

Integrating Omics Data in Genome-Scale Metabolic Modeling: A Methodological Perspective for Precision Medicine



Abstract
1. Introduction
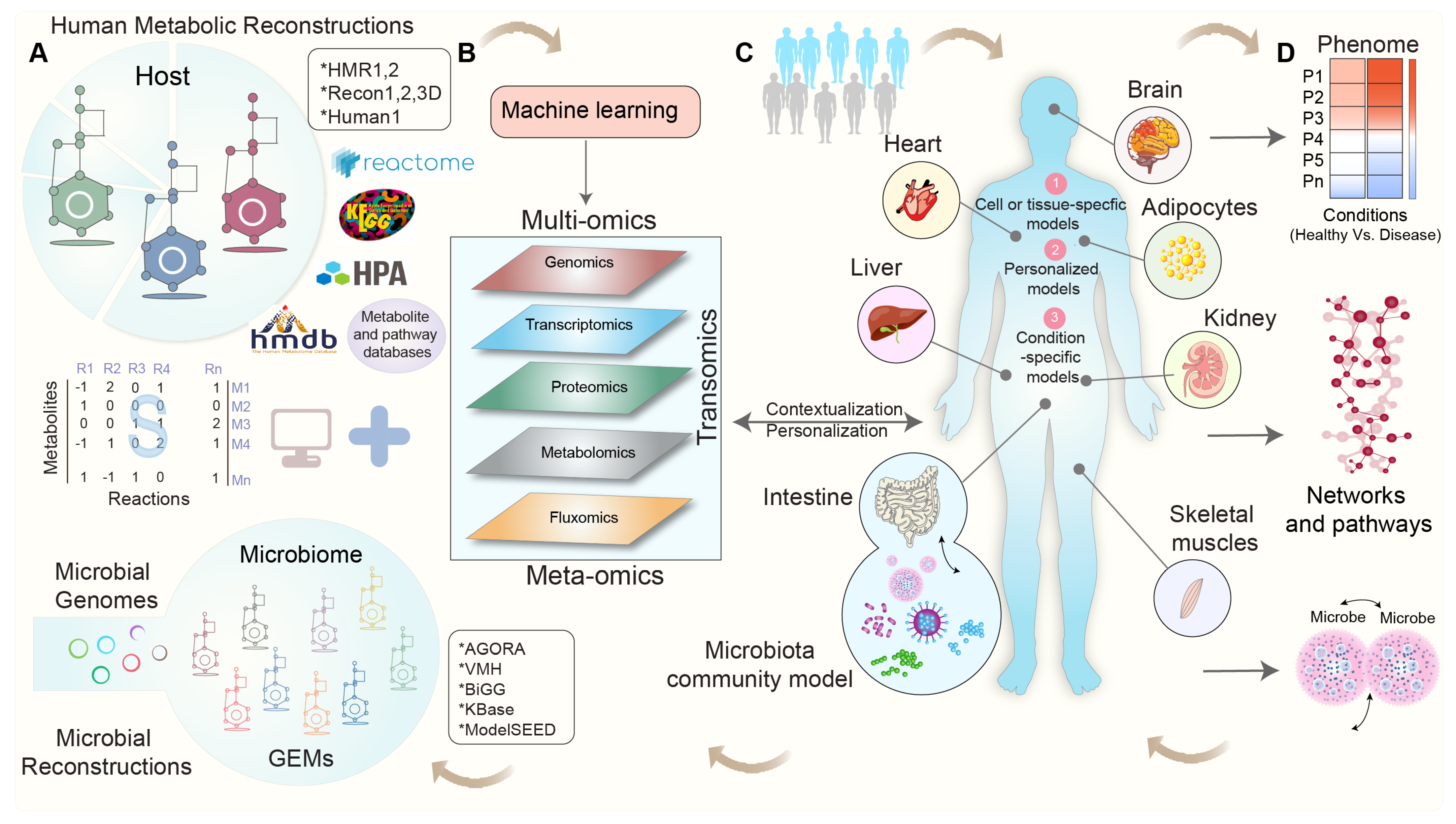
2. Exploring Human Metabolism: The Evolution of Genome-Scale Metabolic Models
3. Integrating Omics Data into Genome-Scale Metabolic Models: Overcoming Challenges and Shaping Perspectives
4. Modeling Tissue-Specific Interactions: Integrating Omics Data for Contextualization
5. Modeling the Interactions between Gut Microbial Communities and Host Metabolism
6. Towards Whole-Body Metabolic Reconstruction: Bridging Precision Medicine and Systems Biology
7. Enhancing the Reproducibility of Genome-Scale Metabolic Models by Addressing Key Challenges
8. Leveraging Machine Learning for Genome-Scale Metabolic Modeling: An Advancing Solution to Address the Key Challenges
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Babu, M.; Snyder, M. Multi-Omics Profiling for Health. Mol. Cell Proteom. 2023, 22, 100561. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Gonzalez, G.A.P.; Aurich, M.K. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272. [Google Scholar] [CrossRef]
- Blazier, A.S.; Papin, J.A. Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 2012, 3, 299. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 2010, 28, 977–982. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef]
- Sen, P.; Andrabi, S.B.A.; Buchacher, T.; Khan, M.M.; Kalim, U.U.; Lindeman, T.M.; Alves, M.A.; Hinkkanen, V.; Kemppainen, E.; Dickens, A.M.; et al. Quantitative genome-scale metabolic modeling of human CD4(+) T cell differentiation reveals subset-specific regulation of glycosphingolipid pathways. Cell Rep. 2021, 37, 109973. [Google Scholar] [CrossRef]
- Sen, P.; Govaere, O.; Sinioja, T.; McGlinchey, A.; Geng, D.; Ratziu, V.; Bugianesi, E.; Schattenberg, J.M.; Vidal-Puig, A.; Allison, M.; et al. Quantitative modeling of human liver reveals dysregulation of glycosphingolipid pathways in nonalcoholic fatty liver disease. iScience 2022, 25, 104949. [Google Scholar] [CrossRef]
- Cook, D.J.; Nielsen, J. Genome-scale metabolic models applied to human health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1393. [Google Scholar] [CrossRef]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.O. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 2012, 10, 291–305. [Google Scholar] [CrossRef]
- Esvap, E.; Ulgen, K.O. Advances in Genome-Scale Metabolic Modeling toward Microbial Community Analysis of the Human Microbiome. ACS Synth. Biol. 2021, 10, 2121–2137. [Google Scholar] [CrossRef]
- Heinken, A.; Basile, A.; Hertel, J.; Thinnes, C.; Thiele, I. Genome-Scale Metabolic Modeling of the Human Microbiome in the Era of Personalized Medicine. Annu. Rev. Microbiol. 2021, 75, 199–222. [Google Scholar] [CrossRef]
- Shoaie, S.; Nielsen, J. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 2014, 5, 86. [Google Scholar] [CrossRef]
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using genome-scale models to predict biological capabilities. Cell 2015, 161, 971–987. [Google Scholar] [CrossRef]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Hertel, J.; Acharya, G.; Ravcheev, D.A.; Nyga, M.; Okpala, O.E.; Hogan, M.; Magnusdottir, S.; Martinelli, F.; Nap, B.; et al. Genome-scale metabolic reconstruction of 7302 human microorganisms for personalized medicine. Nat. Biotechnol. 2023, 1–12. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.J.; Lerman, J.A.; Chang, R.L.; Hyduke, D.R.; Palsson, B.O. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 2013, 9, 693. [Google Scholar] [CrossRef] [PubMed]
- Förster, J.; Famili, I.; Fu, P.; Palsson, B.Ø.; Nielsen, J. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 2003, 13, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Patil, K.R.; Rocha, I.; Förster, J.; Nielsen, J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinform. 2005, 6, 308. [Google Scholar] [CrossRef] [PubMed]
- Suthers, P.F.; Zomorrodi, A.; Maranas, C.D. Genome-scale gene/reaction essentiality and synthetic lethality analysis. Mol. Syst. Biol. 2009, 5, 301. [Google Scholar] [CrossRef] [PubMed]
- Pharkya, P.; Burgard, A.P.; Maranas, C.D. OptStrain: A computational framework for redesign of microbial production systems. Genome Res. 2004, 14, 2367–2376. [Google Scholar] [CrossRef]
- Pharkya, P.; Maranas, C.D. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab. Eng. 2006, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.O. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef]
- Ma, H.; Sorokin, A.; Mazein, A.; Selkov, A.; Selkov, E.; Demin, O.; Goryanin, I. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol. Syst. Biol. 2007, 3, 135. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Swainston, N.; Smallbone, K.; Hefzi, H.; Dobson, P.D.; Brewer, J.; Hanscho, M.; Zielinski, D.C.; Ang, K.S.; Gardiner, N.J.; Gutierrez, J.M. Recon 2.2: From reconstruction to model of human metabolism. Metabolomics 2016, 12, 109. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Nookaew, I.; Jacobson, P.; Walley, A.J.; Froguel, P.; Carlsson, L.M.; Uhlen, M.; et al. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 2013, 9, 649. [Google Scholar] [CrossRef]
- Väremo, L.; Scheele, C.; Broholm, C.; Mardinoglu, A.; Kampf, C.; Asplund, A.; Nookaew, I.; Uhlén, M.; Pedersen, B.K.; Nielsen, J. Proteome-and Transcriptome-Driven Reconstruction of the Human Myocyte Metabolic Network and Its Use for Identification of Markers for Diabetes. Cell Rep. 2016, 14, 1567. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Liu, Z.; Klevstig, M.; Mukhopadhyay, B.; Bergentall, M.; Cinar, R.; Stahlman, M.; Sikanic, N.; Park, J.K.; et al. Network analyses identify liver-specific targets for treating liver diseases. Mol. Syst. Biol. 2017, 13, 938. [Google Scholar] [CrossRef]
- Bidkhori, G.; Benfeitas, R.; Klevstig, M.; Zhang, C.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. USA 2018, 115, E11874–E11883. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Wang, C.; Fessler, J.; DeTomaso, D.; Avila-Pacheco, J.; Kaminski, J.; Zaghouani, S.; Christian, E.; Thakore, P.; Schellhaass, B.; et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 2021, 184, 4168–4185. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Kocabas, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, L.; Kelly, L. Bringing microbiome-drug interaction research into the clinic. eBioMedicine 2019, 44, 708–715. [Google Scholar] [CrossRef]
- Sahoo, S.; Haraldsdottir, H.S.; Fleming, R.M.; Thiele, I. Modeling the effects of commonly used drugs on human metabolism. FEBS J. 2015, 282, 297–317. [Google Scholar] [CrossRef]
- Patil, K.R.; Nielsen, J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. USA 2005, 102, 2685–2689. [Google Scholar] [CrossRef]
- Picard, M.; Scott-Boyer, M.P.; Bodein, A.; Perin, O.; Droit, A. Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 2021, 19, 3735–3746. [Google Scholar] [CrossRef]
- Gomez-Cabrero, D.; Abugessaisa, I.; Maier, D.; Teschendorff, A.; Merkenschlager, M.; Gisel, A.; Ballestar, E.; Bongcam-Rudloff, E.; Conesa, A.; Tegner, J. Data integration in the era of omics: Current and future challenges. BMC Syst. Biol. 2014, 8 (Suppl. S2), I1. [Google Scholar] [CrossRef]
- Misra, B.B.; Langefeld, C.D.; Olivier, M.; Cox, L.A. Integrated Omics: Tools, Advances, and Future Approaches. J. Mol. Endocrinol. 2018, 62, R21–R45. [Google Scholar] [CrossRef]
- Flores, J.E.; Claborne, D.M.; Weller, Z.D.; Webb-Robertson, B.M.; Waters, K.M.; Bramer, L.M. Missing data in multi-omics integration: Recent advances through artificial intelligence. Front. Artif. Intell. 2023, 6, 1098308. [Google Scholar] [CrossRef]
- Valikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Walach, J.; Filzmoser, P.; Hron, K. Chapter Seven—Data Normalization and Scaling: Consequences for the Analysis in Omics Sciences. In Comprehensive Analytical Chemistry; Jaumot, J., Bedia, C., Tauler, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 82, pp. 165–196. [Google Scholar]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Sysi-Aho, M.; Katajamaa, M.; Yetukuri, L.; Oresic, M. Normalization method for metabolomics data using optimal selection of multiple internal standards. BMC Bioinform. 2007, 8, 93. [Google Scholar] [CrossRef]
- Ejigu, B.A.; Valkenborg, D.; Baggerman, G.; Vanaerschot, M.; Witters, E.; Dujardin, J.C.; Burzykowski, T.; Berg, M. Evaluation of normalization methods to pave the way towards large-scale LC-MS-based metabolomics profiling experiments. OMICS A J. Integr. Biol. 2013, 17, 473–485. [Google Scholar] [CrossRef]
- Becker, S.A.; Feist, A.M.; Mo, M.L.; Hannum, G.; Palsson, B.O.; Herrgard, M.J. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox. Nat. Protoc. 2007, 2, 727–738. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdottir, H.S.; Keating, S.M.; Vlasov, V.; Wachowiak, J. Creation and analysis of biochemical constraint-based models: The COBRA Toolbox v3.0. arXiv 2017, arXiv:1710.04038. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, J.; Que, R.; Fleming, R.M.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed]
- Baldini, F.; Heinken, A.; Heirendt, L.; Magnusdottir, S.; Fleming, R.M.T.; Thiele, I. The Microbiome Modeling Toolbox: From microbial interactions to personalized microbial communities. Bioinformatics 2019, 35, 2332–2334. [Google Scholar] [CrossRef] [PubMed]
- Li, G.H.; Dai, S.; Han, F.; Li, W.; Huang, J.; Xiao, W. FastMM: An efficient toolbox for personalized constraint-based metabolic modeling. BMC Bioinform. 2020, 21, 67. [Google Scholar] [CrossRef] [PubMed]
- Thorleifsson, S.G.; Thiele, I. rBioNet: A COBRA toolbox extension for reconstructing high-quality biochemical networks. Bioinformatics 2011, 27, 2009–2010. [Google Scholar] [CrossRef]
- Agren, R.; Liu, L.; Shoaie, S.; Vongsangnak, W.; Nookaew, I.; Nielsen, J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 2013, 9, e1002980. [Google Scholar] [CrossRef]
- Wang, H.; Marcisauskas, S.; Sanchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2015, 44, D515–D522. [Google Scholar] [CrossRef]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Danielsdottir, A.D.; Krecke, M.; Merten, D.; Haraldsdottir, H.S.; et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 2019, 47, D614–D624. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef]
- Pornputtapong, N.; Nookaew, I.; Nielsen, J. Human metabolic atlas: An online resource for human metabolism. Database 2015, 2015, bav068. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2013, 42, D199–D205. [Google Scholar] [CrossRef]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Jeske, L.; Ulbrich, M.; Placzek, S.; Chang, A.; Schomburg, D. The BRENDA enzyme information system—From a database to an expert system. J. Biotechnol. 2017, 261, 194–206. [Google Scholar] [CrossRef]
- D’Eustachio, P. Reactome knowledgebase of human biological pathways and processes. Methods Mol. Biol. 2011, 694, 49–61. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Samaras, P.; Schmidt, T.; Frejno, M.; Gessulat, S.; Reinecke, M.; Jarzab, A.; Zecha, J.; Mergner, J.; Giansanti, P.; Ehrlich, H.C.; et al. ProteomicsDB: A multi-omics and multi-organism resource for life science research. Nucleic Acids Res. 2020, 48, D1153–D1163. [Google Scholar] [CrossRef]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2011, 39, D52–D57. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, H.; Kapushesky, M.; Shojatalab, M.; Abeygunawardena, N.; Coulson, R.; Farne, A.; Holloway, E.; Kolesnykov, N.; Lilja, P.; Lukk, M.; et al. ArrayExpress—A public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007, 35, D747–D750. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, I.; Almeida-King, J.; Kumanduri, V.; Senf, A.; Spalding, J.D.; Ur-Rehman, S.; Saunders, G.; Kandasamy, J.; Caccamo, M.; Leinonen, R.; et al. The European Genome-phenome Archive of human data consented for biomedical research. Nat. Genet. 2015, 47, 692–695. [Google Scholar] [CrossRef]
- GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Franzen, O.; Ermel, R.; Cohain, A.; Akers, N.K.; Di Narzo, A.; Talukdar, H.A.; Foroughi-Asl, H.; Giambartolomei, C.; Fullard, J.F.; Sukhavasi, K.; et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science 2016, 353, 827–830. [Google Scholar] [CrossRef]
- Li, C.; Donizelli, M.; Rodriguez, N.; Dharuri, H.; Endler, L.; Chelliah, V.; Li, L.; He, E.; Henry, A.; Stefan, M.I.; et al. BioModels Database: An enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst. Biol. 2010, 4, 92. [Google Scholar] [CrossRef]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef]
- Becker, S.A.; Palsson, B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef]
- Bordbar, A.; Mo, M.L.; Nakayasu, E.S.; Schrimpe-Rutledge, A.C.; Kim, Y.M.; Metz, T.O.; Jones, M.B.; Frank, B.C.; Smith, R.D.; Peterson, S.N.; et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol. Syst. Biol. 2012, 8, 558. [Google Scholar] [CrossRef] [PubMed]
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.A.; Papin, J.A. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 2011, 27, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Colijn, C.; Brandes, A.; Zucker, J.; Lun, D.S.; Weiner, B.; Farhat, M.R.; Cheng, T.Y.; Moody, D.B.; Murray, M.; Galagan, J.E. Interpreting expression data with metabolic flux models: Predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 2009, 5, e1000489. [Google Scholar] [CrossRef]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef]
- Angione, C.; Lio, P. Predictive analytics of environmental adaptability in multi-omic network models. Sci. Rep. 2015, 5, 15147. [Google Scholar] [CrossRef]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef]
- Vlassis, N.; Pacheco, M.P.; Sauter, T. Fast reconstruction of compact context-specific metabolic network models. PLoS Comput. Biol. 2014, 10, e1003424. [Google Scholar] [CrossRef]
- Pacheco, M.P.; Bintener, T.; Ternes, D.; Kulms, D.; Haan, S.; Letellier, E.; Sauter, T. Identifying and targeting cancer-specific metabolism with network-based drug target prediction. eBioMedicine 2019, 43, 98–106. [Google Scholar] [CrossRef]
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst. Biol. 2012, 6, 153. [Google Scholar] [CrossRef]
- Yizhak, K.; Gaude, E.; Le Devedec, S.; Waldman, Y.Y.; Stein, G.Y.; van de Water, B.; Frezza, C.; Ruppin, E. Phenotype-based cell-specific metabolic modeling reveals metabolic liabilities of cancer. eLife 2014, 3, e03641. [Google Scholar] [CrossRef] [PubMed]
- Robaina Estevez, S.; Nikoloski, Z. Context-Specific Metabolic Model Extraction Based on Regularized Least Squares Optimization. PLoS ONE 2015, 10, e0131875. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Qutub, A.A. Reconstruction of Tissue-Specific Metabolic Networks Using CORDA. PLoS Comput. Biol. 2016, 12, e1004808. [Google Scholar] [CrossRef] [PubMed]
- Moskon, M.; Rezen, T. Context-Specific Genome-Scale Metabolic Modelling and Its Application to the Analysis of COVID-19 Metabolic Signatures. Metabolites 2023, 13, 126. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Arkin, A.P.; Stevens, R.L.; Cottingham, R.W.; Maslov, S.; Henry, C.S.; Dehal, P.; Ware, D.; Perez, F.; Harris, N.L.; Canon, S. The DOE Systems Biology Knowledgebase (KBase). bioRxiv 2016, 096354. [Google Scholar] [CrossRef]
- Hyotylainen, T.; Jerby, L.; Petaja, E.M.; Mattila, I.; Jantti, S.; Auvinen, P.; Gastaldelli, A.; Yki-Jarvinen, H.; Ruppin, E.; Oresic, M. Genome-scale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat. Commun. 2016, 7, 8994. [Google Scholar] [CrossRef]
- Jerby, L.; Shlomi, T.; Ruppin, E. Computational reconstruction of tissue-specific metabolic models: Application to human liver metabolism. Mol. Syst. Biol. 2010, 6, 401. [Google Scholar] [CrossRef]
- Baloni, P.; Funk, C.C.; Readhead, B.; Price, N.D. Systems modeling of metabolic dysregulation in neurodegenerative diseases. Curr. Opin. Pharmacol. 2021, 60, 59–65. [Google Scholar] [CrossRef]
- Lewis, N.E.; Schramm, G.; Bordbar, A.; Schellenberger, J.; Andersen, M.P.; Cheng, J.K.; Patel, N.; Yee, A.; Lewis, R.A.; Eils, R. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010, 28, 1279. [Google Scholar] [CrossRef]
- Kishk, A.; Pacheco, M.P.; Heurtaux, T.; Sinkkonen, L.; Pang, J.; Fritah, S.; Niclou, S.P.; Sauter, T. Review of Current Human Genome-Scale Metabolic Models for Brain Cancer and Neurodegenerative Diseases. Cells 2022, 11, 2486. [Google Scholar] [CrossRef] [PubMed]
- Baloni, P.; Funk, C.C.; Yan, J.; Yurkovich, J.T.; Kueider-Paisley, A.; Nho, K.; Heinken, A.; Jia, W.; Mahmoudiandehkordi, S.; Louie, G.; et al. Metabolic Network Analysis Reveals Altered Bile Acid Synthesis and Metabolism in Alzheimer’s Disease. Cell Rep. Med. 2020, 1, 100138. [Google Scholar] [CrossRef] [PubMed]
- Baloni, P.; Arnold, M.; Buitrago, L.; Nho, K.; Moreno, H.; Huynh, K.; Brauner, B.; Louie, G.; Kueider-Paisley, A.; Suhre, K.; et al. Multi-Omic analyses characterize the ceramide/sphingomyelin pathway as a therapeutic target in Alzheimer’s disease. Commun. Biol. 2022, 5, 1074. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, J. Reconstruction and analysis of human heart-specific metabolic network based on transcriptome and proteome data. Biochem. Biophys. Res. Commun. 2011, 415, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Thiele, I. Predicting the impact of diet and enzymopathies on human small intestinal epithelial cells. Human. Mol. Genet. 2013, 22, 2705–2722. [Google Scholar] [CrossRef]
- Sahoo, S.; Aurich, M.K.; Jonsson, J.J.; Thiele, I. Membrane transporters in a human genome-scale metabolic knowledgebase and their implications for disease. Front. Physiol. 2014, 5, 91. [Google Scholar] [CrossRef]
- Gustafsson, J.; Anton, M.; Roshanzamir, F.; Jornsten, R.; Kerkhoven, E.J.; Robinson, J.L.; Nielsen, J. Generation and analysis of context-specific genome-scale metabolic models derived from single-cell RNA-Seq data. Proc. Natl. Acad. Sci. USA 2023, 120, e2217868120. [Google Scholar] [CrossRef]
- Do Rosario Martins Conde, P.; Sauter, T.; Pfau, T. Constraint Based Modeling Going Multicellular. Front. Mol. Biosci. 2016, 3, 3. [Google Scholar] [CrossRef]
- Bordbar, A.; Feist, A.M.; Usaite-Black, R.; Woodcock, J.; Palsson, B.O.; Famili, I. A multi-tissue type genome-scale metabolic network for analysis of whole-body systems physiology. BMC Syst. Biol. 2011, 5, 180. [Google Scholar] [CrossRef]
- Foguet, C.; Xu, Y.; Ritchie, S.C.; Lambert, S.A.; Persyn, E.; Nath, A.P.; Davenport, E.E.; Roberts, D.J.; Paul, D.S.; Di Angelantonio, E.; et al. Genetically personalised organ-specific metabolic models in health and disease. Nat. Commun. 2022, 13, 7356. [Google Scholar] [CrossRef]
- Pascal Andreu, V.; Augustijn, H.E.; Chen, L.; Zhernakova, A.; Fu, J.; Fischbach, M.A.; Dodd, D.; Medema, M.H. gutSMASH predicts specialized primary metabolic pathways from the human gut microbiota. Nat. Biotechnol. 2023, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gille, C.; Bolling, C.; Hoppe, A.; Bulik, S.; Hoffmann, S.; Hubner, K.; Karlstadt, A.; Ganeshan, R.; Konig, M.; Rother, K.; et al. HepatoNet1: A comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol. Syst. Biol. 2010, 6, 411. [Google Scholar] [CrossRef] [PubMed]
- Karlstadt, A.; Fliegner, D.; Kararigas, G.; Ruderisch, H.S.; Regitz-Zagrosek, V.; Holzhutter, H.G. CardioNet: A human metabolic network suited for the study of cardiomyocyte metabolism. BMC Syst. Biol. 2012, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jimenez, C.A.; Salazar-Barreto, D.; Barreto, G.E.; Gonzalez, J. Genome-Scale Reconstruction of the Human Astrocyte Metabolic Network. Front. Aging Neurosci. 2017, 9, 23. [Google Scholar] [CrossRef]
- Sohrabi-Jahromi, S.; Marashi, S.A.; Kalantari, S. A kidney-specific genome-scale metabolic network model for analyzing focal segmental glomerulosclerosis. Mamm. Genome 2016, 27, 158–167. [Google Scholar] [CrossRef]
- Quek, L.E.; Dietmair, S.; Hanscho, M.; Martinez, V.S.; Borth, N.; Nielsen, L.K. Reducing Recon 2 for steady-state flux analysis of HEK cell culture. J. Biotechnol. 2014, 184, 172–178. [Google Scholar] [CrossRef]
- Bordbar, A.; Lewis, N.E.; Schellenberger, J.; Palsson, B.O.; Jamshidi, N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol. Syst. Biol. 2010, 6, 422. [Google Scholar] [CrossRef]
- Sen, P.; Dickens, A.M.; Lopez-Bascon, M.A.; Lindeman, T.; Kemppainen, E.; Lamichhane, S.; Ronkko, T.; Ilonen, J.; Toppari, J.; Veijola, R.; et al. Metabolic alterations in immune cells associate with progression to type 1 diabetes. Diabetologia 2020, 63, 1017–1031. [Google Scholar] [CrossRef]
- Thiele, I.; Sahoo, S.; Heinken, A.; Hertel, J.; Heirendt, L.; Aurich, M.K.; Fleming, R.M. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol. Syst. Biol. 2020, 16, e8982. [Google Scholar] [CrossRef]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009, 19, 1141–1152. [Google Scholar] [CrossRef]
- Ji, B.; Nielsen, J. New insight into the gut microbiome through metagenomics. Adv. Genom. Genet. 2015, 5, 77–91. [Google Scholar]
- Bauer, E.; Thiele, I. From Network Analysis to Functional Metabolic Modeling of the Human Gut Microbiota. mSystems 2018, 3, 157–197. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Karlsson, F.; Mardinoglu, A.; Nookaew, I.; Bordel, S.; Nielsen, J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 2013, 3, 2532. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ji, B.; Babaei, P.; Das, P.; Lappa, D.; Ramakrishnan, G.; Fox, T.E.; Haque, R.; Petri, W.A., Jr.; Bäckhed, F. Gut microbiota dysbiosis is associated with malnutrition and reduced plasma amino acid levels: Lessons from genome-scale metabolic modeling. Metab. Eng. 2018, 49, 128–142. [Google Scholar] [CrossRef]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; de Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying Diet-Induced Metabolic Changes of the Human Gut Microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Oresic, M. Metabolic Modeling of Human Gut Microbiota on a Genome Scale: An Overview. Metabolites 2019, 9, 22. [Google Scholar] [CrossRef]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Alves, M.A.; Harkonen, T.; Honkanen, J.; Vatanen, T.; Xavier, R.J.; Hyotylainen, T.; Knip, M.; et al. Dysregulation of secondary bile acid metabolism precedes islet autoimmunity and type 1 diabetes. Cell Rep. Med. 2022, 3, 100762. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T. A human gut microbial gene catalog established by metagenomic sequencing. Nature 2010, 464, 59. [Google Scholar] [CrossRef]
- Harcombe, W.R.; Riehl, W.J.; Dukovski, I.; Granger, B.R.; Betts, A.; Lang, A.H.; Bonilla, G.; Kar, A.; Leiby, N.; Mehta, P.; et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014, 7, 1104–1115. [Google Scholar] [CrossRef]
- Bauer, E.; Zimmermann, J.; Baldini, F.; Thiele, I.; Kaleta, C. BacArena: Individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 2017, 13, e1005544. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Islam, M.M.; Maranas, C.D. d-OptCom: Dynamic multi-level and multi-objective metabolic modeling of microbial communities. ACS Synth. Biol. 2014, 3, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Biggs, M.B.; Papin, J.A. Novel multiscale modeling tool applied to Pseudomonas aeruginosa biofilm formation. PLoS ONE 2013, 8, e78011. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, K.; Izallalen, M.; Mouser, P.; Richter, H.; Risso, C.; Mahadevan, R.; Lovley, D.R. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 2011, 5, 305. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; Doebeli, M. Calibration and analysis of genome-based models for microbial ecology. eLife 2015, 4, e08208. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Butcher, J.; Stintzi, A.; Figeys, D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome 2019, 7, 154. [Google Scholar] [CrossRef]
- Segata, N.; Boernigen, D.; Tickle, T.L.; Morgan, X.C.; Garrett, W.S.; Huttenhower, C. Computational meta’omics for microbial community studies. Mol. Syst. Biol. 2013, 9, 666. [Google Scholar] [CrossRef]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef]
- Heinken, A.; Sahoo, S.; Fleming, R.M.; Thiele, I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 2013, 4, 28–40. [Google Scholar] [CrossRef]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Oresic, M.; Bertram, H.C. Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe. Methods 2018, 149, 3–12. [Google Scholar] [CrossRef]
- Lewis, J.E.; Forshaw, T.E.; Boothman, D.A.; Furdui, C.M.; Kemp, M.L. Personalized Genome-Scale Metabolic Models Identify Targets of Redox Metabolism in Radiation-Resistant Tumors. Cell Syst. 2021, 12, 68–81.E11. [Google Scholar] [CrossRef] [PubMed]
- Turanli, B.; Zhang, C.; Kim, W.; Benfeitas, R.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Discovery of therapeutic agents for prostate cancer using genome-scale metabolic modeling and drug repositioning. eBioMedicine 2019, 42, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J., III. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014, 15, 107–120. [Google Scholar] [CrossRef]
- Sen, P.; Vial, H.J.; Radulescu, O. Kinetic modelling of phospholipid synthesis in Plasmodium knowlesi unravels crucial steps and relative importance of multiple pathways. BMC Syst. Biol. 2013, 7, 123. [Google Scholar] [CrossRef]
- Chan, S.H.J.; Friedman, E.S.; Wu, G.D.; Maranas, C.D. Predicting the Longitudinally and Radially Varying Gut Microbiota Composition using Multi-Scale Microbial Metabolic Modeling. Processes 2019, 7, 394. [Google Scholar] [CrossRef]
- Dinh, H.V.; Sarkar, D.; Maranas, C.D. Quantifying the propagation of parametric uncertainty on flux balance analysis. Metab. Eng. 2022, 69, 26–39. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef]
- Reznik, E.; Mehta, P.; Segre, D. Flux imbalance analysis and the sensitivity of cellular growth to changes in metabolite pools. PLoS Comput. Biol. 2013, 9, e1003195. [Google Scholar] [CrossRef]
- Tervo, C.J.; Reed, J.L. Expanding Metabolic Engineering Algorithms Using Feasible Space and Shadow Price Constraint Modules. Metab. Eng. Commun. 2014, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.J.; Kerkhoven, E.J.; Nielsen, J. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 2017, 13, 935. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M. Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling. Metabolites 2018, 8, 4. [Google Scholar] [CrossRef]
- Angione, C. Human Systems Biology and Metabolic Modelling: A Review-From Disease Metabolism to Precision Medicine. Biomed. Res. Int. 2019, 2019, 8304260. [Google Scholar] [CrossRef]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep Learning Accurately Predicts Estrogen Receptor Status in Breast Cancer Metabolomics Data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef]
- Lewis, J.E.; Kemp, M.L. Integration of machine learning and genome-scale metabolic modeling identifies multi-omics biomarkers for radiation resistance. Nat. Commun. 2021, 12, 2700. [Google Scholar] [CrossRef]
- Sen, P.; Lamichhane, S.; Mathema, V.B.; McGlinchey, A.; Dickens, A.M.; Khoomrung, S.; Oresic, M. Deep learning meets metabolomics: A methodological perspective. Brief. Bioinform. 2021, 22, 1531–1542. [Google Scholar] [CrossRef]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef]
- Zampieri, G.; Vijayakumar, S.; Yaneske, E.; Angione, C. Machine and deep learning meet genome-scale metabolic modeling. PLoS Comput. Biol. 2019, 15, e1007084. [Google Scholar] [CrossRef]
- Pearcy, N.; Hu, Y.; Baker, M.; Maciel-Guerra, A.; Xue, N.; Wang, W.; Kaler, J.; Peng, Z.; Li, F.; Dottorini, T. Genome-Scale Metabolic Models and Machine Learning Reveal Genetic Determinants of Antibiotic Resistance in Escherichia coli and Unravel the Underlying Metabolic Adaptation Mechanisms. mSystems 2021, 6, e0091320. [Google Scholar] [CrossRef]
- Rana, P.; Berry, C.; Ghosh, P.; Fong, S.S. Recent advances on constraint-based models by integrating machine learning. Curr. Opin. Biotechnol. 2019, 64, 85–91. [Google Scholar] [CrossRef]
- Medlock, G.L.; Papin, J.A. Guiding the Refinement of Biochemical Knowledgebases with Ensembles of Metabolic Networks and Machine Learning. Cell Syst. 2020, 10, 109–119.E3. [Google Scholar] [CrossRef]
- Kotera, M.; Tabei, Y.; Yamanishi, Y.; Tokimatsu, T.; Goto, S. Supervised de novo reconstruction of metabolic pathways from metabolome-scale compound sets. Bioinformatics 2013, 29, i135–i144. [Google Scholar] [CrossRef] [PubMed]
- Liberal, R.; Pinney, J.W. Simple topological properties predict functional misannotations in a metabolic network. Bioinformatics 2013, 29, i154–i161. [Google Scholar] [CrossRef] [PubMed]
- Zelezniak, A.; Vowinckel, J.; Capuano, F.; Messner, C.B.; Demichev, V.; Polowsky, N.; Mulleder, M.; Kamrad, S.; Klaus, B.; Keller, M.A.; et al. Machine Learning Predicts the Yeast Metabolome from the Quantitative Proteome of Kinase Knockouts. Cell Syst. 2018, 7, 269–283.e6. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Wright, S.N.; Hamblin, M.; McCloskey, D.; Alcantar, M.A.; Schrubbers, L.; Lopatkin, A.J.; Satish, S.; Nili, A.; Palsson, B.O.; et al. A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action. Cell 2019, 177, 1649–1661. [Google Scholar] [CrossRef]
- Guo, W.; Xu, Y.; Feng, X. DeepMetabolism: A deep learning system to predict phenotype from genome sequencing. arXiv 2017, arXiv:1705.03094. [Google Scholar] [CrossRef]
- Antonakoudis, A.; Barbosa, R.; Kotidis, P.; Kontoravdi, C. The era of big data: Genome-scale modelling meets machine learning. Comput. Struct. Biotechnol. J. 2020, 18, 3287–3300. [Google Scholar] [CrossRef]


{kind=link}
| Databases or Repositories | Description | References |
|---|---|---|
| BiGG database | A publicly accessible repository for benchmark GEMs with open access. | [59] |
| Virtual Metabolic Human (VMH) | A freely accessible database for human and gut microbial metabolic reconstructions (GEMs) with open access. | [60] |
| ModelSEED | A web-based platform for metabolic modeling and analysis. | [61] |
| Human Metabolic Atlas (HMA) | An open access web-based platform for studying human metabolism. | [33,62] |
| HumanCyc | A curated database of experimentally validated metabolic pathways for studying human metabolism. | [63] |
| KEGG | A comprehensive resource comprising databases of large-scale molecular datasets and detailed pathway information. | [64,65] |
| LIPID MAPS | A database providing information on lipid structures, pathways, and lipid-related genes; enzymes; and metabolites. | [66] |
| Human Metabolome Database (HMDB) | A comprehensive resource that provides information on the chemical composition, biological roles, and disease associations of metabolites found in the human body. | [67] |
| BRENDA | An enzyme- and ligand-focused information retrieval system. | [68] |
| REACTOME | An open access database for biological pathways. | [69] |
| UniProt | An open access database for curated protein information. | [70] |
| Human Protein Atlas (HPA) | A comprehensive resource providing information on the expression and localization of proteins in human tissues and cells. | [71] |
| ProteomicsDB | A comprehensive resource for exploring and analyzing protein expression data from a variety of organisms and tissues. | [72] |
| Entrez gene | Gene-centered information, including gene sequences, annotations, functional data, and genetic variations. | [73] |
| Gene Expression Omnibus (GEO) | A public repository that provides access to a vast collection of gene expression data from various experiments and studies. | [74] |
| Array Express (AE) | A public database of functional genomics experiments and gene expression profiles. | [75] |
| European Genome-phenome Archive (EGA) | A secure and controlled-access database for hosting and sharing human genetic and phenotypic data. | [76] |
| Genotype-Tissue Expression (GTEx) | A catalog of genetic variants and their influence on gene expression across multiple human tissues. | [77] |
| Stockholm-Tartu Atherosclerosis Reverse Networks Engineering Task (STARNET) | A computational method for reconstructing cell lineage trees from single-cell transcriptomic data. | [78] |
| BioModels | A collection of biological models that encompasses various organisms and biological processes. | [79] |
| Tissue or Cell-Type | Human Metabolic Reconstructions Used for the Contextualization | Omics or Diet Data Type(s) | Phenotypes Modeled | References |
|---|---|---|---|---|
| Liver | HepatoNet1 | G | Liver metabolism | [113] |
| HMR2 (iHepatocytes2322) | T, P, and M | NAFLD | [6] | |
| HMR2 | T and M | NAFLD | [8] | |
| Recon1 | T, M, and F | NAFLD | [98] | |
| Adipocytes | HMR1 (iAdipocytes1809) | T, P, and F | Adipocyte metabolism | [28] |
| Skeletal muscles | HMR2 (iMyocyte2419) | T and P | T2D | [29] |
| –– | T and P | CVD | [105] | |
| Heart | Recon1 (CardioNet) | G | Cardiac metabolism | [114] |
| Brain | Astrocyte metabolic network | T | Ischemic and normal conditions | [115] |
| Recon3D | T and M | AD | [103,104] | |
| Recon1 (iNL403) | G and P | AD | [101] | |
| Kidney | –– | T and P | Focal segmental glomerulosclerosis | [116] |
| Kidney | Recon2 (HEK cell culture) | –– | Metabolism of HEK cells | [117] |
| Alveolar macrophage | Recon1 (iAB-AMØ-1410) | G | Host-pathogen interactions in MTB | [118] |
| CD4+ T-cells | HMR2 | G, T, and M | CD4+ T-cells activation and differentiation | [7] |
| PBMCs | HMR2 | G, T, and M | T1D | [119] |
| Human small intestinal epithelial cells | Recon1 (hs_sIEC611) | American diet and a balanced diet | Intestinal metabolism and IEMs | [106] |
| Whole-body metabolic reconstructions | Recon3D (WBM) | T, P, and M | Human metabolism and host-microbiome co-metabolism | [120] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, P.; Orešič, M. Integrating Omics Data in Genome-Scale Metabolic Modeling: A Methodological Perspective for Precision Medicine. Metabolites 2023, 13, 855. https://doi.org/10.3390/metabo13070855
Sen P, Orešič M. Integrating Omics Data in Genome-Scale Metabolic Modeling: A Methodological Perspective for Precision Medicine. Metabolites. 2023; 13(7):855. https://doi.org/10.3390/metabo13070855
Chicago/Turabian StyleSen, Partho, and Matej Orešič. 2023. "Integrating Omics Data in Genome-Scale Metabolic Modeling: A Methodological Perspective for Precision Medicine" Metabolites 13, no. 7: 855. https://doi.org/10.3390/metabo13070855
APA StyleSen, P., & Orešič, M. (2023). Integrating Omics Data in Genome-Scale Metabolic Modeling: A Methodological Perspective for Precision Medicine. Metabolites, 13(7), 855. https://doi.org/10.3390/metabo13070855