

Optimizing MS-Based Multi-Omics: Comparative Analysis of Protein, Metabolite, and Lipid Extraction Techniques


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Cell Lysis
2.3. Procedure—Proteomics
2.3.1. FASP Method
2.3.2. S-Trap Method
2.4. Procedure—Metabolomics/Lipidomics
2.4.1. 80% MeOH Method
2.4.2. Chloroform/MeOH-Based Method
2.4.3. MTBE-Based Method
2.5. LC-MS Analysis
2.5.1. Chromatographic/Mass Spec Conditions—Proteomics
2.5.2. Chromatographic/Mass Spec Conditions—Metabolomics
2.5.3. Chromatographic/Mass Spec Conditions—Lipidomics
2.6. Data Processing/Statistical Analysis
2.6.1. Proteomics
2.6.2. Metabolomics
2.6.3. Lipidomics
3. Results
3.1. Proteomics
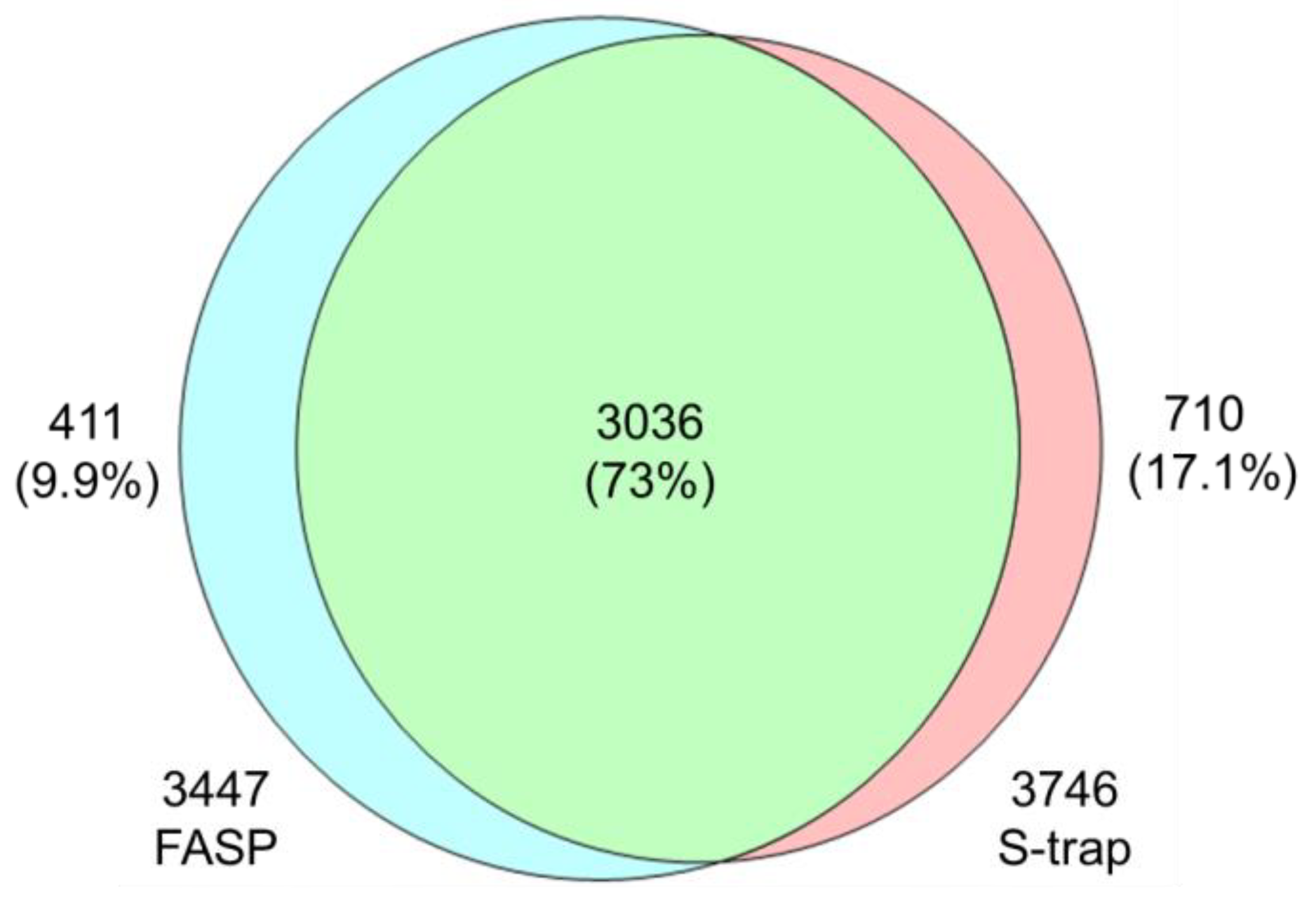
3.1.1. Comparison of FASP and S-Trap Digestion Methods Using KEGG Analysis
3.1.2. Comparison of Profiling Characteristics of FASP and S-Trap Methods
3.2. Metabolomics
3.2.1. VIP Score upon PLS-DA Classification
3.2.2. ANOVA
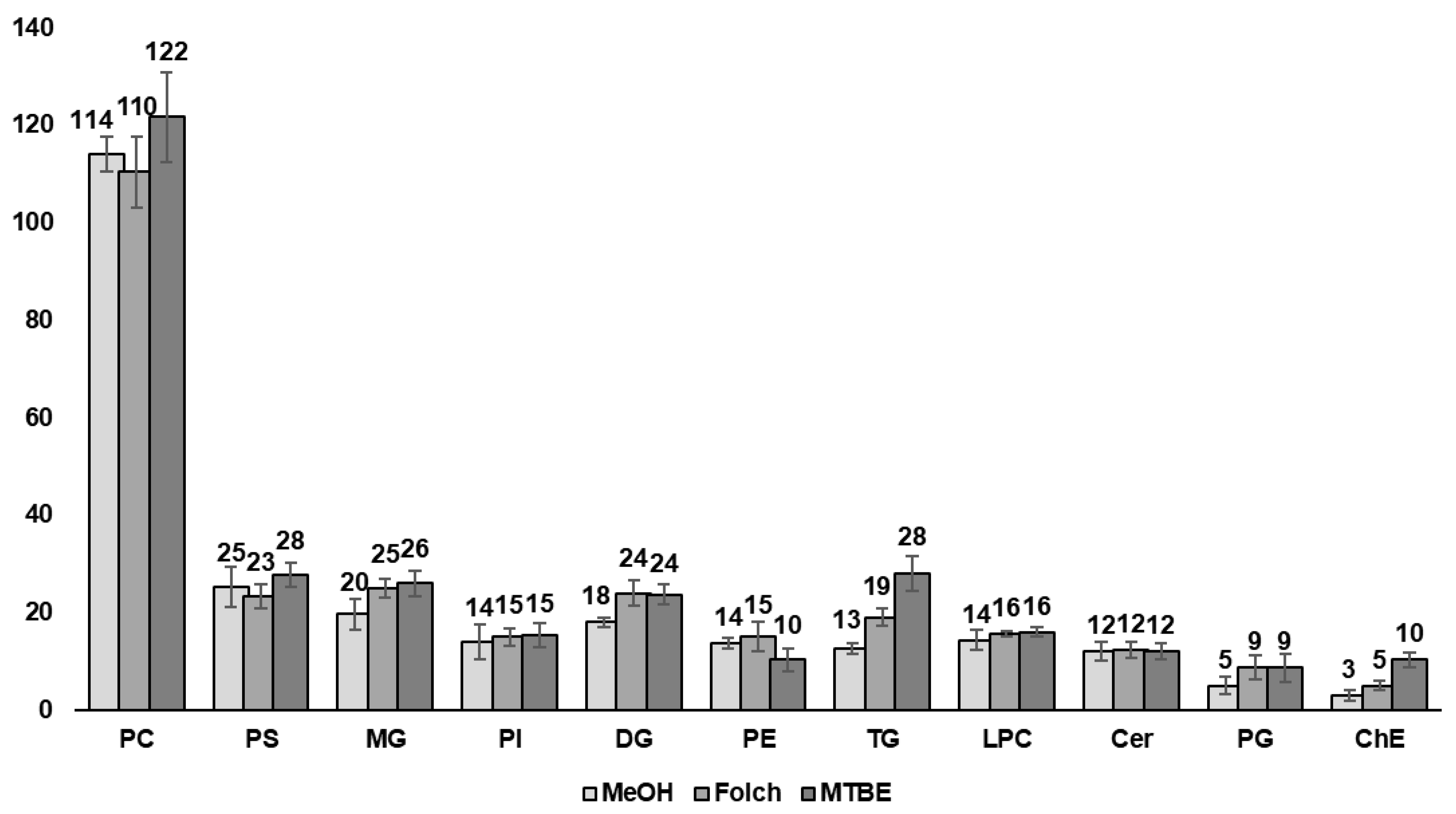
3.3. Lipidomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Bordbar, A.; Mo, M.L.; Nakayasu, E.S.; Schrimpe-Rutledge, A.C.; Kim, Y.M.; Metz, T.O.; Jones, M.B.; Frank, B.C.; Smith, R.D.; Peterson, S.N. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol. Syst. Biol. 2012, 8, 558. [Google Scholar] [CrossRef]
- Chen, R.; Mias, G.I.; Li-Pook-Than, J.; Jiang, L.; Lam, H.Y.; Chen, R.; Miriami, E.; Karczewski, K.J.; Hariharan, M.; Dewey, F.E. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 2012, 148, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Hultman, J.; Waldrop, M.P.; Mackelprang, R.; David, M.M.; McFarland, J.; Blazewicz, S.J.; Harden, J.; Turetsky, M.R.; McGuire, A.D.; Shah, M.B. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 2015, 521, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Coman, C.; Solari, F.A.; Hentschel, A.; Sickmann, A.; Zahedi, R.P.; Ahrends, R. Simultaneous metabolite, protein, lipid extraction (SIMPLEX): A combinatorial multimolecular omics approach for systems biology. Mol. Cell. Proteom. 2016, 15, 1435–1466. [Google Scholar] [CrossRef] [PubMed]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef]
- Begum, F.; Ghosh, D.; Tseng, G.C.; Feingold, E. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. 2012, 40, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A. Genomewide association studies and assessment of the risk of disease. New Engl. J. Med. 2010, 363, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Nakayasu, E.S.; Nicora, C.D.; Sims, A.C.; Burnum-Johnson, K.E.; Kim, Y.-M.; Kyle, J.E.; Matzke, M.M.; Shukla, A.K.; Chu, R.K.; Schepmoes, A.A. MPLEx: A robust and universal protocol for single-sample integrative proteomic, metabolomic, and lipidomic analyses. MSystems 2016, 1, e00043-16. [Google Scholar] [CrossRef]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the field in multi-omics research: From computational needs to data mining and sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]
- Kowalczyk, T.; Ciborowski, M.; Kisluk, J.; Kretowski, A.; Barbas, C. Mass spectrometry based proteomics and metabolomics in personalized oncology. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165690. [Google Scholar] [CrossRef] [PubMed]
- Veras, M.A.; Lim, Y.J.; Kuljanin, M.; Lajoie, G.A.; Urquhart, B.L.; Séguin, C.A. Protocol for parallel proteomic and metabolomic analysis of mouse intervertebral disc tissues. JOR Spine 2020, 3, e1099. [Google Scholar] [CrossRef] [PubMed]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A critical review of bottom-up proteomics: The good, the bad, and the future of this field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Bowness, P.; Kessler, B.M. Two birds with one stone: Doing metabolomics with your proteomics kit. Proteomics 2013, 13, 3371–3386. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.A.; Jüppner, J.; Bajdzienko, K.; Giavalisco, P. Protocol: A fast, comprehensive and reproducible one-step extraction method for the rapid preparation of polar and semi-polar metabolites, lipids, proteins, starch and cell wall polymers from a single sample. Plant Methods 2016, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Nicora, C.D.; Burnum-Johnson, K.E.; Nakayasu, E.S.; Casey, C.P.; White III, R.A.; Chowdhury, T.R.; Kyle, J.E.; Kim, Y.-M.; Smith, R.D.; Metz, T.O. The MPLEx protocol for multi-omic analyses of soil samples. JoVE (J. Vis. Exp.) 2018, 135, e57343. [Google Scholar]
- Kang, J.; David, L.; Li, Y.; Cang, J.; Chen, S. Three-in-One simultaneous extraction of proteins, metabolites and lipids for multi-omics. Front. Genet. 2021, 12, 635971. [Google Scholar] [CrossRef] [PubMed]
- Sapcariu, S.C.; Kanashova, T.; Weindl, D.; Ghelfi, J.; Dittmar, G.; Hiller, K. Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX 2014, 1, 74–80. [Google Scholar] [CrossRef]
- Godzien, J.; Ciborowski, M.; Armitage, E.G.; Jorge, I.; Camafeita, E.; Burillo, E.; Martín-Ventura, J.L.; Ruperez, F.J.; Vazquez, J.; Barbas, C. A single in-vial dual extraction strategy for the simultaneous lipidomics and proteomics analysis of HDL and LDL fractions. J. Proteome Res. 2016, 15, 1762–1775. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid. Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Sostare, J.; Di Guida, R.; Kirwan, J.; Chalal, K.; Palmer, E.; Dunn, W.B.; Viant, M.R. Comparison of modified Matyash method to conventional solvent systems for polar metabolite and lipid extractions. Anal. Chim. Acta 2018, 1037, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, C.Z.; Jones, C.M.; Yost, R.A.; Garrett, T.J.; Bowden, J.A. Optimization of Folch, Bligh-Dyer, and Matyash sample-to-extraction solvent ratios for human plasma-based lipidomics studies. Anal. Chim. Acta 2018, 1037, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Orsburn, B.C. Proteome discoverer—A community enhanced data processing suite for protein informatics. Proteomes 2021, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef]
- Xue, J.; Domingo-Almenara, X.; Guijas, C.; Palermo, A.; Rinschen, M.M.; Isbell, J.; Benton, H.P.; Siuzdak, G. Enhanced in-source fragmentation annotation enables novel data independent acquisition and autonomous METLIN molecular identification. Anal. Chem. 2020, 92, 6051–6059. [Google Scholar] [CrossRef]
- Weitkunat, K.; Schumann, S.; Nickel, D.; Hornemann, S.; Petzke, K.J.; Schulze, M.B.; Pfeiffer, A.F.; Klaus, S. Odd-chain fatty acids as a biomarker for dietary fiber intake: A novel pathway for endogenous production from propionate. Am. J. Clin. Nutr. 2017, 105, 1544–1551. [Google Scholar] [CrossRef]
- Zhukova, N.V. Lipids and fatty acids of nudibranch mollusks: Potential sources of bioactive compounds. Mar. Drugs 2014, 12, 4578–4592. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-P.; Lei, Q.-Y. Metabolite sensing and signaling in cell metabolism. Signal Transduct. Target. Ther. 2018, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Mok, J.-H.; Joo, M.; Duong, V.-A.; Cho, S.; Park, J.-M.; Eom, Y.-S.; Song, T.-H.; Lim, H.-J.; Lee, H. Proteomic and Metabolomic Analyses of Maggots in Porcine Corpses for Post-Mortem Interval Estimation. Appl. Sci. 2021, 11, 7885. [Google Scholar] [CrossRef]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion–switching, targeted mass spectrometry–based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [Google Scholar] [CrossRef]
- Ludwig, K.R.; Schroll, M.M.; Hummon, A.B. Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. J. Proteome Res. 2018, 17, 2480–2490. [Google Scholar] [CrossRef]
- Zhang, X. Less is More: Membrane Protein Digestion Beyond Urea–Trypsin Solution for Next-level Proteomics*. Mol. Cell. Proteom. 2015, 14, 2441–2453. [Google Scholar] [CrossRef]
- Loo, R.O.; Dales, N.; Andrews, P. Surfactant effects on protein structure examined by electrospray ionization mass spectrometry. Protein Sci. 1994, 3, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Zhang, Y.; Gui, S.Q.; Feng, Y.R.; Han, H.C.; Mao, S.H.; Lu, F.P. Electro-ultrafiltration to remove sodium dodecyl sulfate in proteins extracted for proteomics. RSC Adv. 2017, 7, 25144–25148. [Google Scholar] [CrossRef]
- Loo, R.R.; Dales, N.; Andrews, P.C. The effect of detergents on proteins analyzed by electrospray ionization. Methods Mol. Biol. 1996, 61, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Li, Y.; Hu, Q.; Zhu, L.; Huang, Z.; Yi, J.; Yang, X.; Hu, J.; Feng, X. Preparation and Drug Release Study of Novel Nanopharmaceuticals with Polysorbate 80 Surface Adsorption. J. Nanomater. 2018, 2018, 4718045. [Google Scholar] [CrossRef]
- Elshaier, Y.A.M.M.; Mostafa, A.; Valerio-Pascua, F.; Tesch, M.L.; Costin, J.M.; Rahaghi, F.F. Chlorpheniramine Maleate Displays Multiple Modes of Antiviral Action Against SARS-CoV-2: A Mechanistic Study. bioRxiv 2023. [Google Scholar] [CrossRef]
- Chen, S.; Hoene, M.; Li, J.; Li, Y.; Zhao, X.; Häring, H.-U.; Schleicher, E.D.; Weigert, C.; Xu, G.; Lehmann, R. Simultaneous extraction of metabolome and lipidome with methyl tert-butyl ether from a single small tissue sample for ultra-high performance liquid chromatography/mass spectrometry. J. Chromatogr. A 2013, 1298, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Kind, T.; Yoon, Y.-R.; Fiehn, O.; Liu, K.-H. Comparative evaluation of extraction methods for simultaneous mass-spectrometric analysis of complex lipids and primary metabolites from human blood plasma. Anal. Bioanal. Chem. 2014, 406, 7275–7286. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | p-Value | Gene Name | |
|---|---|---|---|
| FASP | Shigellosis | 3.00 × 10−6 | BECN1, GSK3B, ROCK1, PXN, UBE2D1, TRAF2, WIPI2, MAPK14, ATG12, NFKB1, MYL12B, SEPTIN8, RPTOR, MAPK9, CYTH2, H3C15, H3-4, RRAGB, PLCG1, H3-3A |
| Non-alcoholic fatty liver disease | 2.01 × 10−4 | GSK3B, UQCRB, INSR, NDUFA10, TRAF2, COX6C, SDHA, MAPK14, NFKB1, MAPK9, NDUFAB1, UQCRFS1, UQCRC2 | |
| Thermogenesis | 7.91 × 10−4 | COA3, UQCRB, NDUFA10, ACSL4, COX6C, SDHA, MAPK14, ARID1B, RPTOR, RPS6KA3, CREB1, NDUFAB1, COX11, UQCRFS1, UQCRC2 | |
| Alzheimer’s disease | 1.11 × 10−3 | BECN1, RTN3, GSK3B, FZD2, UQCRB, APAF1, INSR, NDUFA10, TRAF2, WIPI2, COX6C, SDHA, NFKB1, MAPK9, PPP3R1, PSMA4, NDUFAB1, UQCRFS1, CTNNB1, UQCRC2 | |
| RNA degradation | 2.02 × 10−3 | HSPA9, EXOSC5, CNOT7, EXOSC9, CNOT10, PABPC1, EXOSC2, EXOSC1 | |
| S-Trap | RNA degradation | 7.35 × 10−4 | HSPA9, CNOT7, EXOSC9, EXOSC8, PARN, PABPC1, NUDT16, ENO3, EXOSC2, EXOSC1, LSM3 |
| Basal transcription factors | 1.36 × 10−3 | TAF6L, GTF2A2, TAF15, TBP, ERCC2, TAF5, GTF2H3, MNAT1 | |
| Parkinson’s disease | 2.58 × 10−3 | NDUFA8, NDUFB8, MT-ND5, NDUFA6, TUBAL3, APAF1, NDUFB3, HTRA2, ITPR2, SDHC, SDHA, MT-ND2, SLC39A14, MAPK8, PSMA4, NDUFS6, MFN2, PLCG1, UQCRC2, NDUFV2, SLC39A1 | |
| Huntington’s disease | 2.80 × 10−3 | DCTN5, NDUFA8, NDUFB8, MT-ND5, NDUFA6, TBP, TUBAL3, APAF1, NDUFB3, GPX7, SLC1A3, SDHC, SDHA, MT-ND2, AP2A2, MAPK8, PSMA4, NDUFS6, EP300, UQCRC2, NDUFV2, POLR2I, POLR2J | |
| mRNA surveillance pathway | 3.53 × 10−3 | PPP2CA, CPSF4, FIP1L1, NCBP2, CSTF2T, PPP2R5A, PABPC1, MSI2, BCL2L2-PABPN1, SMG5, SMG6 |
| Term | Count | p-Value | |
|---|---|---|---|
| FASP | late endosome membrane (GO:0031902) | 11 | 6.02 × 10−8 |
| cytosol (GO:0005829) | 68 | 6.10 × 10−8 | |
| Golgi apparatus (GO:0005794) | 25 | 5.77 × 10−7 | |
| ubiquitin ligase complex (GO:0000151) | 9 | 8.68 × 10−7 | |
| endoplasmic reticulum (GO:0005783) | 24 | 2.52 × 10−6 | |
| macromolecular complex (GO:0032991) | 18 | 3.40 × 10−6 | |
| intracellular membrane-bounded organelle (GO:0043231) | 21 | 1.16 × 10−5 | |
| membrane (GO:0016020) | 46 | 1.61 × 10−5 | |
| trans-Golgi network (GO:0005802) | 9 | 4.28 × 10−5 | |
| lysosome (GO:0005764) | 11 | 4.87 × 10−5 | |
| S-Trap | nucleoplasm (GO:0005654) | 80 | 8.39 × 10−13 |
| nuclear speck (GO:0016607) | 23 | 7.65 × 10−11 | |
| nuclear membrane (GO:0031965) | 18 | 2.22 × 10−10 | |
| centrosome (GO:0005813) | 25 | 3.80 × 10−10 | |
| mitochondrial respiratory chain complex I (GO:0005747) | 9 | 1.79 × 10−8 | |
| kinetochore (GO:0000776) | 13 | 2.78 × 10−8 | |
| cytosol (GO:0005829) | 86 | 5.28 × 10−8 | |
| mitochondrial inner membrane (GO:0005743) | 19 | 4.50 × 10−7 | |
| nuclear envelope (GO:0005635) | 12 | 2.57 × 10−6 | |
| actin cytoskeleton (GO:0015629) | 13 | 5.96 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mok, J.-H.; Joo, M.; Cho, S.; Duong, V.-A.; Song, H.; Park, J.-M.; Lee, H. Optimizing MS-Based Multi-Omics: Comparative Analysis of Protein, Metabolite, and Lipid Extraction Techniques. Metabolites 2024, 14, 34. https://doi.org/10.3390/metabo14010034
Mok J-H, Joo M, Cho S, Duong V-A, Song H, Park J-M, Lee H. Optimizing MS-Based Multi-Omics: Comparative Analysis of Protein, Metabolite, and Lipid Extraction Techniques. Metabolites. 2024; 14(1):34. https://doi.org/10.3390/metabo14010034
Chicago/Turabian StyleMok, Jeong-Hun, Minjoong Joo, Seonghyeon Cho, Van-An Duong, Haneul Song, Jong-Moon Park, and Hookeun Lee. 2024. "Optimizing MS-Based Multi-Omics: Comparative Analysis of Protein, Metabolite, and Lipid Extraction Techniques" Metabolites 14, no. 1: 34. https://doi.org/10.3390/metabo14010034
APA StyleMok, J.-H., Joo, M., Cho, S., Duong, V.-A., Song, H., Park, J.-M., & Lee, H. (2024). Optimizing MS-Based Multi-Omics: Comparative Analysis of Protein, Metabolite, and Lipid Extraction Techniques. Metabolites, 14(1), 34. https://doi.org/10.3390/metabo14010034