


Metabolite Measurement in Index Substrate Drug Interaction Studies: A Review of the Literature and Recent New Drug Application Reviews

Abstract
:1. Introduction
2. Methods
3. Results
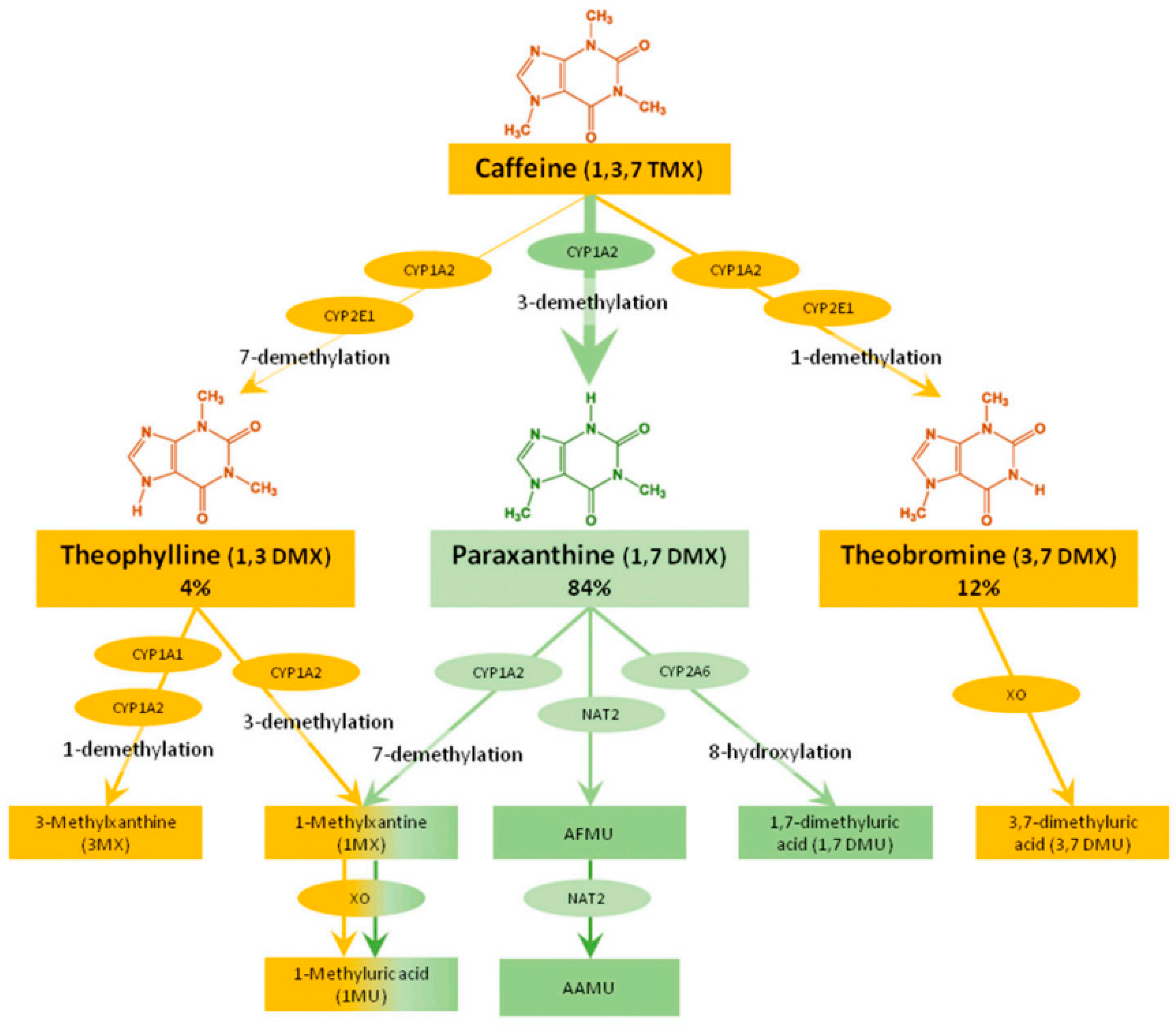
3.1. CYP1A2 Caffeine
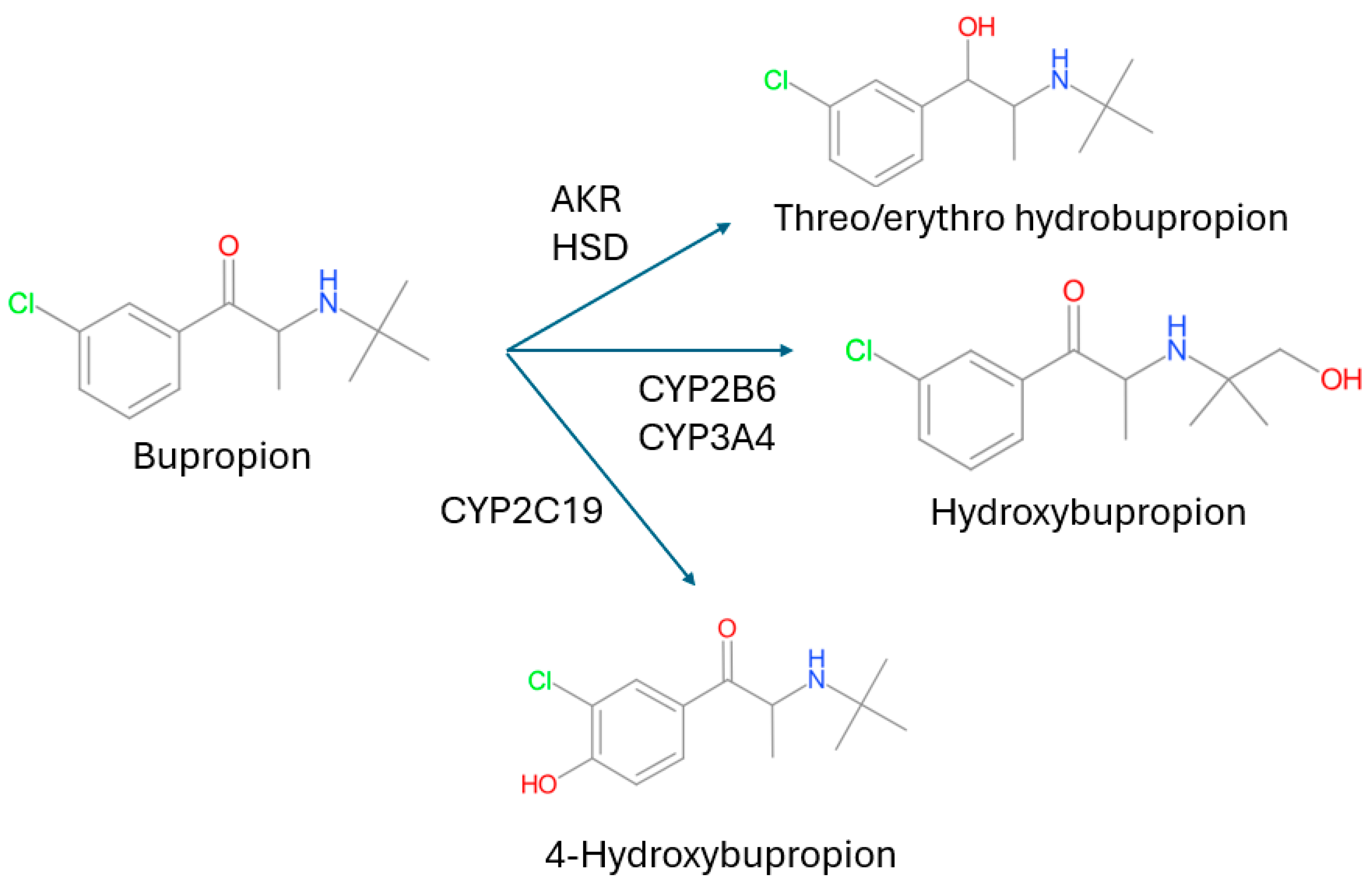
3.2. CYP2B6 Bupropion
3.3. CYP2C8 Repaglinide
3.4. CYP2C9 (S)-Warfarin and Flurbiprofen
3.4.1. (S)-Warfarin
3.4.2. Flurbiprofen
3.5. CYP2C19 Omeprazole
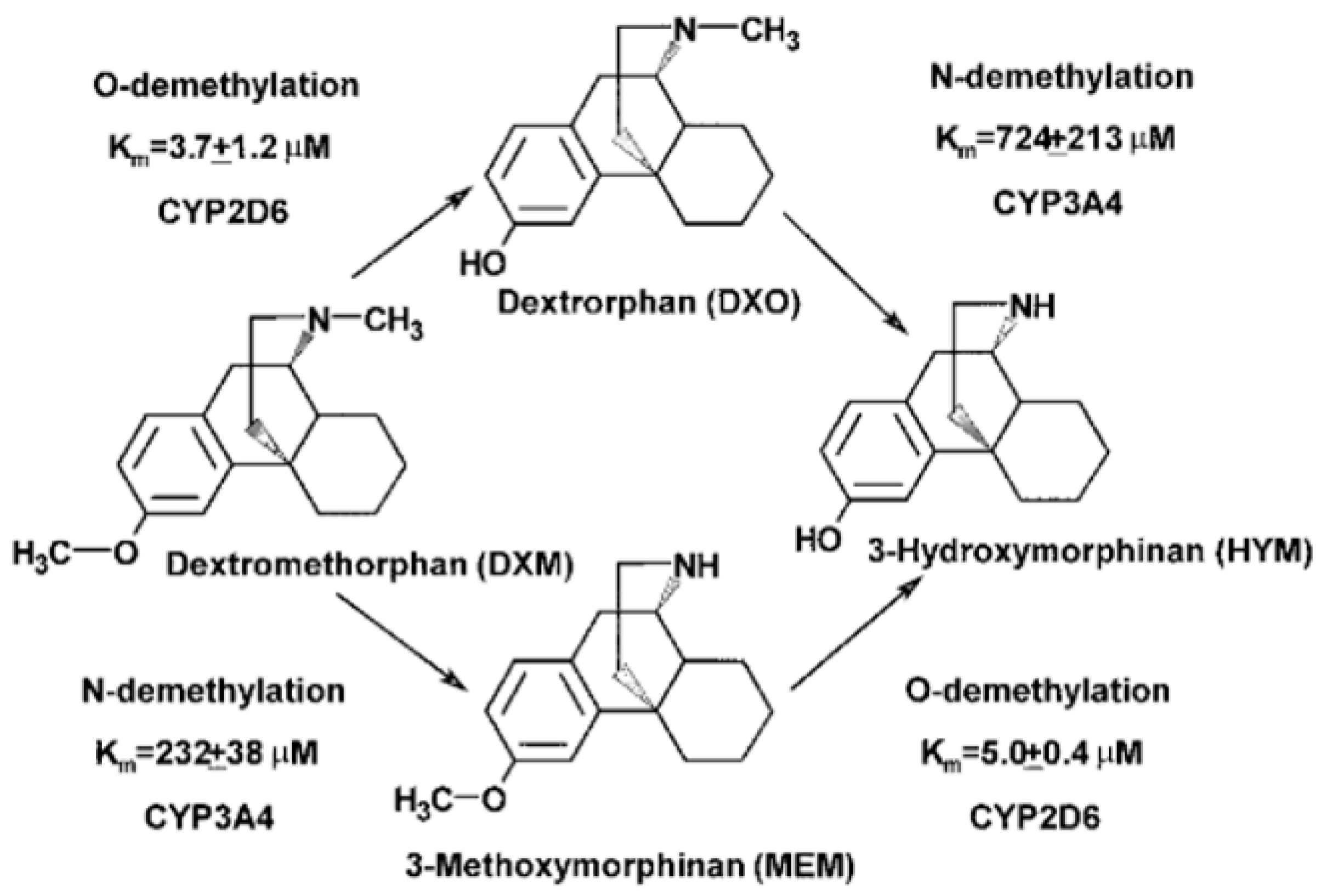
3.6. CYP2D6 Desipramine, Dextromethorphan, and Nebivolol
3.6.1. Desipramine
3.6.2. Dextromethorphan
3.6.3. Nebivolol
3.7. CYP3A Midazolam and Triazolam
3.7.1. Midazolam
3.7.2. Triazolam
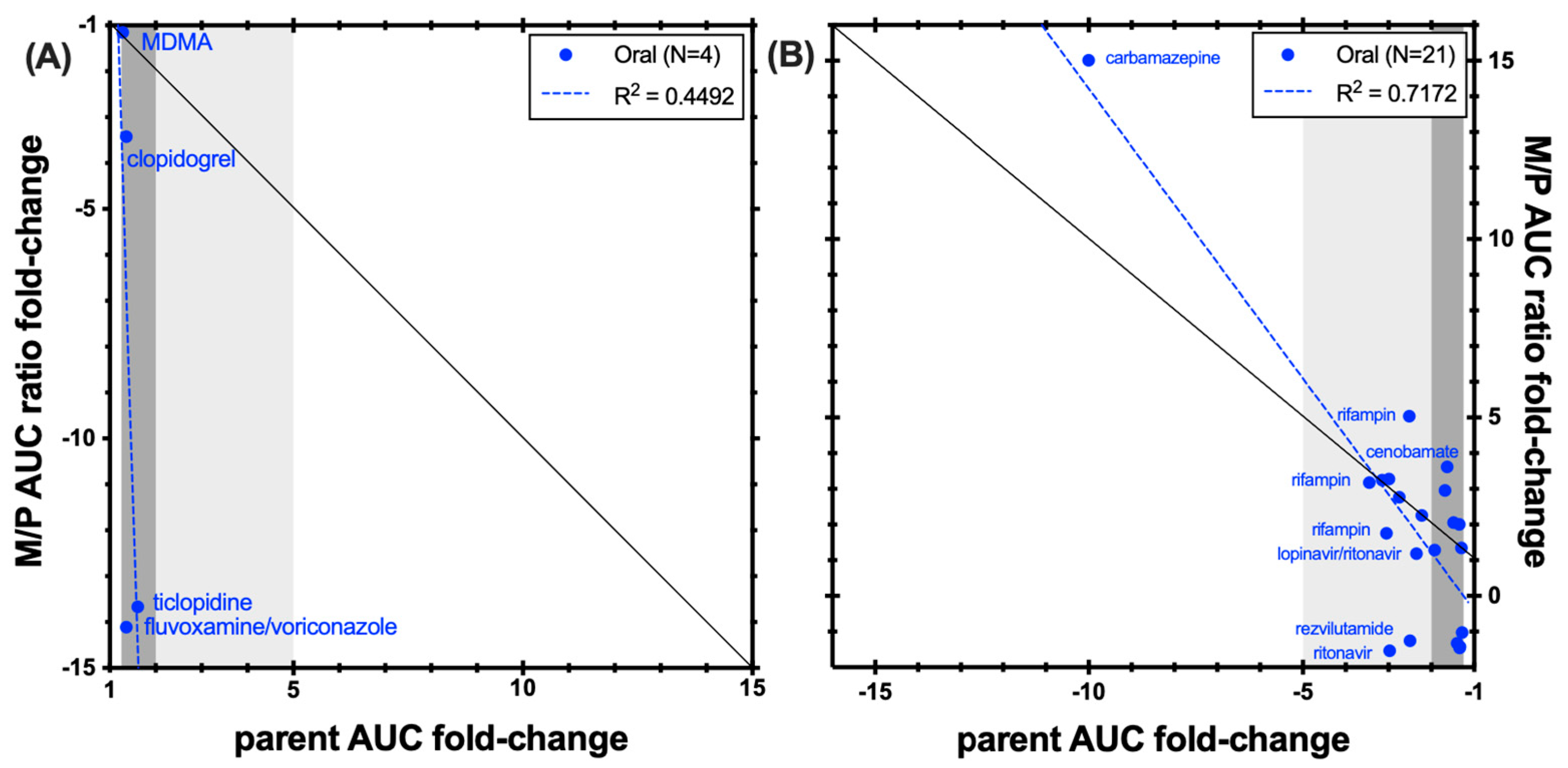
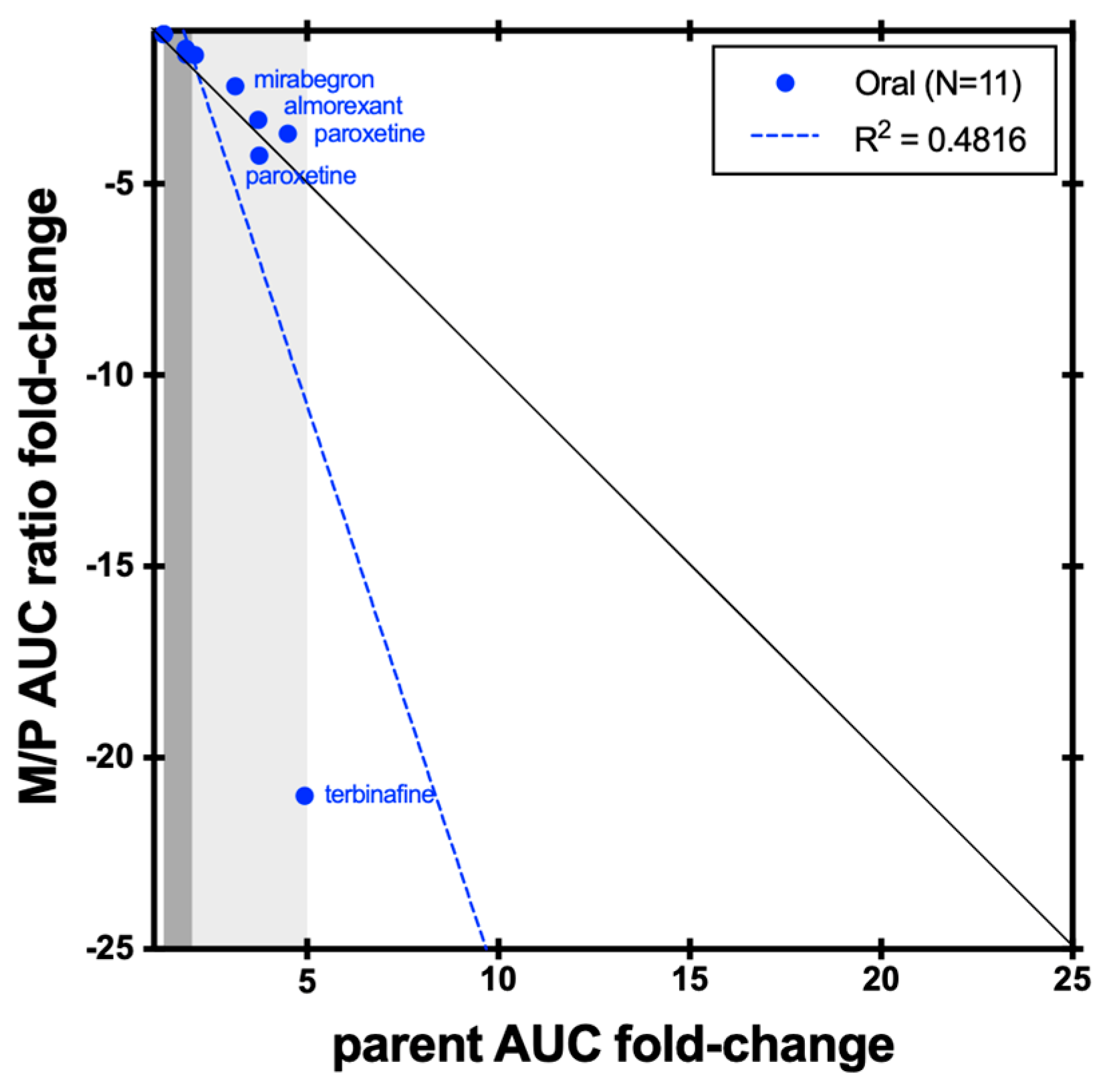
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ICH Harmonised Guideline on Drug Interaction Studies M12. Available online: https://database.ich.org/sites/default/files/ICH_M12_Step4_Guideline_2024_0521.pdf (accessed on 1 June 2024).
- VandenBrink, B.M.; Isoherranen, N. The role of metabolites in predicting drug-drug interactions: Focus on irreversible cytochrome P450 inhibition. Curr. Opin. Drug Discov. Devel. 2010, 13, 66–77. [Google Scholar] [PubMed]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical Studies on Drug-Drug Interactions Involving Metabolism and Transport: Methodology, Pitfalls, and Interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef]
- Magliocca, M.; Berger, B.; Lemoine, V.; Kaufmann, P.; Dingemanse, J. Value of Assessing 1-Hydroxymidazolam in Drug-Drug Interaction Studies with Midazolam as a Substrate of Cytochrome P450 3A. J. Clin. Pharmacol. 2024, 64, 1123–1129. [Google Scholar] [CrossRef]
- Nehlig, A. Interindividual Differences in Caffeine Metabolism and Factors Driving Caffeine Consumption. Pharmacol. Rev. 2018, 70, 384–411. [Google Scholar] [CrossRef]
- Grant, D.M.; Tang, B.K.; Campbell, M.E.; Kalow, W. Effect of allopurinol on caffeine disposition in man. Br. J. Clin. Pharmacol. 1986, 21, 454–458. [Google Scholar] [CrossRef]
- Fuhr, U.; Rost, K.L.; Engelhardt, R.; Sachs, M.; Liermann, D.; Belloc, C.; Beaune, P.; Janezic, S.; Grant, D.; Meyer, U.A.; et al. Evaluation of caffeine as a test drug for CYP1A2, NAT2 and CYP2E1 phenotyping in man by in vivo versus in vitro correlations. Pharmacogenetics 1996, 6, 159–176. [Google Scholar] [CrossRef]
- Carrillo, J.A.; Christensen, M.; Ramos, S.I.; Alm, C.; Dahl, M.L.; Benitez, J.; Bertilsson, L. Evaluation of caffeine as an in vivo probe for CYP1A2 using measurements in plasma, saliva, and urine. Ther. Drug Monit. 2000, 22, 409–417. [Google Scholar] [CrossRef]
- Tucker, G.T.; Rostami-Hodjegan, A.; Jackson, P.R. Determination of drug-metabolizing enzyme activity in vivo: Pharmacokinetic and statistical issues. Xenobiotica 1998, 28, 1255–1273. [Google Scholar] [CrossRef]
- Culm-Merdek, K.E.; von Moltke, L.L.; Harmatz, J.S.; Greenblatt, D.J. Fluvoxamine impairs single-dose caffeine clearance without altering caffeine pharmacodynamics. Br. J. Clin. Pharmacol. 2005, 60, 486–493. [Google Scholar] [CrossRef]
- Mitchell, D.C.; Knight, C.A.; Hockenberry, J.; Teplansky, R.; Hartman, T.J. Beverage caffeine intakes in the U.S. Food Chem. Toxicol. 2014, 63, 136–142. [Google Scholar] [CrossRef]
- Mendes, V.M.; Coelho, M.; Tomé, A.R.; Cunha, R.A.; Manadas, B. Validation of an LC-MS/MS Method for the Quantification of Caffeine and Theobromine Using Non-Matched Matrix Calibration Curve. Molecules 2019, 24, 2863. [Google Scholar] [CrossRef] [PubMed]
- ICH Harmonised Guideline on Bioanalytical Method Validation and Study Sample Analysis M10. Available online: https://database.ich.org/sites/default/files/M10_Guideline_Step4_2022_0524.pdf (accessed on 1 May 2024).
- Gufford, B.T.; Metzger, I.F.; Bamfo, N.O.; Benson, E.A.; Masters, A.R.; Lu, J.B.L.; Desta, Z. Influence of CYP2B6 Pharmacogenetics on Stereoselective Inhibition and Induction of Bupropion Metabolism by Efavirenz in Healthy Volunteers. J. Pharmacol. Exp. Ther. 2022, 382, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Price, L.S.; Isoherranen, N. Stereoselective Metabolism of Bupropion to OH-bupropion, Threohydrobupropion, Erythrohydrobupropion, and 4′-OH-bupropion in vitro. Drug Metab. Dispos. 2016, 44, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Kharasch, E.D.; Mitchell, D.; Coles, R. Stereoselective bupropion hydroxylation as an in vivo phenotypic probe for cytochrome P4502B6 (CYP2B6) activity. J. Clin. Pharmacol. 2008, 48, 464–474. [Google Scholar] [CrossRef]
- Turpeinen, M.; Tolonen, A.; Uusitalo, J.; Jalonen, J.; Pelkonen, O.; Laine, K. Effect of clopidogrel and ticlopidine on cytochrome P450 2B6 activity as measured by bupropion hydroxylation. Clin. Pharmacol. Ther. 2005, 77, 553–559. [Google Scholar] [CrossRef]
- FDA Drug Approval Package: PRANDIN (repaglinide). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/020741s044lbl.pdf (accessed on 1 June 2024).
- Bidstrup, T.B.; Bjørnsdottir, I.; Sidelmann, U.G.; Thomsen, M.S.; Hansen, K.T. CYP2C8 and CYP3A4 are the principal enzymes involved in the human in vitro biotransformation of the insulin secretagogue repaglinide. Br. J. Clin. Pharmacol. 2003, 56, 305–314. [Google Scholar] [CrossRef]
- Säll, C.; Houston, J.B.; Galetin, A. A comprehensive assessment of repaglinide metabolic pathways: Impact of choice of in vitro system and relative enzyme contribution to in vitro clearance. Drug Metab. Dispos. 2012, 40, 1279–1289. [Google Scholar] [CrossRef]
- Honkalammi, J.; Niemi, M.; Neuvonen, P.J.; Backman, J.T. Dose-dependent interaction between gemfibrozil and repaglinide in humans: Strong inhibition of CYP2C8 with subtherapeutic gemfibrozil doses. Drug Metab. Dispos. 2011, 39, 1977–1986. [Google Scholar] [CrossRef]
- Kalliokoski, A.; Backman, J.T.; Kurkinen, K.J.; Neuvonen, P.J.; Niemi, M. Effects of gemfibrozil and atorvastatin on the pharmacokinetics of repaglinide in relation to SLCO1B1 polymorphism. Clin. Pharmacol. Ther. 2008, 84, 488–496. [Google Scholar] [CrossRef]
- Tornio, A.; Niemi, M.; Neuvonen, M.; Laitila, J.; Kalliokoski, A.; Neuvonen, P.J.; Backman, J.T. The effect of gemfibrozil on repaglinide pharmacokinetics persists for at least 12 h after the dose: Evidence for mechanism-based inhibition of CYP2C8 in vivo. Clin. Pharmacol. Ther. 2008, 84, 403–411. [Google Scholar] [CrossRef]
- Backman, J.T.; Honkalammi, J.; Neuvonen, M.; Kurkinen, K.J.; Tornio, A.; Niemi, M.; Neuvonen, P.J. CYP2C8 activity recovers within 96 hours after gemfibrozil dosing: Estimation of CYP2C8 half-life using repaglinide as an in vivo probe. Drug Metab. Dispos. 2009, 37, 2359–2366. [Google Scholar] [CrossRef] [PubMed]
- Honkalammi, J.; Niemi, M.; Neuvonen, P.J.; Backman, J.T. Gemfibrozil is a strong inactivator of CYP2C8 in very small multiple doses. Clin. Pharmacol. Ther. 2012, 91, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Rettie, A.E.; Korzekwa, K.R.; Kunze, K.L.; Lawrence, R.F.; Eddy, A.C.; Aoyama, T.; Gelboin, H.V.; Gonzalez, F.J.; Trager, W.F. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: A role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem. Res. Toxicol. 1992, 5, 54–59. [Google Scholar] [CrossRef]
- Kaminsky, L.S.; Zhang, Z.Y. Human P450 metabolism of warfarin. Pharmacol. Ther. 1997, 73, 67–74. [Google Scholar] [CrossRef]
- FDA Drug Approval Package: COUMADIN (warfarin). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/009218s118lbl.pdf (accessed on 1 June 2024).
- FDA Drug Approval Package: ANSAID (flurbiprofen). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/018766s020lbl.pdf (accessed on 1 June 2024).
- Tracy, T.S.; Marra, C.; Wrighton, S.A.; Gonzalez, F.J.; Korzekwa, K.R. Studies of flurbiprofen 4′-hydroxylation. Additional evidence suggesting the sole involvement of cytochrome P450 2C9. Biochem. Pharmacol. 1996, 52, 1305–1309. [Google Scholar] [CrossRef]
- Tracy, T.S.; Rosenbluth, B.W.; Wrighton, S.A.; Gonzalez, F.J.; Korzekwa, K.R. Role of cytochrome P450 2C9 and an allelic variant in the 4′-hydroxylation of (R)- and (S)-flurbiprofen. Biochem. Pharmacol. 1995, 49, 1269–1275. [Google Scholar] [CrossRef]
- Kuehl, G.E.; Lampe, J.W.; Potter, J.D.; Bigler, J. Glucuronidation of nonsteroidal anti-inflammatory drugs: Identifying the enzymes responsible in human liver microsomes. Drug Metab. Dispos. 2005, 33, 1027–1035. [Google Scholar] [CrossRef]
- Mano, Y.; Usui, T.; Kamimura, H. Predominant contribution of UDP-glucuronosyltransferase 2B7 in the glucuronidation of racemic flurbiprofen in the human liver. Drug Metab. Dispos. 2007, 35, 1182–1187. [Google Scholar] [CrossRef]
- Kumar, V.; Brundage, R.C.; Oetting, W.S.; Leppik, I.E.; Tracy, T.S. Differential genotype dependent inhibition of CYP2C9 in humans. Drug Metab. Dispos. 2008, 36, 1242–1248. [Google Scholar] [CrossRef]
- Greenblatt, D.J.; von Moltke, L.L.; Perloff, E.S.; Luo, Y.; Harmatz, J.S.; Zinny, M.A. Interaction of flurbiprofen with cranberry juice, grape juice, tea, and fluconazole: In vitro and clinical studies. Clin. Pharmacol. Ther. 2006, 79, 125–133. [Google Scholar] [CrossRef]
- Hanley, M.J.; Masse, G.; Harmatz, J.S.; Cancalon, P.F.; Dolnikowski, G.G.; Court, M.H.; Greenblatt, D.J. Effect of blueberry juice on clearance of buspirone and flurbiprofen in human volunteers. Br. J. Clin. Pharmacol. 2013, 75, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Hanley, M.J.; Masse, G.; Harmatz, J.S.; Court, M.H.; Greenblatt, D.J. Pomegranate juice and pomegranate extract do not impair oral clearance of flurbiprofen in human volunteers: Divergence from in vitro results. Clin. Pharmacol. Ther. 2012, 92, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Bosilkovska, M.; Samer, C.F.; Déglon, J.; Rebsamen, M.; Staub, C.; Dayer, P.; Walder, B.; Desmeules, J.A.; Daali, Y. Geneva cocktail for cytochrome p450 and P-glycoprotein activity assessment using dried blood spots. Clin. Pharmacol. Ther. 2014, 96, 349–359. [Google Scholar] [CrossRef]
- Daali, Y.; Samer, C.; Déglon, J.; Thomas, A.; Chabert, J.; Rebsamen, M.; Staub, C.; Dayer, P.; Desmeules, J. Oral flurbiprofen metabolic ratio assessment using a single-point dried blood spot. Clin. Pharmacol. Ther. 2012, 91, 489–496. [Google Scholar] [CrossRef]
- Bachmann, F.; Duthaler, U.; Krähenbühl, S. Effect of deglucuronidation on the results of the Basel phenotyping cocktail. Br. J. Clin. Pharmacol. 2021, 87, 4608–4618. [Google Scholar] [CrossRef]
- Bachmann, F.; Duthaler, U.; Meyer Zu Schwabedissen, H.E.; Puchkov, M.; Huwyler, J.; Haschke, M.; Krähenbühl, S. Metamizole is a Moderate Cytochrome P450 Inducer Via the Constitutive Androstane Receptor and a Weak Inhibitor of CYP1A2. Clin. Pharmacol. Ther. 2021, 109, 1505–1516. [Google Scholar] [CrossRef]
- Zahner, C.; Kruttschnitt, E.; Uricher, J.; Lissy, M.; Hirsch, M.; Nicolussi, S.; Krähenbühl, S.; Drewe, J. No Clinically Relevant Interactions of St. John’s Wort Extract Ze 117 Low in Hyperforin With Cytochrome P450 Enzymes and P-glycoprotein. Clin. Pharmacol. Ther. 2019, 106, 432–440. [Google Scholar] [CrossRef]
- Haberer, L.J.; McSherry, I.; Cargill, A.; McCarthy, L. Effects of vercirnon on the activity of CYP3A4, CYP2C19 and CYP2C8 enzymes and BCRP and OATP1B1 transporters using probe substrates. Eur. J. Clin. Pharmacol. 2014, 70, 37–45. [Google Scholar] [CrossRef]
- Yu, A.; Haining, R.L. Comparative contribution to dextromethorphan metabolism by cytochrome P450 isoforms in vitro: Can dextromethorphan be used as a dual probe for both CTP2D6 and CYP3A activities? Drug Metab. Dispos. 2001, 29, 1514–1520. [Google Scholar]
- Strauch, K.; Lutz, U.; Bittner, N.; Lutz, W.K. Dose-response relationship for the pharmacokinetic interaction of grapefruit juice with dextromethorphan investigated by human urinary metabolite profiles. Food Chem. Toxicol. 2009, 47, 1928–1935. [Google Scholar] [CrossRef]
- Stage, T.B.; Graff, M.; Wong, S.; Rasmussen, L.L.; Nielsen, F.; Pottegård, A.; Brøsen, K.; Kroetz, D.L.; Khojasteh, S.C.; Damkier, P. Dicloxacillin induces CYP2C19, CYP2C9 and CYP3A4 in vivo and in vitro. Br. J. Clin. Pharmacol. 2018, 84, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Pope, L.E.; Khalil, M.H.; Berg, J.E.; Stiles, M.; Yakatan, G.J.; Sellers, E.M. Pharmacokinetics of dextromethorphan after single or multiple dosing in combination with quinidine in extensive and poor metabolizers. J. Clin. Pharmacol. 2004, 44, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- FDA Drug Approval Package: NUEDEXTA (Dextromethorphan Hydrobromide and Quinidine Sulfate). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021879s014lbl.pdf (accessed on 1 June 2024).
- Hu, X.; Lan, T.; Dai, D.; Xu, R.A.; Yuan, L.; Zhou, Q.; Li, Y.; Cai, J.; Hu, G. Evaluation of 24 CYP2D6 Variants on the Metabolism of Nebivolol In Vitro. Drug Metab. Dispos. 2016, 44, 1828–1831. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.; Kharasch, E.D.; Thummel, K.T.; Narang, V.S.; Hoffer, C.J.; Unadkat, J.D. Simultaneous measurement of in vivo P-glycoprotein and cytochrome P450 3A activities. J. Clin. Pharmacol. 2006, 46, 1313–1319. [Google Scholar] [CrossRef]
- Heizmann, P.; Ziegler, W.H. Excretion and metabolism of 14C-midazolam in humans following oral dosing. Arzneimittelforschung 1981, 31, 2220–2223. [Google Scholar]
- Dundee, J.W.; Halliday, N.J.; Harper, K.W.; Brogden, R.N. Midazolam. A review of its pharmacological properties and therapeutic use. Drugs 1984, 28, 519–543. [Google Scholar] [CrossRef]
- Tian, D.D.; Leonowens, C.; Cox, E.J.; González-Pérez, V.; Frederick, K.S.; Scarlett, Y.V.; Fisher, M.B.; Paine, M.F. Indinavir Increases Midazolam N-Glucuronidation in Humans: Identification of an Alternate CYP3A Inhibitor Using an In Vitro to In Vivo Approach. Drug Metab. Dispos. 2019, 47, 724–731. [Google Scholar] [CrossRef]
- Seo, K.A.; Bae, S.K.; Choi, Y.K.; Choi, C.S.; Liu, K.H.; Shin, J.G. Metabolism of 1′- and 4-hydroxymidazolam by glucuronide conjugation is largely mediated by UDP-glucuronosyltransferases 1A4, 2B4, and 2B7. Drug Metab. Dispos. 2010, 38, 2007–2013. [Google Scholar] [CrossRef]
- Hyland, R.; Osborne, T.; Payne, A.; Kempshall, S.; Logan, Y.R.; Ezzeddine, K.; Jones, B. In vitro and in vivo glucuronidation of midazolam in humans. Br. J. Clin. Pharmacol. 2009, 67, 445–454. [Google Scholar] [CrossRef]
- Johnson, T.N.; Rostami-Hodjegan, A.; Goddard, J.M.; Tanner, M.S.; Tucker, G.T. Contribution of midazolam and its 1-hydroxy metabolite to preoperative sedation in children: A pharmacokinetic-pharmacodynamic analysis. Br. J. Anaesth. 2002, 89, 428–437. [Google Scholar] [CrossRef]
- Prommer, E. Midazolam: An essential palliative care drug. Palliat. Care Soc. Pract. 2020, 14, 2632352419895527. [Google Scholar] [CrossRef]
- Nguyen, J.T.; Tian, D.D.; Tanna, R.S.; Arian, C.M.; Calamia, J.C.; Rettie, A.E.; Thummel, K.E.; Paine, M.F. An Integrative Approach to Elucidate Mechanisms Underlying the Pharmacokinetic Goldenseal-Midazolam Interaction: Application of In Vitro Assays and Physiologically Based Pharmacokinetic Models to Understand Clinical Observations. J. Pharmacol. Exp. Ther. 2023, 387, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V.; Hariparsad, N.; Li, F.; Desai, P.; Thummel, K.E.; Unadkat, J.D. Cytochrome P450 enzymes and transporters induced by anti-human immunodeficiency virus protease inhibitors in human hepatocytes: Implications for predicting clinical drug interactions. Drug Metab. Dispos. 2007, 35, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, O.A.; Maurer, T.S.; Kish, M.; Cardenas, E.; Boldt, S.; Nettleton, D. A combined model for predicting CYP3A4 clinical net drug-drug interaction based on CYP3A4 inhibition, inactivation, and induction determined in vitro. Drug Metab. Dispos. 2008, 36, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Faucette, S.R.; Wang, H.; LeCluyse, E.L. Modulation of UDP-glucuronosyltransferase 1A1 in primary human hepatocytes by prototypical inducers. J. Biochem. Mol. Toxicol. 2005, 19, 96–108. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Index Substrate | Index DDI Study | Metabolite Measured (%) | Cocktail Study | Metabolite Measured (%) |
|---|---|---|---|---|---|
| CYP1A2 | caffeine | 474 | 300 (63%) | 195 | 132 (68%) |
| CYP2B6 | bupropion | 112 | 92 (82%) | 15 | 15 (100%) |
| CYP2B6 | (R)-bupropion | 13 | 12 (92%) | 8 | 8 (100%) |
| CYP2B6 | (S)-bupropion | 15 | 14 (93%) | 8 | 8 (100%) |
| CYP2C8 | repaglinide | 132 | 38 (29%) | 6 | 0 |
| CYP2C9 | flurbiprofen 1 | 47 | 40 (85%) | 20 | 15 (75%) |
| CYP2C9 | (S)-warfarin | 390 | 41 (11%) | 55 | 10 (18%) |
| CYP2C19 | omeprazole | 331 | 201 (61%) | 143 | 95 (66%) |
| CYP2C19 | (R)-omeprazole | 2 | 2 (100%) | 0 | 0 |
| CYP2C19 | esomeprazole 2 | 20 | 4 (20%) | 0 | 0 |
| CYP2D6 | dextromethorphan | 263 | 215 (82%) | 106 | 78 (74%) |
| CYP2D6 | desipramine | 81 | 43 (53%) | 0 | 0 |
| CYP2D6 | nebivolol | 11 | 7 (63%) | 0 | 0 |
| CYP2D6 | (d)-nebivolol | 13 | 0 | 0 | 0 |
| CYP2D6 | (l)-nebivolol | 13 | 0 | 0 | 0 |
| CYP3A | midazolam | 1283 | 468 (36%) | 241 | 132 (55%) |
| CYP3A | triazolam | 61 | 5 (8%) | 0 | 0 |
| all | 3261 | 1466 (45%) | 797 | 477 (60%) |
| Enzyme | Index Substrate | Clinical Index Study | Metabolite Measured (%) |
|---|---|---|---|
| CYP1A2 | caffeine | 16 | 5 (31%) |
| CYP2B6 | bupropion | 10 | 3 (30%) |
| CYP2C8 | repaglinide | 11 | 0 |
| CYP2C9 | (S)-warfarin | 24 | 0 |
| CYP2C9 | flurbiprofen | 3 | 1 (33%) |
| CYP2C19 | omeprazole | 20 | 4 (20%) |
| CYP2D6 | desipramine | 2 | 0 |
| CYP2D6 | dextromethorphan | 11 | 4 (36%) |
| CYP2D6 | nebivolol | 0 | 0 |
| CYP3A | midazolam | 100 | 35 (35%) |
| CYP3A | triazolam | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Rioux, N.; Gardner, I.; Owens, K.; Ragueneau-Majlessi, I. Metabolite Measurement in Index Substrate Drug Interaction Studies: A Review of the Literature and Recent New Drug Application Reviews. Metabolites 2024, 14, 522. https://doi.org/10.3390/metabo14100522
Yu J, Rioux N, Gardner I, Owens K, Ragueneau-Majlessi I. Metabolite Measurement in Index Substrate Drug Interaction Studies: A Review of the Literature and Recent New Drug Application Reviews. Metabolites. 2024; 14(10):522. https://doi.org/10.3390/metabo14100522
Chicago/Turabian StyleYu, Jingjing, Nathalie Rioux, Iain Gardner, Katie Owens, and Isabelle Ragueneau-Majlessi. 2024. "Metabolite Measurement in Index Substrate Drug Interaction Studies: A Review of the Literature and Recent New Drug Application Reviews" Metabolites 14, no. 10: 522. https://doi.org/10.3390/metabo14100522