1. Introduction
Thyroid hormones (THs) triiodothyronine (T3) and thyroxine (T4) regulate many important biological processes in the body, including the normal development of tissue, cellular metabolism, cell structure, and membrane transport [
1]. THs are synthesized and secreted from thyroid follicular cells in the thyroid gland either as the active T3 or in the inactive T4 form [
2]. After synthesis in the thyroid, T3 and T4 enter the bloodstream, reaching the different target organs, including the brain and liver. The conversion of T4 to the active T3 form is also facilitated in other tissue such as the kidney, brown adipose tissue, pituitary gland, liver, brain, and heart by 5′-deiodinase type 1 (D1) or 5′-deiodinase type 2 (D2) [
3], which are the primary sources of circulating T3. In the liver, T3/T4 can be metabolized species-specifically to either glucuronide or sulfate conjugates and then excreted through the bile duct into the intestine [
4]. T3 can also be deactivated by 5-deiodinase type 3 (D3) in the skin, vascular tissue, and placenta [
5].
The synthesis of THs is regulated by the thyroid-hypothalamus axis [
5]. Circulating T4 reaches the hypothalamus and is converted by D2 to the active T3, inhibiting the secretion of thyrotropin-releasing hormone (TRH). Conversely, TRH secretion can be activated by the signal transducer and activator of transcription STAT3, which is activated by phosphorylation stimulated by leptin, produced in peripheral fat tissue [
5]. The released TRH reaches the anterior pituitary gland stimulating the release of thyroid-stimulating hormone (TSH). TSH circulates to the thyroid, where it binds to the G-protein coupled with the TSH receptor, which mediates the expression of genes and functions involved in THs synthesis [
6], thereby increasing THs production and release.
Disturbances in TH homeostasis are linked to thyroid nodules, thyroiditis, hyperthyroidism, hypothyroidism, autoimmune thyroid disease, and thyroid cancer [
7]. Hypothyroidism, the overt deficiency of THs, can cause several symptoms, such as fatigue, lethargy, cold intolerance, weight gain, and even goiter and death if untreated [
2]. Hyperthyroidism is characterized by excessive production of thyroid hormones (THs) and is typically associated with symptoms such as weight loss, heat intolerance, anxiety, and muscle tremors [
8,
9].
Several drugs are known to influence the TH levels within the body. Propylthiouracil (PTU) directly affects the thyroid by inhibiting the thyroid peroxidase, and deiodinase type 1 is a commonly used antithyroid drug to treat hyperthyroidism [
3,
10]. The overt dosage of PTU administration leads to primary hypothyroidism. Phenytoin, a potent anticonvulsant used to treat and prevent generalized seizures, is a further drug influencing TH levels [
11]. It has been associated with hypothyroidism [
12], though it seems to function indirectly by not acting on the thyroid but by increasing the THs clearance in the liver [
13].
Researchers have made considerable efforts to elucidate the toxic effects of drug-induced hypothyroidism from various perspectives and methods. For example, Edgar et al. found that compared to thyroidectomy-induced hypothyroidism, methimazole-induced hypothyroidism, an antithyroid drug, can cause cellular damage in several organs, such as the liver, lung, and kidney [
14]. Meanwhile, Wu et al. used an untargeted metabolomics approach to identify 13 potential biomarkers related to sphingolipid metabolism, fatty acid transportation, phospholipid metabolism, and phenylalanine metabolism in rat serum after propylthiouracil administration [
15]. They also systematically investigated the urinary metabolic profiling in three classic hypothyroidism models, i.e., methimazole-, propylthiouracil-, and thyroidectomy-induced hypothyroidism, and identified several metabolites related to energy metabolism, amino acid metabolism, sphingolipid metabolism, and purine metabolism [
16]. In another study, Hennicke et al. conducted a metabolomics study on phenytoin-administered rat plasma samples. They found that phenytoin-induced toxicity can cause liver enzyme induction and liver toxicity, even though liver histopathology revealed slight centrilobular hepatocellular hypertrophy [
17]. However, no systematic metabolomics studies have been carried out at the organ level, particularly in the thyroid gland and the liver. Changes in the molecular metabolome level induced by alterations in TH levels facilitated by PTU and phenytoin treatment are still not fully understood. Therefore, we investigated the influence of the two drugs PTU and phenytoin on the metabolome of the different organs involved in regulating TH levels. Specifically, we investigated the metabolomes from the thyroid tissue, where T3 and T4 are synthesized; the liver tissue, where THs are metabolized before excretion; and plasma, where systemic effects on the metabolome can be investigated. In addition, we investigated these metabolic changes induced by PTU and phenytoin administration in a dose- and time-dependent manner. The selection of the drug dosage administered was based on previous studies [
17,
18,
19,
20,
21], with low doses known to induce toxic effects while high doses are known to cause known severe toxicity. Finally, we aimed at determining if any metabolic changes were reversible after ceasing administration of the drugs. The study design facilitated identifying metabolic changes in a concentration and time-dependent manner and which of the detected metabolic changes were reversible.
2. Materials and Methods
2.2. Substances Administration and Samples Collection
The rat strain RjHan:WI was used. Animals were supplied from Janvier Labs, CS 4105 Le Genest-Saint-Isle, F-53941 Saint-Berthevin Cedex. The study was conducted using three subsets, A, B, and C, which represented three planned exposure groups: short-term (2 weeks), long-term (4 weeks), and long-term (4 weeks) with a recovery period (2 weeks). The age at the start of the administration period was 16 weeks (subset A), 14 weeks (subset B), and 12 weeks (subset C). The animals were allocated to the test groups before the start of the administration period based on their weight. The weight variation of the animals did not exceed 20 percent of the mean weight. At the beginning of the administration period, the male animals had a mean weight range of 477.0 to 502.6 g. Each test group consisted of 10 male animals (with 5 animals per cage). Test substances PTU (6-Propyl-2-thiouracil, Sigma-Aldrich, Taufkirchen, Germany) and phenytoin (Sigma-Aldrich, Taufkirchen, Germany) were weighed and mixed with a small amount of food in a laboratory mixer and reached the desired concentration by further mixing with the food.
In the present study, 300 ppm and 2400 ppm were chosen as the low and high doses for phenytoin, whereas 5 ppm and 50 ppm were selected as the low and high doses for PTU. The substance intake was monitored weekly to calculate the mean daily intake and recorded as test substance intake (mg/kg/day). Food intake data were collected for each group since the animals were housed together. To obtain this measure, the following formula was used: Daily food intake (g/day) × nominal substance concentration in diet (mg test item/g vehicle) ÷ Average body weight during the food consumption interval (kg). The calculated daily intake of PTU in subsets A (2 weeks), B (4 weeks), and C (6 weeks) were 0.2 and 2.2 mg/kg/day for low and high-dose groups, respectively. For phenytoin, the intake was 15 mg/kg/day in the low-dose groups in subsets A, B, and C. In the high-dose groups of subsets A, B, and C, the intake was 96, 111, and 121 mg/kg/day, respectively.
Body weight was determined before the administration period to randomize the animals into groups. During the administration period, the body weight was determined on day 0 (the start of the administration period) and after that at weekly intervals.
In the morning (08:00–10:00 h), blood samples were taken from fasted animals by puncturing the retrobulbar venous plexus under isoflurane anesthesia (3.5–4% Isoflurane in air with a flow rate of 160 L/h). Anesthesia was confirmed by a lack of eyelid reflex, lack of intertoe reflex, and flat respiration. Blood sampling and examination were carried out in a randomized sequence (with randomization being compiled with a computer). From each animal, 1 mL of blood was collected with either EDTA-K3 (potassium salt) as an anticoagulant (10 μL of a 10% solution) for plasma metabolome profiling or EDTA-free for serum TSH/T4/T3 concentration measurement. The samples were centrifuged at 14,000× g under 4 °C, and the plasma was separated. The blood was sampled and prepared in original 2 mL Eppendorf tubes. The preparation of the samples was conducted under cooling. All samples were stored covered with an N2 atmosphere at −80 °C. Blood samples were taken on 2 consecutive days sampling 5 animals per group and day for subset A on study days 15/16, subset B on study days 28/29, and subset C on study days 42/43.
The serum concentrations of TSH were determined by direct, competitive radioimmunoassay (RIA), using rat-specific TSH RIA kits (Institute of Isotopes Co., Ltd., Budapest, Hungary, cat.no. RK-554) and a Gamma-Counter (LB 2111, Berthold, Germany) and evaluated with the LBIS501 (version: 4.00) software of the instrument producer. The TSH control kit used is called Control Set rTSH (Trilevel; cat-no RK-554Cset, Izotop, Budapest, Hungary). An automatic smoothed cubic Spline curve fit method was applied (smoothing factor 0.125). Acceptance criteria for the standard curve were a total count of >10,000 cpm and a non-specific binding of <5%. The lowest quantifiable value (LOQ) is 1 μg/L. Values above the linear range (20.80. intercept of the curve) were repeated with diluted samples. At least two levels of quality control were applied in each test run. Intra- test-run and between-test-run precision of TSH values in the range of 3.7–33.5 μg/L were below 16.3 (% coefficient of variation, values from kit provider).
T4 ELISA kits (DRG Instruments, Marburg, Germany, cat.no. EIA-4568) were measured with a Sunrise MTP-reader (Tecan AG, Maennedorf, Switzerland) and evaluated with the Magellan-Software (version: 7.3) of the instrument producer. Bilevel controls were provided within the test kits. A Four-Parameter-Marquardt curve fit was applied. Acceptance criteria for the standard curve were a maximum OD of at least 1.5 and a curve fit correlation of r = 0.999. The linear range of the standard curve is defined as a 10–90 intercept of the curve. Values above the range were repeated with diluted samples. The lowest quantifiable value (LOQ) is 15 nmol/L (7.5 nmol/L for fetuses and PND4 pups). Two levels of quality control were applied on each microtiter plate. Intra-test run and between-test run precision of T4 values in the range of 23–179 nmol/L were below 12.0 (% coefficient of variation, values from in-house validation).
T3 ELISA kits (Diagnostic Biochem Canada Inc., London, Ontario, Canada, cat.no. CAN-T3-4220) were measured with a Sunrise MTP-reader (Tecan AG, Maennedorf, Switzerland) and evaluated with the Magellan-Software (version: 7.3) of the instrument producer. Bilevel controls were provided within the test kits. A Four-Parameter-Marquardt curve fit was applied. Acceptance criteria for the standard curve were a maximum OD of at least 1.2 and a curve fit correlation of r = 0.999. The linear range of the standard curve is defined as a 10–90 intercept of the curve. Values above the range were repeated with diluted samples. The lowest quantifiable value (LOQ) is 0.3 nmol/L. Two levels of quality control were applied on each microtiter plate. Intra-test run and between-test run precision of T3 values in the range of 1.4-2.2 nmol/L were below 11.3 (% coefficient of variation, values from in-house validation).
All animals were sacrificed by decapitation under isoflurane anesthesia (as described above). The exsanguinated animals were necropsied, i.e., a post-mortem examination took place by opening the body and assessing visually (gross necropsy). Organs were removed and weighted before fixation. The liver and left thyroid gland were preserved in a 4% neutral buffered formaldehyde solution and embedded in paraffin. Sections of 4 μm were stained with hematoxylin and eosin (H&E) and examined by light microscopy [
22]. From the right thyroid gland (with parathyroid gland), serial sections were prepared with the cryostat and split alternating in 3 separate samples for the different omics experiments.
2.3. Targeted Metabolomics for Plasma Samples
The standardized protocol for mass spectrometry-based targeted metabolite profiling of blood plasma samples is described below [
23,
24]. All 10 harvested replicates from each treatment group were input into plasma metabolomics experiments.
Targeted metabolites were measured from 60 μL rat plasma by extracting with a mixture of methanol, dichloromethane, water, and toluene (93:47:16,5:1, v/v) buffered with ammonium acetate using a ball mill ( OMNI INTERNATIONAL, 935 Cobb Place Blvd, Kennesaw, GA, USA). Internal standards were added to the extraction mixture to enhance reproducible analysis. After centrifugation (12,000 rpm 10 min@ 12 °C), an aliquot of the extract was subjected to LC-MS/MS analysis using reversed-phase and hydrophilic interaction liquid chromatography (ZIC—HILIC, 2.1 × 100 mm, 3.5 μm, Supelco, Merck KGaA, Darmstadt, Germany) followed by MS/MS detection (AB Sciex QTrap 6500+) using the positive and negative ionization mode. For reverse-phase high-performance liquid chromatography (RP-HPLC, Ascentis Express C18, 5 cm × 2.1 mm, 2.7 μm Supelco), gradient elution was performed with water/methanol/0.1 M ammonium formate (1:1:0.02 w/w) and methyl-tert-butylether/2-propanol/methanol/0.1 M ammonium formate (2:1:0.5:0.035 w/w) with 0.5% (w%) formic acid (0 min 100% A, 0.5 min 75% A, 5.9 min 10% A). HILIC gradient elution was performed with acetonitrile with 1 vol-% water and 0.2 vol-% acetic acid (A) and 0.007 M ammonium acetate with 0.2 vol-% acetic acid (B) (0 min 100% A, 5 min 10% A, 600 μL/min).
A second aliquot of the extract was mixed with water (3.75:1,
v/
v), resulting in phase separation. Both phases were analyzed with gas chromatography-mass spectrometry (GC7890-5975 MSD, Agilent Technologies, Santa Clara, CA, USA) after derivatization, as described in Grossmann et al. [
25]. Briefly, the non-polar fraction was evaporated to dryness and treated with a mixture of 130 μL of dichloromethane, 40 μL of hydrochloric acid, 310 μL of methanol, and 20 μL toluol followed by heating (2 h, 100 °C) and evaporation to dryness. The polar and non-polar fractions were further derivatized with O-methyl-hydroxylamine hydrochloride (20 mg/mL in pyridine, 50 μL for 1.5 h at 60 °C) to convert oxo-groups to O-methylomes. Subsequently, 50 μL of the silylating agent
N-methyl-
N-(trimethylsilyl)-2,2,2-trifluoroacetamide (MSTFA), and 10 μL internal standard mix was added, and the derivatization was carried out for 30 min at 60 °C. The polar fraction was chromatographed on a DB-XLB column (30 m × 0.25 mm × 0.25 μm) using an oven temperature program (70 °C for 2 min, 50 °C/min up to 100 °C, 8 °C/min up to 200 °C, 14 °C/min up to 340 °C for 5 min, 100 °C/min down to 70 °C). The non-polar fraction was chromatographed on an HP-5 MS column (30 m × 0.25 mm × 0.25 μm) using an oven temperature program (70 °C for 3 min, 50 °C/min up to 130 °C, 10 °C/min up to 340 °C for 9 min, 120 °C/min down to 70 °C).
All the samples were analyzed once in a randomized analytical sequence design to avoid artificial results concerning analytical shifts. For GC-MS and LC-MS/MS profiling, data were normalized to the median of reference samples (“rat-ultra pool”). The rat-ultra pool reference material is used as standard in BASF rat plasma studies. It was generated by pooling blood plasma from a large number of animals. This material was aliquoted and freeze-dried, and aliquots are measured in parallel to the study samples to serve as a standard reference for inter- and intra-instrumental variation. A total of 495 metabolites was analyzed using the single peak signal of the respective metabolite and a normalization strategy according to the patent WO2007012643A1 [
26], resulting in ratio values representing the metabolite change in treated versus control animals. Of those 495 analyzed metabolites, 454 are chemically identified, and 41 are structurally unknown. It should be noted that only 420 unique metabolites were considered for further data analysis.
Steroid hormones, catecholamines, and their metabolites were measured from separate plasma aliquots, 50 μL each, by online SPE-LC–MS/MS (Solid phase extraction-LC–MS/MS, Symbiosis Pharma Pro Online SPE UHPLC System, Spark Holland) in positive electrospray mode using stable-isotope-labeled internal standards [
27,
28].
Before steroid analysis, the samples were extracted with 500 μL MTBE (methyl t-butyl ether). After evaporation, the residue was derivatized by dissolving it in 80 μL of acetonitrile/water (9:1) with ultrasonication for 3 min, adding 40 μL of dansyl chloride (1 mg/mL in acetonitrile) and 40 μL of NaHCO3 (0.2 mol/L), then shaking for 20 min at 60 °C at 400 rpm (Thermomixer) and adding 75 μL of water after cooling it to ambient temperature. Chromatography was performed with a YMC Triart C18 ExRs plus 50 × 2.1 mm × 3 μm HPLC Column at 30 °C using 5% acetonitrile in water with 0.1% formic acid (w/w/w) (A) and acetonitrile with 0.1% formic acid (w/w) (B) (0 min 5% B, 5.5 min 65% B, 8 min 100% B, 9.5 min 100% B, 600 μL/min).
For catecholamine analysis, 50 μL of 0.01 M HCl (internal standard solution) was added, and proteins were removed by membrane filter tube centrifugation for 45 min at 13,000 rpm, 4 °C. An amount of 50 μL of each of NaHCO3 (0.2 mol/L), dansyl chloride (2 mg/mL in acetone), and methanol, was added to the filtrate and kept for 12 h at 4 °C before analysis. Chromatography was performed with an ODS 7 pH, 60 mm × 2 mm, 4 μm HPLC Column (Alltech Grom) at 30 °C using 5% acetonitrile in water with 0.5% formic acid (w/w/w) (A) and acetonitrile with 0.5% formic acid (w/w) (B) (0 min 40% B, 4 min 52% B, 4.17 min 70% B, 7 min 75% B, 11.67 min 90% B, 12 min 90% B, 300 μL/min).
2.5. Targeted Metabolomics for Thyroid/liver Samples
The targeted metabolites quantification was conducted by using the MxP Quant 500 kit (Biocrates Life Science, Innsbruck, Austria), which includes the absolute quantification of 630 metabolites, covering 26 biochemical classes.
Further sample processing steps and measurement tasks were executed following the manufacturer’s instructions. Briefly, the sample preparation was done on the provided 96-well plate. A total of 7 calibration standard levels and 3 different quality control standards (QC1, QC2, QC3) were part of the Kit. Additional 4 QC2 samples were distributed over the plate. All other remaining wells were left for the current project samples. The samples were added to an adsorption pad system in the 96-well plate and dried for 30 min under nitrogen flow. Followed by derivatization using 50 μL 5% PITC (phenylisothiocyanate) for 60 min before 60 min drying under nitrogen flow, 300 μL extraction solvent (5 mM ammonium acetate in methanol) was added and shaken for 30 min. The samples were then eluted into the lower well by centrifugation for 30 min at 450 rpm. The extracted elutes were separated into two parts and diluted for FIA and LC measurement.
The metabolite measurements were processed in the QTRAP 5500 system (AB SCIEX) equipped with the LC 1290 Infinity system (Agilent). As for the FIA part, neither oven temperature nor columns were used, and only solvent B (290 mL methanol + 1 ampule FIA Mobile Phase Additive) was used for the injection. The flow rate was stabilized at 0.03 mL/min for 1.6 min, increased to 0.2 mL/min for 0.8 min, maintained for 0.2 min, and then decreased down to 0.03 mL/min in 0.2 min. The compound-specific MRM transitions were determined in positive ionization mode. For the LC part, an MxP Quan 500 Column System (Biocrates Life Science) was equipped for use and heated by setting the oven temperature to 50 °C.
The substance analysis via specific MRM transitions and LC was split into one part measured in positive (LC1) and another in negative ionization mode (LC2). Solvent A (2000 mL water + 4 mL formic acid) and solvent B (2000 mL acetonitrile + 4 mL formic acid) were used as eluents. Gradient LC1: 0–0.25 min (0.8 mL/min, 0% B), 0.25–1.5 min (0.8 mL/min; 0–12% B), 1.5–2.7 min (0.8 mL/min; 12–17.5% B), 2.7–4 min (0.8 mL/min; 17.5–50% B), 4–4.5 min (0.8 mL/min; 50–100% B), 4.5–4.7 min (0.8–1 mL/min; 100% B), 4.7–5 min (1 mL/min; 100% B), 5–5.1 min (1 mL/min; 100–0% B), 5.1–5.8 min (1–0.8 mL/min; 0% B). Gradient LC2: 0–0.25 min (0.8 mL/min, 0% B), 0.25–0.5 min (0.8 mL/min; 0–25% B), 0.5–2 min (0.8 mL/min; 25–50% B), 2–3 min (0.8 mL/min; 50–75% B), 3–3.5 min (0.8 mL/min; 75–100% B), 3.5–4.7 min (0.8–1 mL/min; 100% B), 4.7–5 min (1 mL/min; 100% B), 5–5.1 min (1 mL/min; 100–0% B), 5.1–5.8 min (1–0.8 mL/min; 0% B). For quantification and quality assurance, the MetIDQ software was used according to the manufacturer’s instructions.
4. Discussion
Several studies have investigated the mechanism of hypothyroidism through different experimental designs. These included human plasma [
29], as well as rat models [
15,
16,
30]. A number of drugs like methimazole [
16], PTU [
15,
16,
30], and phenytoin [
31] have been tested in adopted dosages [
15] and exposure courses [
15,
32]. This study, in comparison, was designed to follow metabolomics alterations in a dose- and time-dependent rat model that was exposed to two different drugs, namely PTU, which directly influences TH synthesis in the thyroid [
10], and phenytoin, which influences the excretion of THs from the body [
13]. In this study, both PTU and phenytoin altered the TH levels and affected body weight. Although we observed large changes in the plasma metabolome induced by PTU and phenytoin, on the tissue level, we only observed changes in the metabolome in the thyroid during high-dose exposure to PTU. Nevertheless, the findings of this study should enhance the understanding of how the metabolome is affected by these drugs, and the additional two weeks of recovery should highlight changes in the metabolome are reversible.
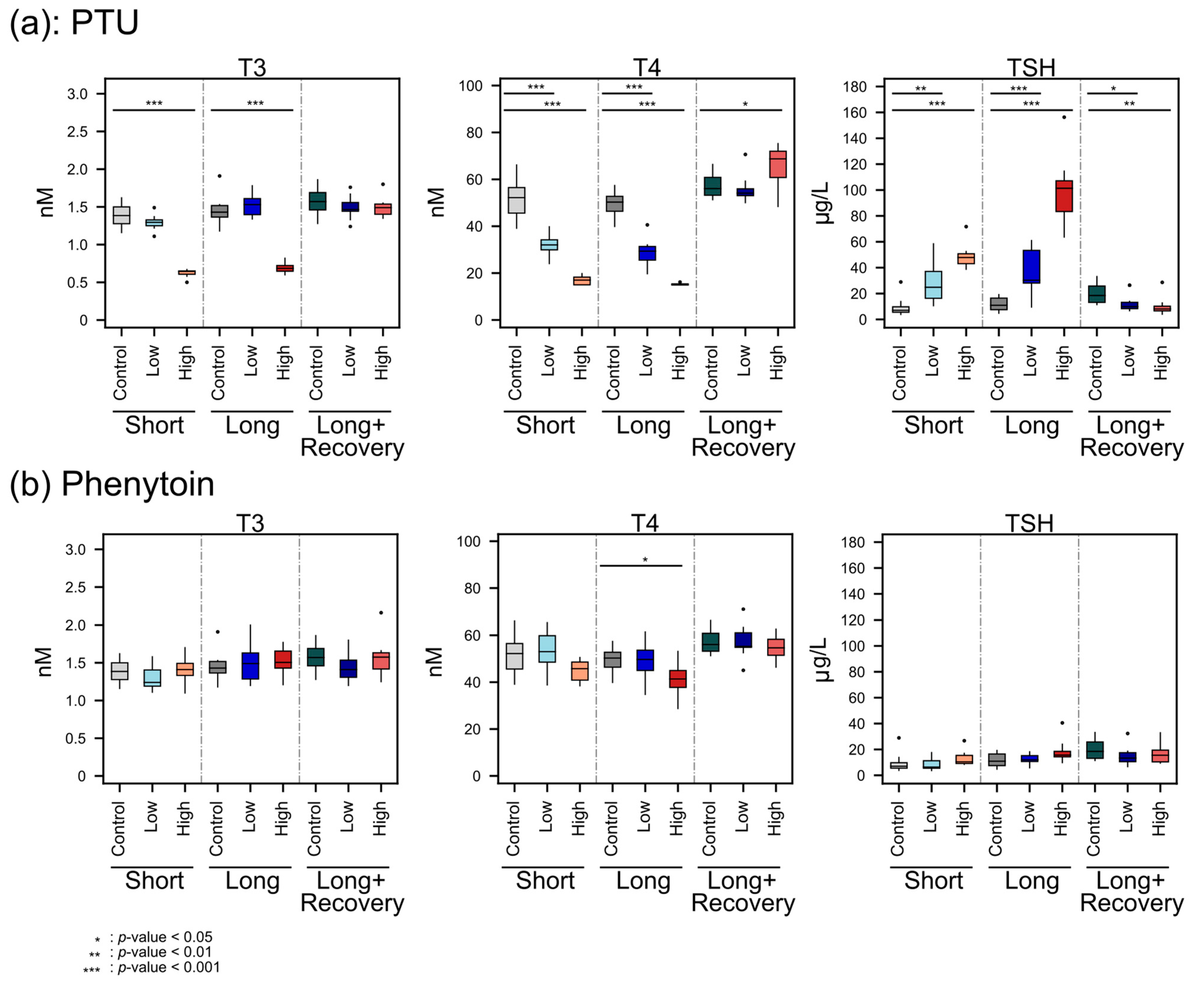
The decreasing T4 concentrations with the higher PTU dose administration indicated the hypothyroid effect of PTU, which may have facilitated a change in energy metabolism and, therefore, a change in body weight. PTU is known to reduce TH levels by inhibiting thyroid TPO and thereby blocking thyroid hormone synthesis, as well as inhibiting the conversion of T4 to T3 by deiodinase type 1 (D1) in peripheral tissues, which is the predominate source of T3 [
3,
10]. However, T3 plasma levels were not decreased during low-dose administration of PTU, possibly because PTU concentrations may have been too low in peripheral tissue. A previous study also reported no effect of PTU on T3 blood levels in low concentrations (0.1 mg/kg/day vs 0.2 mg/kg/day in our study) [
32]. However, when a high dose of PTU was administered, T3 decreased significantly, which is also in line with the findings of O’Connor et al. [
32].
Furthermore, the TH feedback regulation was observed as expected since, with lower THs concentrations, significantly higher TSH concentrations were detected. Moreover, after the recovery phase, the TSH concentration was even lower than the controls. This might be explained by the sensitive negative feedback regulation, where the T4 level significantly increased. Furthermore, the smaller weight gain of high-dose PTU-administered animals compared to the untreated control animal was also in line with previous studies on rats [
33,
34].
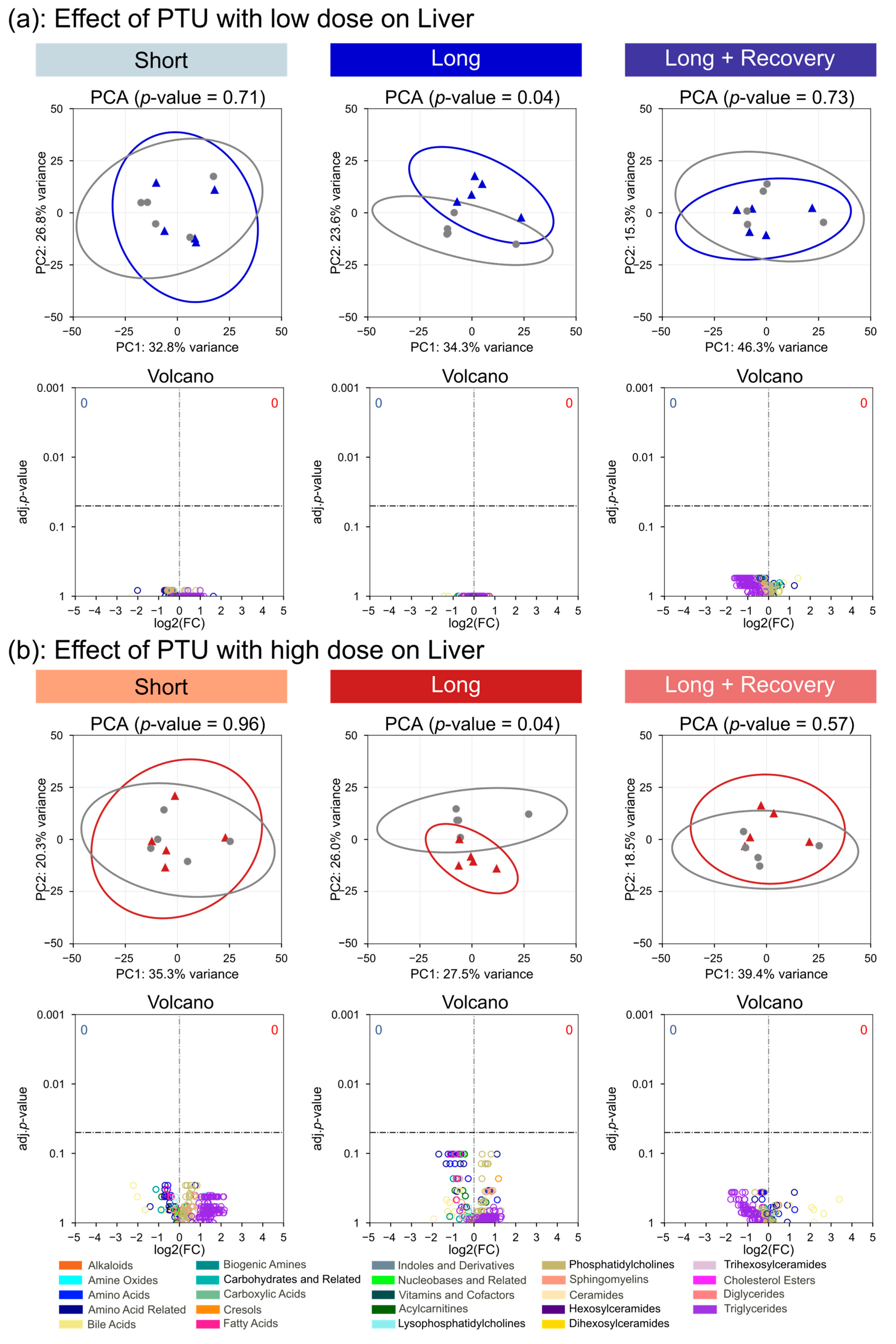
The plasma metabolome of the rats only changed significantly after high-dose administration of PTU, suggesting that the low-dose administration of PTU had little effect on the thyroid. This was further seen for the metabolomes of the thyroid tissue and liver, where the low dose of PTU had no significant impact on metabolite concentrations. In contrast, the changes in metabolome for high-dose PTU administration were already considerable after short-term exposure and were further observed after long-term exposure. Interestingly we observed decreasing triglycerides and acylcarnitines concentrations in the plasma. Decreases in triglyceride blood concentrations have been previously observed in rats [
21,
34,
35,
36], which contrasts with observations in humans [
36]. Triglycerides are important energy sources, and in blood, triglycerides enable the bidirectional transfer of adipose fat and glucose from the liver [
37]. It is widely known that THs regulate fatty acid transportation and metabolism. Lower blood triglyceride levels are probably caused by lower activity of lipoprotein lipases [
38]. Adipose lipoprotein lipases are increased in rats deficient in thyroid hormone [
39]. In humans, T4 levels were associated with acylcarnitine concentrations in plasma, suggesting that T4 could influence
β-oxidation and cellular fatty acid transport [
40].
Furthermore, in the study, an increase in phosphatidylcholines was observed. Our findings revealed plasma lower T4 levels with a decrease in acylcarnitines and an increase in phosphatidylcholines, including lysophosphatidylcholines and sphingomyelins, matching the previously published results. Interestingly, in the thyroid tissue at short-term exposure to the high dose of PTU, the decrease in many triglycerides and an increase in many phosphatidylcholines also suggested the same mechanistic hypothesis. In addition, we observed at short-term high PTU administration higher concentration of carnitine, which could hint at lower levels of carnitine being loaded with acyl moieties, resulting in a decrease in
β-oxidation activity. Though, there seemed to be an adaptation of the thyroid tissue to the lower levels of THs, since, after long-term exposure to high-dose PTU, no lipids in the organ tissue were significantly altered in concentration compared to the control animals. A further interesting finding in the thyroid was the increase in the concentration of the two fatty acids, arachidonic acid and eicosapentaenoic acid, with known metabolic signaling functions. Contrary to most thyroid metabolomes, both these fatty acids were seen at higher concentrations at short-term, high-dose PTU exposure. Especially, eicosapentaenoic acid has been found to decrease cardiometabolic disease risk and levels of circulating triglycerides [
41]. This would suggest that the higher eicosapentaenoic acid in the thyroid might directly contribute to the lower TGs concentrations in plasma and thyroid after high-dose PTU treatment.
Amino acids also exhibited significant changes in concentration when exposed to high-dose PTU. Interestingly, all the amino acids with a significant change were downregulated. This was observed in the plasma already after short-term administration of PTU and was further seen after long-term exposure in the plasma and the liver. One study described a decrease in glutamate during hypothyroidism, which we observed as well, and an increase in tyrosine, tryptophan as well as arginine [
21]. In our study, arginine, tryptophan, and tyrosine were not significantly changed in concentration in the plasma during short and long-term administration of PTU. THs are known to control lipid metabolism, and TH levels are lowered during hypothyroidism, possibly indicating that amino acids are increasingly used as an energy source to compensate for the decrease in lipid utilization through
β-oxidation. Hence, this could lead to the observed decreasing amino acid concentrations. Another potential reason is that amino acids are not being released to the same level from endogenous proteins, which was previously reported in a study investigating rat skeletal muscle [
42], where a lack of THs led to reduced protein breakdown. A study with human participants [
43], however, did not find any associations between T4 hormone levels and amino acid concentrations in plasma. However, T3 levels were not analyzed. Potential support for this hypothesis was provided by a study where hyperthyroidism was induced through the application of T3 in rats [
44]. Therein, Short et al. showed that elevated T3 levels lead to an increased turnover of proteins, including both their synthesis and breakdown. In our study, only in the PTU-treated animals did we observe a decrease in amino acids. Phenytoin administration did not alter T3 levels in the plasma. Taken together, this implies that in our study, the decrease in amino acids was probably associated with a decrease in T3 and not T4.
Phenytoin administration only led to significant changes in plasma T4 levels at high doses, though the effect size was a lot smaller than that for PTU. Neither the plasma concentrations for T3 nor for TSH were significantly altered, which may explain the rather scarce effect on the metabolomes of the thyroid, plasma, and liver, respectively. A previous study found significantly decreased levels of T4 and increased levels of TSH in human serum samples from patients treated long-term with phenytoin [
45]. The T4 findings fit our results. However, we did not observe an increase in TSH. This could be because the other study involved human patients.
Interestingly, after the recovery phase, the metabolomes of the thyroid, liver, and plasma were generally indistinguishable from controls for both drugs tested at high and low doses, suggesting that the observed effects were reversible.
The current study provides valuable results to clarify the effects of drug-induced hypothyroidism on molecular perturbations on the metabolome in plasma, thyroid, and liver tissue in a rat model system. The data were collected using a rat model system. Even though the rat is a proven and accepted model organism, there are species-specific differences that do not allow for a one-to-one transferability of the results to humans, as already mentioned above. The study did not measure the internal exposure levels of the two drugs, though estimates were made based on the calculated drug intake by rats. The tissue metabolome was measured using the MxP Quant 500 kit. However, there is no previous evidence to support the use of this kit on tissue samples, despite the manufacturer’s claim that it is suitable for serum, plasma, urine, and feces. The presented study focuses solely on a metabolomics-based view and given the surprisingly marginal effects that have been observed in most treatment groups, we assume that an additional transcriptomics or proteomics layer would enhance the study, i.e., to evaluate any adaptation in gene expression or protein abundance to counter any metabolic changes from the drugs. The investigation of metabolic fluxes might also be highly beneficial in order to analyze how these drugs affect metabolite turnover rates. This could be performed using stable isotopically labeled substrates to follow fluxes, though this would be expensive to set up in a rat model.



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






