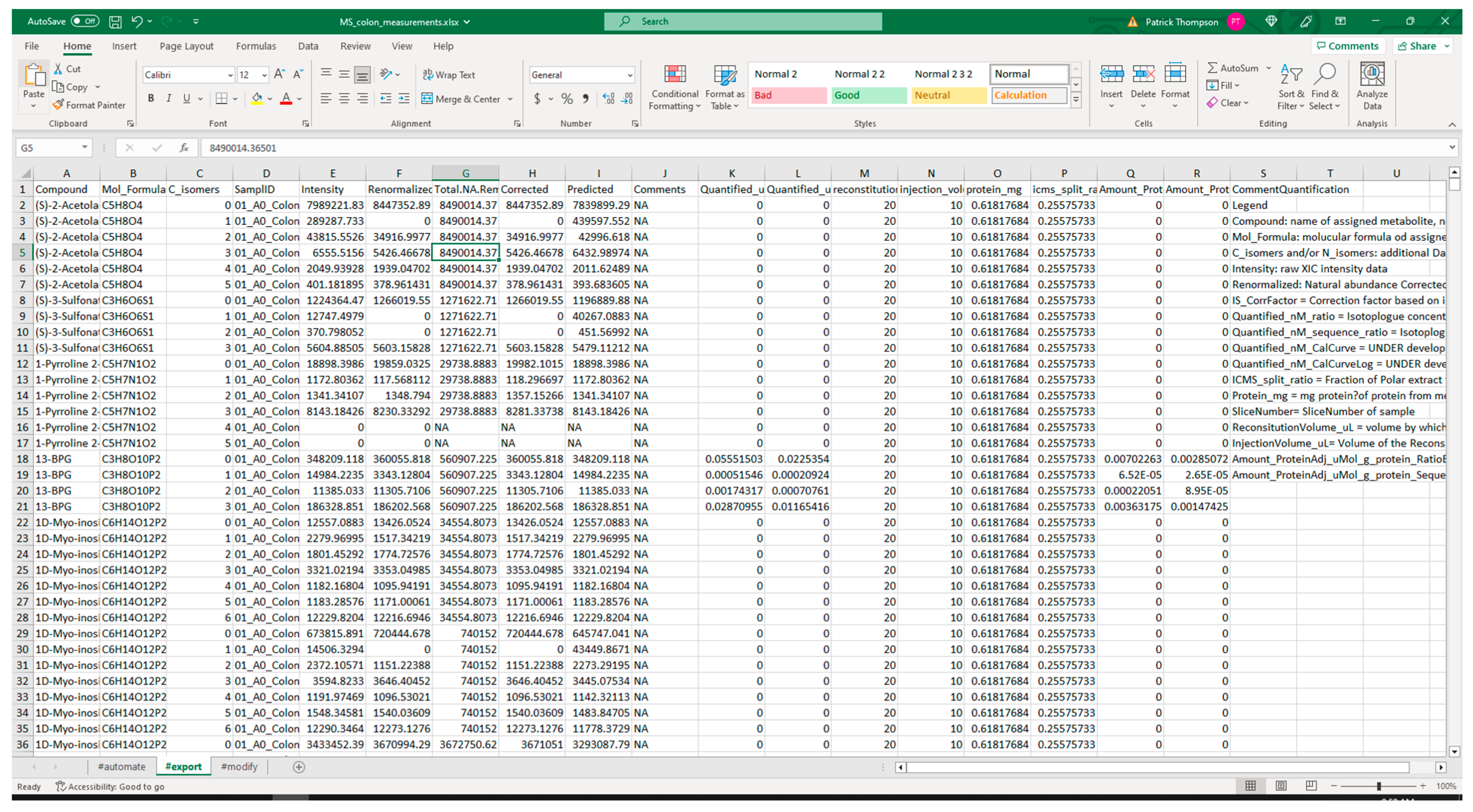
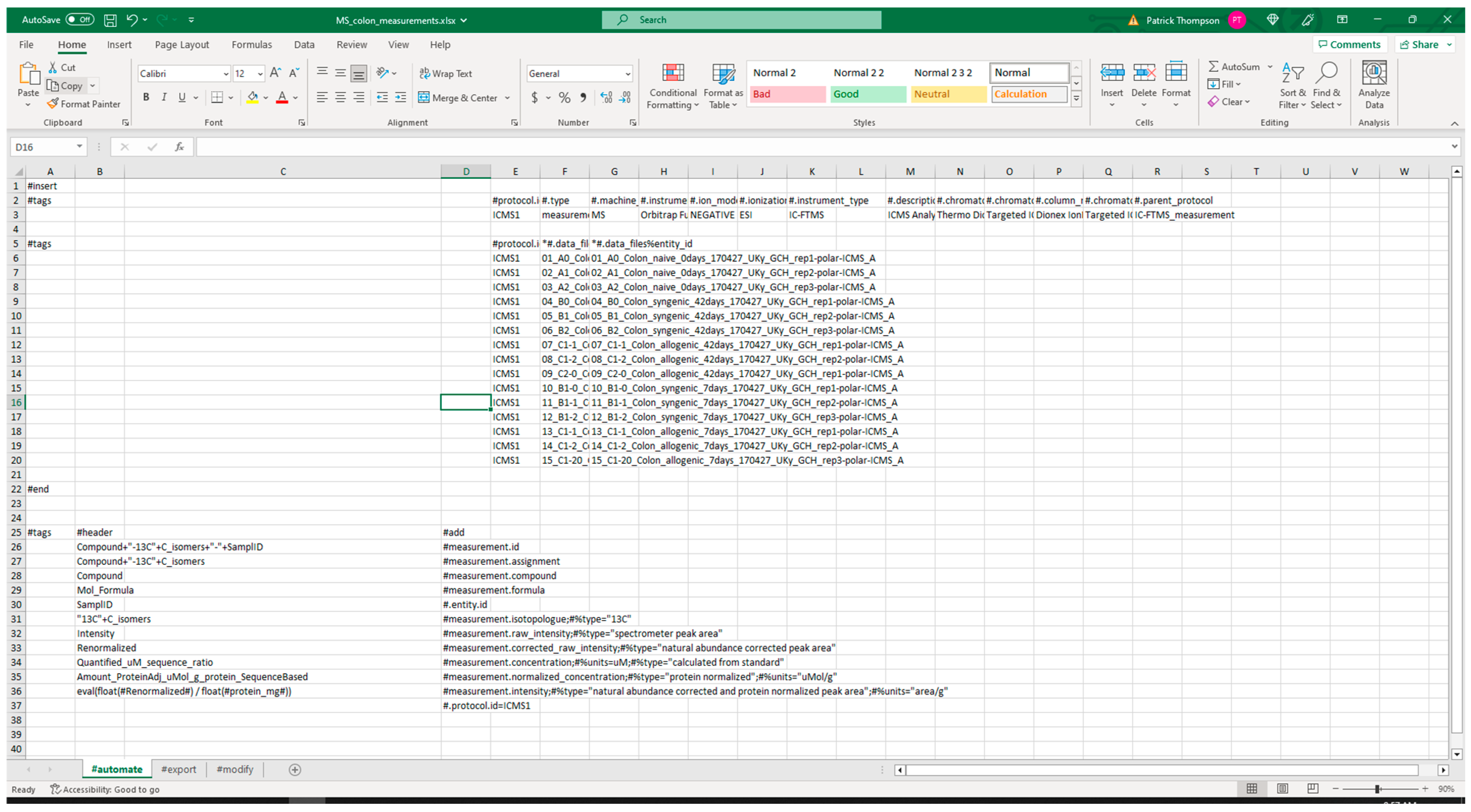
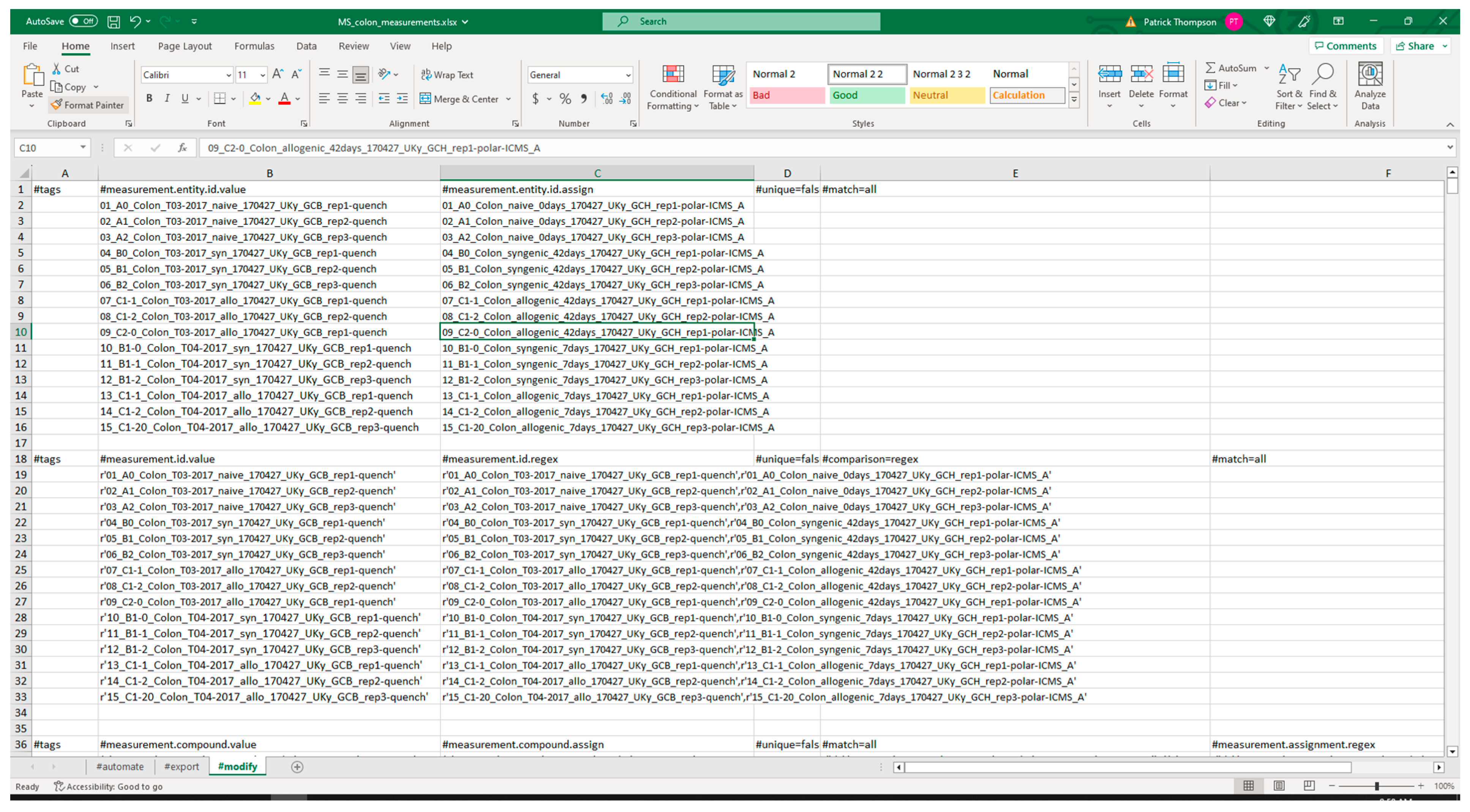





| Package | Version | Utilization | PyPI URL a |
|---|---|---|---|
| docopt | 0.6.2 | Implement CLI. | https://pypi.org/project/docopt/ |
| jsonschema | 3.0.1 | Validate JSON files. | https://pypi.org/project/jsonschema/ |
| pandas | 0.24.2 | Read and write tabular files. | https://pypi.org/project/pandas/ |
| numpy | 1.22.4 | Optimize tabular data algorithms. | https://pypi.org/project/numpy/ |
| openpyxl | 2.6.2 | Write Excel files. | https://pypi.org/project/openpyxl/ |
| xlsxwriter | 3.0.3 | Write Excel files. | https://pypi.org/project/xlsxwriter/ |
| jellyfish | 0.9.0 | Calculate Levenshtein distance. | https://pypi.org/project/jellyfish/ |
| Cython | 3.0.0a11 | Optimize algorithms. | https://pypi.org/project/Cython/ |
| mwtab | 1.2.5 | Create mwTab formatted files. | https://pypi.org/project/mwtab/ |
| Submodule | Description |
|---|---|
| __main__.py | Contains the top-most CLI. |
| extract.py | Implements the extract command and CLI. |
| cythonized_tagSheet.pyx | Cythonized version of the tagSheet method for extract. |
| validate.py | Implements the validate command and CLI. |
| validate_schema.py | Contains the JSON Schema schemas used by the validate command. |
| convert.py | Implements the convert command and CLI. |
| convert_schema.py | Contains the JSON Schema schemas used by the convert command. |
| user_input_checking.py | Validates conversion directives for the convert command. |
| mwtab_conversion_directives.py | Contains the built-in conversion directives for the mwTab format. |
| mwtab_functions.py | Contains functions specific to creating the mwTab format. |
| Table | Entry Description | Entry Information |
|---|---|---|
| project | A research project with multiple analytical datasets derived from one or more experimental designs. | PI Name PI Contact Information Institution Name Address Department Description Title |
| study | One experimental design or analytical experiment inside of the project. | PI Name PI Contact Information Institution Name Address Department Description Title |
| protocol | An operation or set of operations done on a subject or sample entity. | Description Type Instrument Settings Instrument Information Software Settings Software Information Data Files Generated File Detailing Protocol |
| entity | Either subjects or samples that were collected or experimented on. | Type Weight Sex Protocols Underwent Parent Entity Experimental Factor |
| measurement | The results acquired after putting a sample through an assay or analytical instrument such as a mass spectrometer or nuclear magnetic resonance spectrometer as well as any data calculation steps applied to raw measurements to generate usable processed results for downstream analysis. | Measurement Protocol Measurements Acquired Associated Entity Calculations or Statistics Labels Obtained from Measurements |
| factor | A controlled independent variable of the experimental design. Conditions set in the experiment. May be other classifications such as male or female gender. | Discrete Values of the Factor Units of the Values Name of Factor Field Name of Factor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, P.T.; Moseley, H.N.B. MESSES: Software for Transforming Messy Research Datasets into Clean Submissions to Metabolomics Workbench for Public Sharing. Metabolites 2023, 13, 842. https://doi.org/10.3390/metabo13070842
Thompson PT, Moseley HNB. MESSES: Software for Transforming Messy Research Datasets into Clean Submissions to Metabolomics Workbench for Public Sharing. Metabolites. 2023; 13(7):842. https://doi.org/10.3390/metabo13070842
Chicago/Turabian StyleThompson, P. Travis, and Hunter N. B. Moseley. 2023. "MESSES: Software for Transforming Messy Research Datasets into Clean Submissions to Metabolomics Workbench for Public Sharing" Metabolites 13, no. 7: 842. https://doi.org/10.3390/metabo13070842