
Novel Approaches to Studying SLC13A5 Disease
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
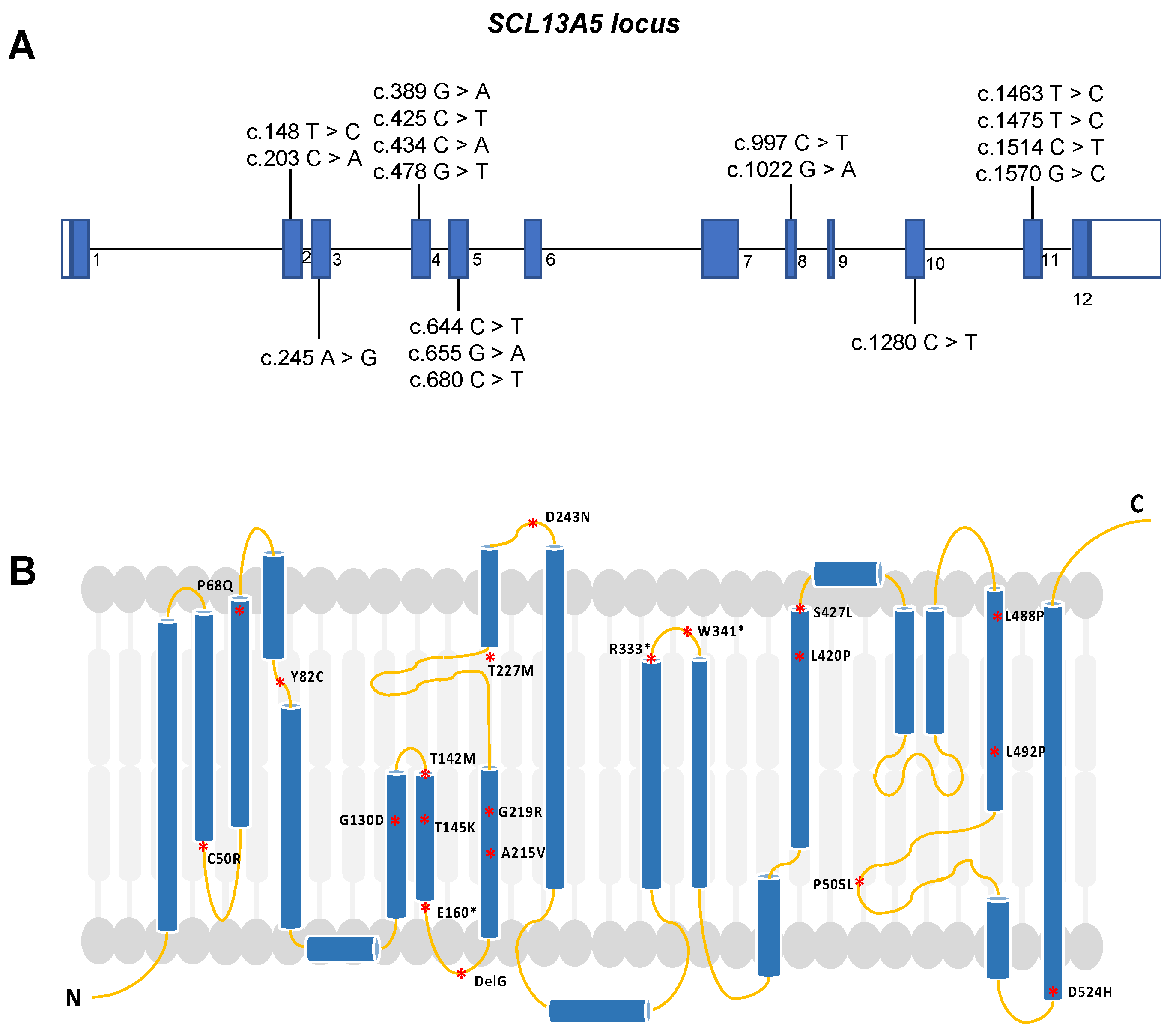
2. SLC13A5 Variants
3. SLC13A5 Expression in Different Cell Types
3.1. SLC13A5 Expression and Function in the Brain
3.2. SLC13A5 Expression and Function in the Liver
3.3. SLC13A5 Expression and Function in Various Cell Types
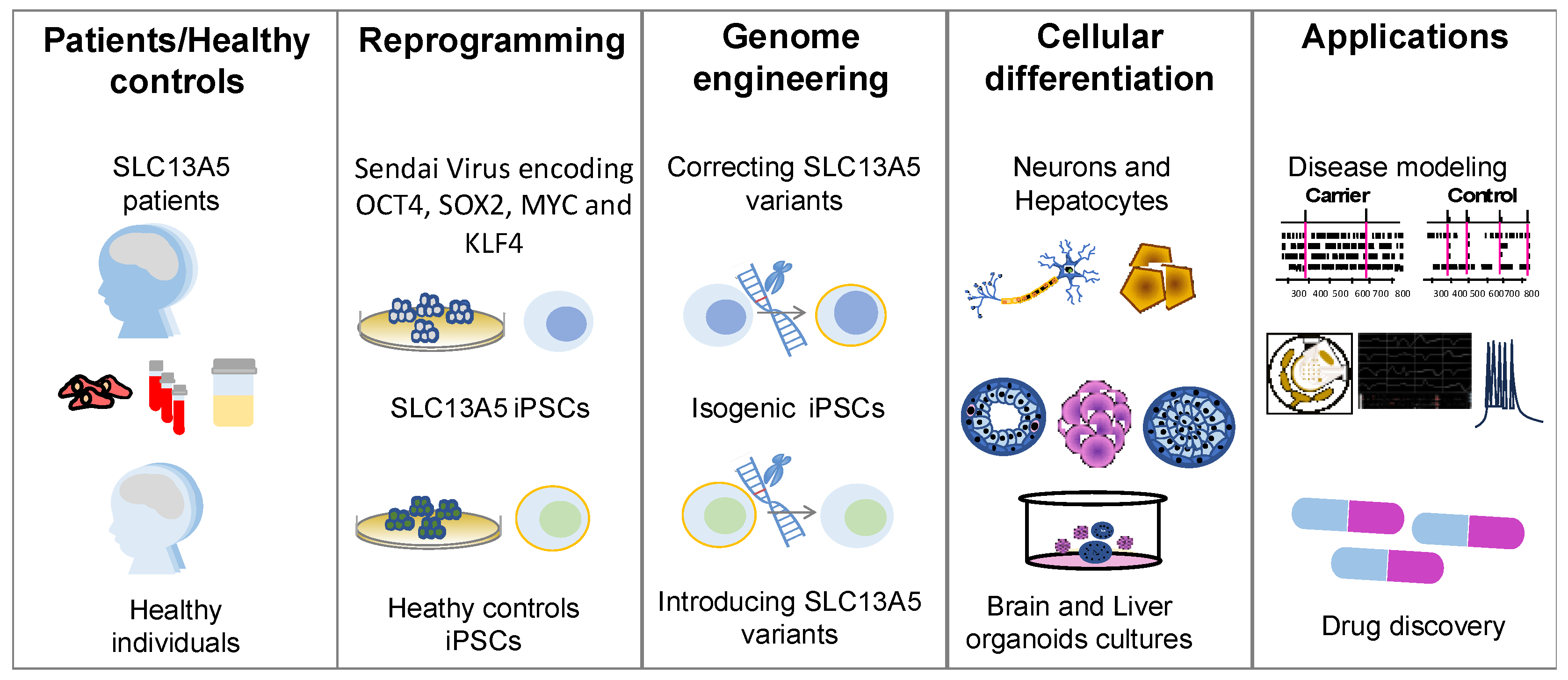
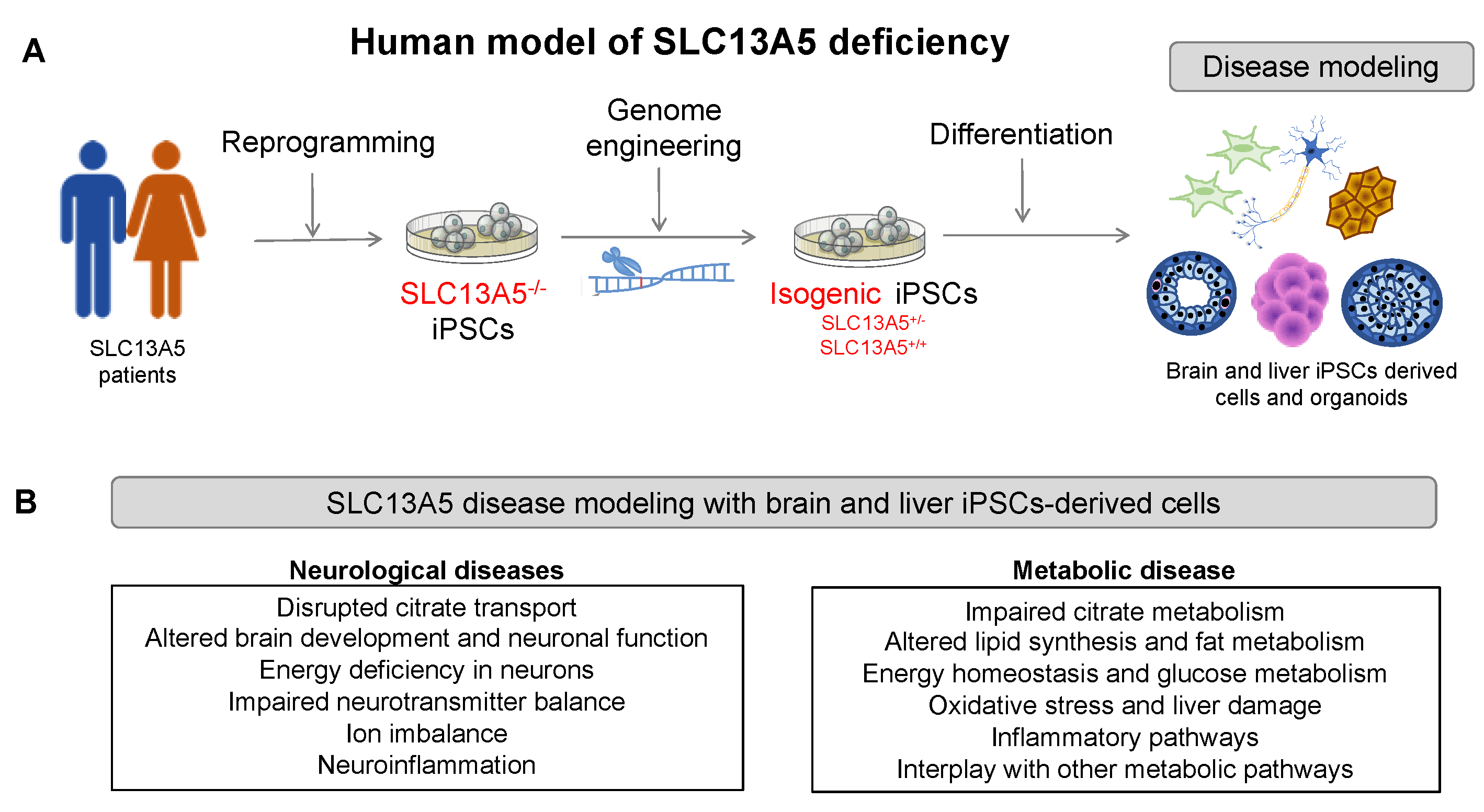
4. Physiologically Relevant Cellular Models to Study Human SLC13A5 Disease
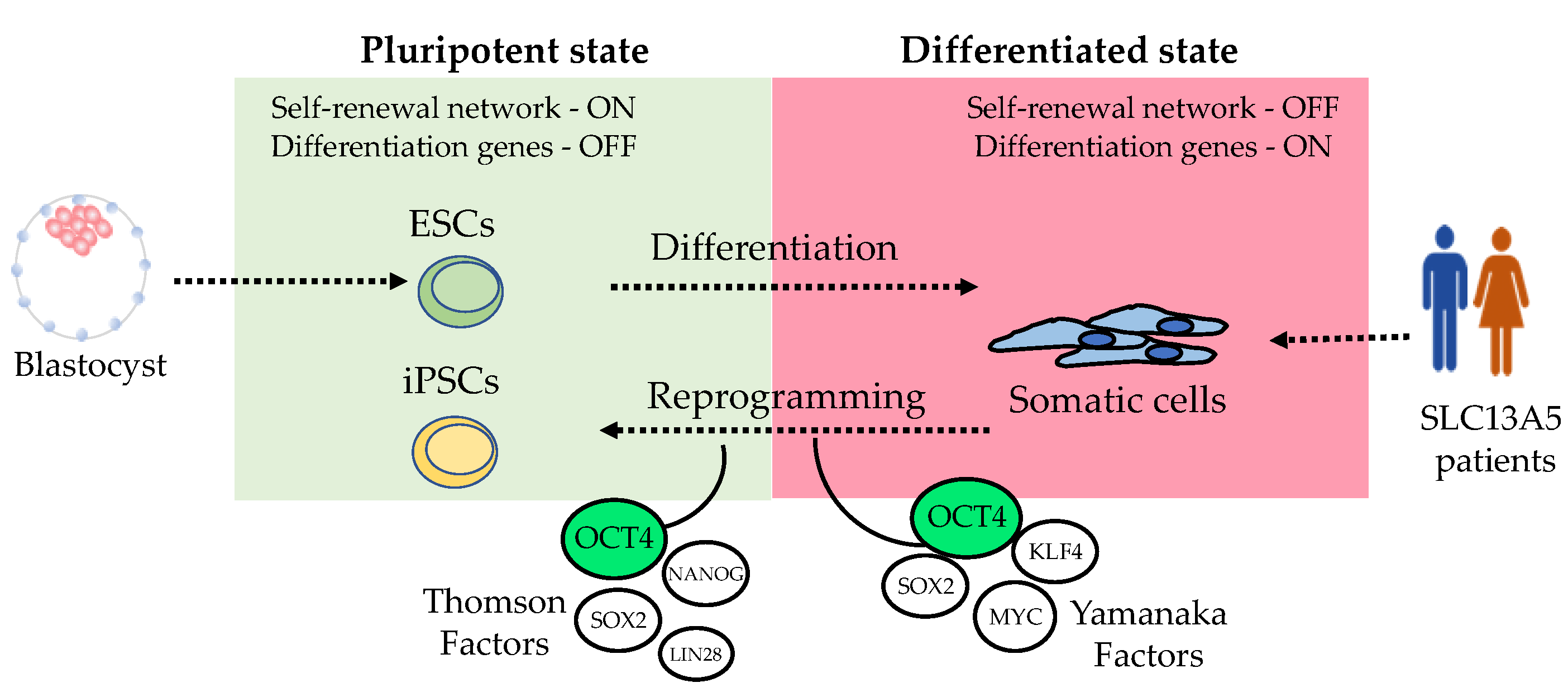
4.1. Induced Pluripotent Stem Cells Models
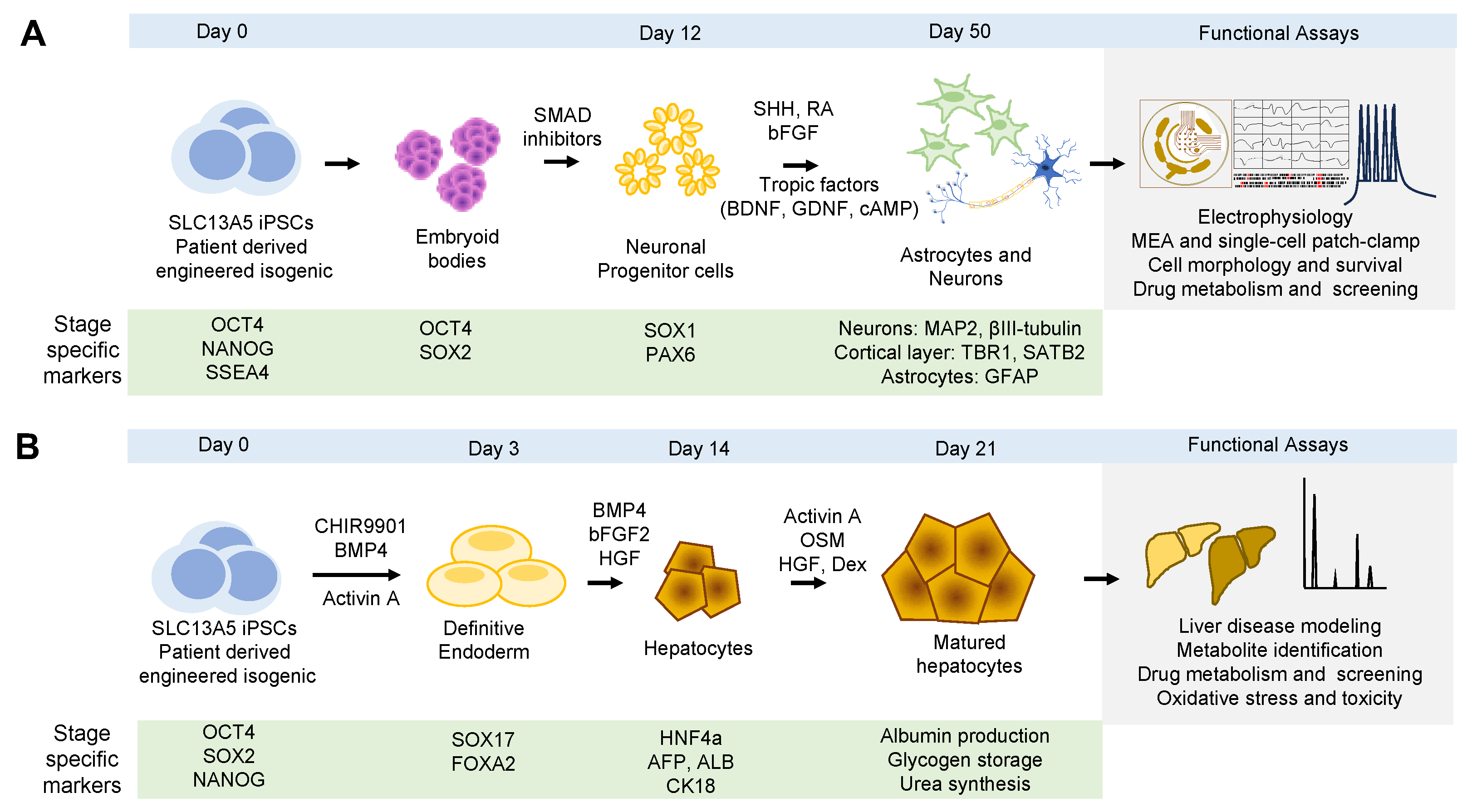
4.2. Human Neurons Derived from Human Pluripotent Stem Cells
4.3. Human Hepatocytes Induced from Induced Pluripotent Stem Cells
4.4. Organoids Models Derived from Pluripotent Stem Cells
4.4.1. Brain Organoids
4.4.2. Liver Organoids
4.4.3. Brain–Liver Organoid Systems
4.5. Challenges and Limitations of iPSC-Derived Model
5. CRISPR Genome Editing of Pluripotent Stem Cells
6. Discussion
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thevenon, J.; Milh, M.; Feillet, F.; St-Onge, J.; Duffourd, Y.; Jugé, C.; Roubertie, A.; Héron, D.; Mignot, C.; Raffo, E.; et al. Mutations in SLC13A5 Cause Autosomal-Recessive Epileptic Encephalopathy with Seizure Onset in the First Days of Life. Am. J. Hum. Genet. 2014, 95, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hardies, K.; De Kovel, C.G.; Weckhuysen, S.; Asselbergh, B.; Geuens, T.; Deconinck, T.; Azmi, A.; May, P.; Zara, F.; Brilstra, E.; et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 2015, 138, 3238–3250. [Google Scholar] [CrossRef]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Human sodium-coupled citrate transporter, the orthologue of Drosophila Indy, as a novel target for lithium action. Biochem. J. 2003, 374 Pt 1, 21–26. [Google Scholar] [CrossRef]
- Inoue, K.; Zhuang, L.; Ganapathy, V. Human Na+ -coupled citrate transporter: Primary structure, genomic organization, and transport function. Biochem. Biophys. Res. Commun. 2002, 299, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, Function, and Expression Pattern of a Novel Sodium-coupled Citrate Transporter (NaCT) Cloned from Mammalian Brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef]
- Rogers, R.P.; Rogina, B. The role of INDY in metabolism, health and longevity. Front. Genet. 2015, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Cordes, T.; Thalacker-Mercer, A.E.; Pajor, A.M.; Murphy, A.N.; Metallo, C.M. NaCT/SLC13A5 facilitates citrate import and metabolism under nutrient-limited conditions. Cell Rep. 2021, 36, 109701. [Google Scholar] [CrossRef]
- Mosaoa, R.; Kasprzyk-Pawelec, A.; Fernandez, H.R.; Avantaggiati, M.L. The Mitochondrial Citrate Carrier SLC25A1/CIC and the Fundamental Role of Citrate in Cancer, Inflammation and Beyond. Biomolecules 2021, 11, 141. [Google Scholar] [CrossRef]
- Spelbrink, E.M.; Brown, T.L.; Brimble, E.; Blanco, K.A.; Nye, K.L.; Porter, B.E. Characterizing a rare neurogenetic disease, SLC13A5 citrate transporter disorder, utilizing clinical data in a cloud-based medical record collection system. Front. Genet. 2023, 14, 1109547. [Google Scholar] [CrossRef]
- Bainbridge, M.N.; Cooney, E.; Miller, M.; Kennedy, A.D.; Wulff, J.E.; Donti, T.; Jhangiani, S.N.; Gibbs, R.A.; Elsea, S.H.; Porter, B.E.; et al. Analyses of SLC13A5 -epilepsy patients reveal perturbations of TCA cycle. Mol. Genet. Metab. 2017, 121, 314–319. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V. Citrate--new functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Kopel, J.J.; Lawrence, J.J.; Neugebauer, V.; Ganapathy, V. Plasma Membrane Na+-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules 2017, 22, 378. [Google Scholar] [CrossRef]
- Gopal, E.; Miyauchi, S.; Martin, P.M.; Ananth, S.; Srinivas, S.R.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G402–G408. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, H.; Garzel, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 2015, 87, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Lee, H.Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Shulman, G.I.; et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Von Loeffelholz, C.; Lieske, S.; Neuschäfer-Rube, F.; Willmes, D.M.; Raschzok, N.; Sauer, I.M.; König, J.; Fromm, M.F.; Horn, P.; Chatzigeorgiou, A.; et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 2017, 66, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Neuschäfer-Rube, F.; Lieske, S.; Kuna, M.; Henkel, J.; Perry, R.J.; Erion, D.M.; Pesta, D.; Willmes, D.M.; Brachs, S.; von Loeffelholz, C.; et al. The Mammalian INDY Homolog Is Induced by CREB in a Rat Model of Type 2 Diabetes. Diabetes 2014, 63, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Mansvelder, H.D.; Verhoog, M.B.; Goriounova, N.A. Synaptic plasticity in human cortical circuits: Cellular mechanisms of learning and memory in the human brain? Curr. Opin. Neurobiol. 2019, 54, 186–193. [Google Scholar] [CrossRef]
- Mason, J.O.; Price, D.J. Building brains in a dish: Prospects for growing cerebral organoids from stem cells. Neuroscience 2016, 334, 105–118. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Kouroupi, G.; Zygogianni, O.; Matsas, R. Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: An overview of induced pluripotent stem-cell-based disease models. Open Biol. 2018, 8, 180138. [Google Scholar] [CrossRef] [PubMed]
- Javaid, M.S.; Tan, T.; Dvir, N.; Anderson, A.; O’brien, T.J.; Kwan, P.; Antonic-Baker, A. Human In Vitro Models of Epilepsy Using Embryonic and Induced Pluripotent Stem Cells. Cells 2022, 11, 3957. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.L.; Nye, K.L.; Porter, B.E. Growth and Overall Health of Patients with SLC13A5 Citrate Transporter Disorder. Metabolites 2021, 11, 746. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, A.R.; Yan, G.; Zeng, Q.; Lucchesi, J.; Hamang, M.J.; Ma, Y.L.; Rong, J.X. Defective enamel and bone development in sodium-dependent citrate transporter (NaCT) Slc13a5 deficient mice. PLoS ONE 2017, 12, e0175465. [Google Scholar] [CrossRef] [PubMed]
- Klotz, J.; Porter, B.E.; Colas, C.; Schlessinger, A.; Pajor, A.M. Mutations in the Na+/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 2016, 22, 310–321. [Google Scholar] [CrossRef]
- Yang, Q.-Z.; Spelbrink, E.M.; Nye, K.L.; Hsu, E.R.; Porter, B.E. Epilepsy and EEG Phenotype of SLC13A5 Citrate Transporter Disorder. Child Neurol. Open 2020, 7, 1–7. [Google Scholar] [CrossRef]
- Goodspeed, K.; Liu, J.S.; Nye, K.L.; Prasad, S.; Sadhu, C.; Tavakkoli, F.; Bilder, D.A.; Minassian, B.A.; Bailey, R.M. SLC13A5 Deficiency Disorder: From Genetics to Gene Therapy. Genes 2022, 13, 1655. [Google Scholar] [CrossRef]
- Sauer, D.B.; Song, J.; Wang, B.; Hilton, J.K.; Karpowich, N.K.; Mindell, J.A.; Rice, W.J.; Wang, D.-N. Structure and inhibition mechanism of the human citrate transporter NaCT. Nature 2021, 591, 157–161. [Google Scholar] [CrossRef]
- Jaramillo-Martinez, V.; Ganapathy, V.; Urbatsch, I.L. A home run for human NaCT/SLC13A5/INDY: Cryo-EM structure and homology model to predict transport mechanisms, inhibitor interactions and mutational defects. Biochem. J. 2021, 478, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Selch, S.; Chafai, A.; Sticht, H.; Birkenfeld, A.L.; Fromm, M.F.; König, J. Analysis of naturally occurring mutations in the human uptake transporter NaCT important for bone and brain development and energy metabolism. Sci. Rep. 2018, 8, 11330. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Martinez, V.; Urbatsch, I.L.; Ganapathy, V. Functional Distinction between Human and Mouse Sodium-Coupled Citrate Transporters and Its Biologic Significance: An Attempt for Structural Basis Using a Homology Modeling Approach. Chem. Rev. 2021, 121, 5359–5377. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, H. Molecular Mechanisms of the SLC13A5 Gene Transcription. Metabolites 2021, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Yodoya, E.; Wada, M.; Shimada, A.; Katsukawa, H.; Okada, N.; Yamamoto, A.; Ganapathy, V.; Fujita, T. Functional and molecular identification of sodium-coupled dicarboxylate transporters in rat primary cultured cerebrocortical astrocytes and neurons. J. Neurochem. 2006, 97, 162–173. [Google Scholar] [CrossRef]
- Inoue, K.; Fei, Y.-J.; Huang, W.; Zhuang, L.; Chen, Z.; Ganapathy, V. Functional identity of Drosophila melanogaster Indy as a cation-independent, electroneutral transporter for tricarboxylic acid-cycle intermediates. Biochem. J. 2002, 367 Pt 2, 313–319. [Google Scholar] [CrossRef]
- Bergeron, M.; Clémençon, B.; Hediger, M.; Markovich, D. SLC13 family of Na+-coupled di- and tri-carboxylate/sulfate transporters. Mol. Asp. Med. 2013, 34, 299–312. [Google Scholar] [CrossRef]
- Henke, C.; Töllner, K.; van Dijk, R.M.; Miljanovic, N.; Cordes, T.; Twele, F.; Bröer, S.; Ziesak, V.; Rohde, M.; Hauck, S.M.; et al. Disruption of the sodium-dependent citrate transporter SLC13A5 in mice causes alterations in brain citrate levels and neuronal network excitability in the hippocampus. Neurobiol. Dis. 2020, 143, 105018. [Google Scholar] [CrossRef]
- Milosavljevic, S.; Glinton, K.E.; Li, X.; Medeiros, C.; Gillespie, P.; Seavitt, J.R.; Graham, B.H.; Elsea, S.H. Untargeted Metabolomics of Slc13a5 Deficiency Reveal Critical Liver–Brain Axis for Lipid Homeostasis. Metabolites 2022, 12, 351. [Google Scholar] [CrossRef]
- Rigby, M.J.; Orefice, N.S.; Lawton, A.J.; Ma, M.; Shapiro, S.L.; Yi, S.Y.; Dieterich, I.A.; Frelka, A.; Miles, H.N.; Puglielli, L.; et al. SLC13A5/sodium-citrate co-transporter overexpression causes disrupted white matter integrity and an autistic-like phenotype. Brain Commun. 2022, 4, fcac002. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Vicario, N.; Costa, A.S.; Kwok, C.K.; Leonardi, T.; Booty, L.M.; Bicci, I.; Balzarotti, B.; Volpe, G.; et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell 2018, 22, 355–368.e13. [Google Scholar] [CrossRef] [PubMed]
- Srisawang, P.; Chatsudthipong, A.; Chatsudthipong, V. Modulation of succinate transport in Hep G2 cell line by PKC. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 1378–1388. [Google Scholar] [CrossRef][Green Version]
- Li, Z.; Li, L.; Heyward, S.; Men, S.; Xu, M.; Sueyoshi, T.; Wang, H. Phenobarbital Induces SLC13A5 Expression through Activation of PXR but Not CAR in Human Primary Hepatocytes. Cells 2021, 10, 3381. [Google Scholar] [CrossRef] [PubMed]
- Poolsri, W.-A.; Phokrai, P.; Suwankulanan, S.; Phakdeeto, N.; Phunsomboon, P.; Pekthong, D.; Richert, L.; Pongcharoen, S.; Srisawang, P. Combination of Mitochondrial and Plasma Membrane Citrate Transporter Inhibitors Inhibits De Novo Lipogenesis Pathway and Triggers Apoptosis in Hepatocellular Carcinoma Cells. BioMed Res. Int. 2018, 2018, 3683026. [Google Scholar] [CrossRef] [PubMed]
- Huard, K.; Brown, J.; Jones, J.C.; Cabral, S.; Futatsugi, K.; Gorgoglione, M.; Lanba, A.; Vera, N.B.; Zhu, Y.; Yan, Q.; et al. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5). Sci. Rep. 2015, 5, 17391. [Google Scholar] [CrossRef]
- Bainbridge, W.A.; Dilks, D.D.; Oliva, A. Memorability: A stimulus-driven perceptual neural signature distinctive from memory. NeuroImage 2017, 149, 141–152. [Google Scholar] [CrossRef]
- Karner, C.M.; Esen, E.; Okunade, A.L.; Patterson, B.W.; Long, F. Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. J. Clin. Investig. 2015, 125, 551–562. [Google Scholar] [CrossRef]
- Ma, C.; Tian, X.; Kim, J.P.; Xie, D.; Ao, X.; Shan, D.; Lin, Q.; Hudock, M.R.; Bai, X.; Yang, J. Citrate-based materials fuel human stem cells by metabonegenic regulation. Proc. Natl. Acad. Sci. USA 2018, 115, E11741–E11750. [Google Scholar] [CrossRef]
- Kannan, K.; Rogina, B. The Role of Citrate Transporter INDY in Metabolism and Stem Cell Homeostasis. Metabolites 2021, 11, 705. [Google Scholar] [CrossRef]
- François, C.M.; Pihl, T.; de Segonzac, M.D.; Hérault, C.; Hudry, B. Metabolic regulation of proteome stability via N-terminal acetylation controls male germline stem cell differentiation and reproduction. Nat. Commun. 2023, 14, 6737. [Google Scholar] [CrossRef]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248–268. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Rezende, N.C.; Scotland, K.B.; Shaffer, S.M.; Persson, J.L.; Gudas, L.J.; Mongan, N.P.; Ran, X.; Xiao, C.-H.; Xiang, G.-M.; et al. Regulation of Stem Cell Pluripotency and Differentiation Involves a Mutual Regulatory Circuit of the Nanog, OCT4, and SOX2 Pluripotency Transcription Factors with Polycomb Repressive Complexes and Stem Cell microRNAs. Stem Cells Dev. 2009, 18, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I.; Tomlinson, S.R. The transcriptional foundation of pluripotency. Development 2009, 136, 2311–2322. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of Human Peripheral Blood Cells to Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Jang, J. Directed Differentiation of Pluripotent Stem Cells by Transcription Factors. Mol. Cells 2019, 42, 200–209. [Google Scholar] [CrossRef]
- Bharathan, S.P.; Manian, K.V.; Aalam, S.M.M.; Palani, D.; Deshpande, P.A.; Pratheesh, M.D.; Srivastava, A.; Velayudhan, S.R. Systematic evaluation of markers used for the identification of human induced pluripotent stem cells. Biol. Open 2017, 6, 100–108. [Google Scholar] [CrossRef]
- Godini, R.; Lafta, H.Y.; Fallahi, H. Epigenetic modifications in the embryonic and induced pluripotent stem cells. Gene Expr. Patterns 2018, 29, 1–9. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Lashen, S.; Cenciarelli, C.; Hasan, A. Genetically unmatched human iPSC and ESC exhibit equivalent gene expression and neuronal differentiation potential. Sci. Rep. 2017, 7, 17504. [Google Scholar] [CrossRef]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef]
- McKenna, D.H.; Perlingeiro, R.C.R. Development of allogeneic iPS cell-based therapy: From bench to bedside. EMBO Mol. Med. 2023, 15, e15315. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S.; Takahashi, K. Induction of pluripotent stem cells from mouse fibroblast cultures. Tanpakushitsu Kakusan Koso. 2006, 51, 2346–2351. [Google Scholar]
- Malik, N.; Rao, M.S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [PubMed]
- Patel, M.; Yang, S. Advances in Reprogramming Somatic Cells to Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2010, 6, 367–380. [Google Scholar] [CrossRef]
- Loh, Y.-H.; Agarwal, S.; Park, I.-H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An Efficient Nonviral Method to Generate Integration-Free Human-Induced Pluripotent Stem Cells from Cord Blood and Peripheral Blood Cells. Stem Cells 2013, 31, 458–466. [Google Scholar] [CrossRef]
- Chou, B.-K.; Mali, P.; Huang, X.; Ye, Z.; Dowey, S.N.; Resar, L.M.; Zou, C.; Zhang, Y.A.; Tong, J.; Cheng, L. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011, 21, 518–529. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Yu, J.; Chau, K.F.; Vodyanik, M.A.; Jiang, J.; Jiang, Y. Efficient Feeder-Free Episomal Reprogramming with Small Molecules. PLoS ONE 2011, 6, e17557. [Google Scholar] [CrossRef]
- Haase, A.; Glienke, W.; Engels, L.; Göhring, G.; Esser, R.; Arseniev, L.; Martin, U. GMP-compatible manufacturing of three iPS cell lines from human peripheral blood. Stem Cell Res. 2019, 35, 101394. [Google Scholar] [CrossRef]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of Induced Pluripotent Stem Cells from Human Terminally Differentiated Circulating T Cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef]
- Guan, J.; Wang, G.; Wang, J.; Zhang, Z.; Fu, Y.; Cheng, L.; Meng, G.; Lyu, Y.; Zhu, J.; Li, Y.; et al. Chemical reprogramming of human somatic cells to pluripotent stem cells. Nature 2022, 605, 325–331. [Google Scholar] [CrossRef]
- Cousin, M.A.; Creighton, B.A.; Breau, K.A.; Spillmann, R.C.; Torti, E.; Dontu, S.; Tripathi, S.; Ajit, D.; Edwards, R.J.; Afriyie, S.; et al. Pathogenic SPTBN1 variants cause an autosomal dominant neurodevelopmental syndrome. Nat. Genet. 2021, 53, 1006–1021. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.G.; Beltran, A.A.; Beltran, A.S. Generation of an integration-free induced pluripotent stem cell line (UNC001-A) from blood of a healthy individual. Stem Cell Res. 2020, 49, 102015. [Google Scholar] [CrossRef] [PubMed]
- Beltran, A.A.; Molina, S.G.; Marquez, A.; Munoz, L.J.; Olivares, J.F.; Beltran, A.S. Generation of an induced pluripotent stem cell line (UNCCi002-A) from a healthy donor using a non-integration system to study Cerebral Cavernous Malformation (CCM). Stem Cell Res. 2021, 54, 102421. [Google Scholar] [CrossRef] [PubMed]
- Lund, R.J.; Nikula, T.; Rahkonen, N.; Närvä, E.; Baker, D.; Harrison, N.; Andrews, P.; Otonkoski, T.; Lahesmaa, R. High-throughput karyotyping of human pluripotent stem cells. Stem Cell Res. 2012, 9, 192–195. [Google Scholar] [CrossRef][Green Version]
- D’Antonio, M.; Woodruff, G.; Nathanson, J.L.; D’Antonio-Chronowska, A.; Arias, A.; Matsui, H.; Williams, R.; Herrera, C.; Reyna, S.M.; Yeo, G.W.; et al. High-Throughput and Cost-Effective Characterization of Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 1101–1111. [Google Scholar] [CrossRef]
- Wang, G.; Weng, R.; Lan, Y.; Guo, X.; Liu, Q.; Liu, X.; Lu, C.; Kang, J. Synergetic effects of DNA methylation and histone modification during mouse induced pluripotent stem cell generation. Sci. Rep. 2017, 7, e39527. [Google Scholar] [CrossRef]
- Lee, D.-S.; Shin, J.-Y.; Tonge, P.D.; Puri, M.C.; Lee, S.; Park, H.; Lee, W.-C.; Hussein, S.M.I.; Bleazard, T.; Yun, J.-Y.; et al. An epigenomic roadmap to induced pluripotency reveals DNA methylation as a reprogramming modulator. Nat. Commun. 2014, 5, 5619. [Google Scholar] [CrossRef]
- Churko, J.M.; Lee, J.; Ameen, M.; Gu, M.; Venkatasubramanian, M.; Diecke, S.; Sallam, K.; Im, H.; Wang, G.; Gold, J.D.; et al. Transcriptomic and epigenomic differences in human induced pluripotent stem cells generated from six reprogramming methods. Nat. Biomed. Eng. 2017, 1, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Hasaart, K.A.; Manders, F.; Ubels, J.; Verheul, M.; van Roosmalen, M.J.; Groenen, N.M.; Oka, R.; Kuijk, E.; Lopes, S.M.C.d.S.; van Boxtel, R. Human induced pluripotent stem cells display a similar mutation burden as embryonic pluripotent cells in vivo. iScience 2022, 25, 103736. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Nardelli, J. Cellular and molecular introduction to brain development. Neurobiol. Dis. 2016, 92 Pt A, 3–17. [Google Scholar] [CrossRef]
- Ebert, A.D.; Liang, P.; Wu, J.C. Induced Pluripotent Stem Cells as a Disease Modeling and Drug Screening Platform. J. Cardiovasc. Pharmacol. 2012, 60, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, R.; Faridounnia, M.; Beltran, A.; Robinson, J.; Kinghorn, K.; Ezzell, J.A.; Bharucha-Goebel, D.; Bonnemann, C.; Hooper, J.E.; Snider, N.; et al. Intermediate filament dysregulation in astrocytes in the human disease model of KLHL16 mutation in giant axonal neuropathy (GAN). Mol. Biol. Cell 2023, 34, ar121. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, R.A.; Beltran, A.S.; Delic, S.; Dumitru, R.; Robinson, J.A.; Kabiraj, P.; Herring, L.E.; Madden, V.J.; Ravinder, N.; Snider, N.T. Site-specific phosphorylation and caspase cleavage of GFAP are new markers of Alexander disease severity. eLife 2019, 8, e47789. [Google Scholar] [CrossRef]
- Guo, N.-N.; Liu, L.-P.; Zheng, Y.-W.; Li, Y.-M. Inducing human induced pluripotent stem cell differentiation through embryoid bodies: A practical and stable approach. World J. Stem Cells 2020, 12, 25–34. [Google Scholar] [CrossRef]
- Stiles, J.; Jernigan, T.L. The Basics of Brain Development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef]
- Briscoe, J.; Ericson, J. Specification of neuronal fates in the ventral neural tube. Curr. Opin. Neurobiol. 2001, 11, 43–49. [Google Scholar] [CrossRef]
- Lee, K.J.; Jessell, T.M. The Specification of Dorsal Cell Fates in the Vertebrate Central Nervous System. Annu. Rev. Neurosci. 1999, 22, 261–294. [Google Scholar] [CrossRef]
- Wilson, S.I.; Edlund, T. Neural induction: Toward a unifying mechanism. Nat. Neurosci. 2001, 4, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Streit, A.; Berliner, A.J.; Papanayotou, C.; Sirulnik, A.; Stern, C.D. Initiation of neural induction by FGF signalling before gastrulation. Nature 2000, 406, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Dhara, S.K.; Hasneen, K.; Machacek, D.W.; Boyd, N.L.; Rao, R.R.; Stice, S.L. Human neural progenitor cells derived from embryonic stem cells in feeder-free cultures. Differentiation 2008, 76, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007, 8, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Erceg, S.; Laínez, S.; Ronaghi, M.; Stojkovic, P.; Pérez-Aragó, M.A.; Moreno-Manzano, V.; Moreno-Palanques, R.; Planells-Cases, R.; Stojkovic, M. Differentiation of Human Embryonic Stem Cells to Regional Specific Neural Precursors in Chemically Defined Medium Conditions. PLoS ONE 2008, 3, e2122. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid Single-Step Induction of Functional Neurons from Human Pluripotent Stem Cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef]
- Busskamp, V.; Lewis, N.E.; Guye, P.; Ng, A.H.; Shipman, S.L.; Byrne, S.M.; Sanjana, N.E.; Murn, J.; Li, Y.; Li, S.; et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014, 10, 760. [Google Scholar] [CrossRef]
- Mertens, J.; Paquola, A.C.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef]
- Victor, M.B.; Richner, M.; Olsen, H.E.; Lee, S.W.; Monteys, A.M.; Ma, C.; Huh, C.J.; Zhang, B.; Davidson, B.L.; Yang, X.W.; et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 2018, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Greig, L.C.; Woodworth, M.B.; Galazo, M.J.; Padmanabhan, H.; Macklis, J.D. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 2013, 14, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Xie, P.; Liu, L.; Kuang, X.; Wang, Y.; Qu, L.; Gong, H.; Jiang, S.; Li, A.; Ruan, Z.; et al. Morphological diversity of single neurons in molecularly defined cell types. Nature 2021, 598, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Obien, M.E.J.; Deligkaris, K.; Bullmann, T.; Bakkum, D.J.; Frey, U. Revealing neuronal function through microelectrode array recordings. Front. Neurosci. 2014, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Garma, L.D.; Matino, L.; Melle, G.; Moia, F.; De Angelis, F.; Santoro, F.; Dipalo, M. Cost-effective and multifunctional acquisition system for in vitro electrophysiological investigations with multi-electrode arrays. PLoS ONE 2019, 14, e0214017. [Google Scholar] [CrossRef] [PubMed]
- Mossink, B.; Verboven, A.H.; van Hugte, E.J.; Gunnewiek, T.M.K.; Parodi, G.; Linda, K.; Schoenmaker, C.; Kleefstra, T.; Kozicz, T.; van Bokhoven, H.; et al. Human neuronal networks on micro-electrode arrays are a highly robust tool to study disease-specific genotype-phenotype correlations in vitro. Stem Cell Rep. 2021, 16, 2182–2196. [Google Scholar] [CrossRef] [PubMed]
- Quraishi, I.H.; Stern, S.; Mangan, K.P.; Zhang, Y.; Ali, S.R.; Mercier, M.R.; Marchetto, M.C.; McLachlan, M.J.; Jones, E.M.; Kaczmarek, L.K. An Epilepsy-Associated KCNT1 Mutation Enhances Excitability of Human iPSC-Derived Neurons by Increasing Slack K(Na) Currents. J. Neurosci. 2019, 39, 7438–7449. [Google Scholar] [CrossRef]
- Tidball, A.M.; Lopez-Santiago, L.F.; Yuan, Y.; Glenn, T.W.; Margolis, J.L.; Walker, J.C.; Kilbane, E.G.; Miller, C.A.; Bebin, E.M.; Perry, M.S.; et al. Variant-specific changes in persistent or resurgent sodium current in SCN8A-related epilepsy patient-derived neurons. Brain 2020, 143, 3025–3040. [Google Scholar] [CrossRef]
- Simkin, D.; Ambrosi, C.; Marshall, K.A.; Williams, L.A.; Eisenberg, J.; Gharib, M.; Dempsey, G.T.; George, A.L.; McManus, O.B.; Kiskinis, E. ‘Channeling’ therapeutic discovery for epileptic encephalopathy through iPSC technologies. Trends Pharmacol. Sci. 2022, 43, 392–405. [Google Scholar] [CrossRef]
- Stafstrom, C.E. Persistent Sodium Current and Its Role in Epilepsy. Epilepsy Curr. 2007, 7, 15–22. [Google Scholar] [CrossRef]
- Varghese, D.S.; Alawathugoda, T.T.; Ansari, S.A. Fine Tuning of Hepatocyte Differentiation from Human Embryonic Stem Cells: Growth Factor vs. Small Molecule-Based Approaches. Stem Cells Int. 2019, 2019, 5968236. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Siller, R.; Greenhough, S.; Naumovska, E.; Sullivan, G.J. Small-Molecule-Driven Hepatocyte Differentiation of Human Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 939–952. [Google Scholar] [CrossRef]
- Ölander, M.; Wegler, C.; Flörkemeier, I.; Treyer, A.; Handin, N.; Pedersen, J.M.; Vildhede, A.; Mateus, A.; LeCluyse, E.L.; Urdzik, J.; et al. Hepatocyte size fractionation allows dissection of human liver zonation. J. Cell. Physiol. 2021, 236, 5885–5894. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.-P.; Zhang, F.; Atkinson-Dell, R.; Rowena, S.-Y.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef]
- Ardalani, H.; Sengupta, S.; Harms, V.; Vickerman, V.; Thomson, J.A.; Murphy, W.L. 3-D culture and endothelial cells improve maturity of human pluripotent stem cell-derived hepatocytes. Acta Biomater. 2019, 95, 371–381. [Google Scholar] [CrossRef]
- Carpentier, A.; Nimgaonkar, I.; Chu, V.; Xia, Y.; Hu, Z.; Liang, T.J. Hepatic differentiation of human pluripotent stem cells in miniaturized format suitable for high-throughput screen. Stem Cell Res. 2016, 16, 640–650. [Google Scholar] [CrossRef]
- Carberry, C.K.; Ferguson, S.S.; Beltran, A.S.; Fry, R.C.; Rager, J.E. Using liver models generated from human-induced pluripotent stem cells (iPSCs) for evaluating chemical-induced modifications and disease across liver developmental stages. Toxicol. Vitr. 2022, 83, 105412. [Google Scholar] [CrossRef]
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541. [Google Scholar] [CrossRef]
- Sauer, V.; Roy-Chowdhury, N.; Guha, C.; Roy-Chowdhury, J. Induced Pluripotent Stem Cells as a Source of Hepatocytes. Curr. Pathobiol. Rep. 2014, 2, 11–20. [Google Scholar] [CrossRef]
- Behbahan, I.S.; Duan, Y.; Lam, A.; Khoobyari, S.; Ma, X.; Ahuja, T.P.; Zern, M.A. New Approaches in the Differentiation of Human Embryonic Stem Cells and Induced Pluripotent Stem Cells toward Hepatocytes. Stem Cell Rev. Rep. 2011, 7, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Freyer, N.; Knöspel, F.; Strahl, N.; Amini, L.; Schrade, P.; Bachmann, S.; Damm, G.; Seehofer, D.; Jacobs, F.; Monshouwer, M.; et al. Hepatic Differentiation of Human Induced Pluripotent Stem Cells in a Perfused Three-Dimensional Multicompartment Bioreactor. BioResearch Open Access 2016, 5, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Snykers, S.; De Kock, J.; Rogiers, V.; Vanhaecke, T. In Vitro Differentiation of Embryonic and Adult Stem Cells into Hepatocytes: State of the Art. Stem Cells 2009, 27, 577–605. [Google Scholar] [CrossRef] [PubMed]
- Rambhatla, L.; Chiu, C.-P.; Kundu, P.; Peng, Y.; Carpenter, M.K. Generation of hepatocyte-like cells from human embryonic stem cells. Cell Transplant. 2003, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Ma, X.; Zou, W.; Wang, C.; Bahbahan, I.S.; Ahuja, T.P.; Tolstikov, V.; Zern, M.A. Differentiation and Characterization of Metabolically Functioning Hepatocytes from Human Embryonic Stem Cells. Stem Cells 2010, 28, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Solanki, A.; Lee, K.-B. Nanotechnology-Based Approaches for Guiding Neural Regeneration. Accounts Chem. Res. 2016, 49, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Quinti, L.; Tanzi, R.E.; Kim, D.Y. 3D culture models of Alzheimer’s disease: A road map to a ”cure-in-a-dish”. Mol. Neurodegener. 2016, 11, 75. [Google Scholar] [CrossRef]
- Papaspyropoulos, A.; Tsolaki, M.; Foroglou, N.; Pantazaki, A.A. Modeling and Targeting Alzheimer’s Disease with Organoids. Front. Pharmacol. 2020, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-T.; Liu, D.; Kang, H.-C.; Lu, L.; Huang, H.-Z.; Ai, W.-Q.; Zhou, Y.; Deng, M.-F.; Li, H.; Liu, Z.-Q.; et al. Tau pathology epigenetically remodels the neuron-glial cross-talk in Alzheimer’s disease. Sci. Adv. 2023, 9, eabq7105. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Woo, H.J.; Kim, E.H.; Kim, H.S.; Suh, H.N.; Kim, S.H.; Song, J.-J.; Wulansari, N.; Kang, M.; Lee, S.H. Neural stem cells derived from human midbrain organoids as a stable source for treating Parkinson’s disease: Midbrain organoid-NSCs (Og-NSC) as a stable source for PD treatment. Prog. Neurobiol. 2021, 204, 102086. [Google Scholar] [CrossRef] [PubMed]
- Sabate-Soler, S.; Nickels, S.L.; Saraiva, C.; Berger, E.; Dubonyte, U.; Barmpa, K.; Lan, Y.J.; Kouno, T.; Jarazo, J.; Robertson, G.; et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia 2022, 70, 1267–1288. [Google Scholar] [CrossRef] [PubMed]
- Teli, P.; Kale, V.; Vaidya, A. Beyond animal models: Revolutionizing neurodegenerative disease modeling using 3D in vitro organoids, microfluidic chips, and bioprinting. Cell Tissue Res. 2023, 394, 75–91. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef]
- Maroof, A.M.; Keros, S.; Tyson, J.A.; Ying, S.-W.; Ganat, Y.M.; Merkle, F.T.; Liu, B.; Goulburn, A.; Stanley, E.G.; Elefanty, A.G.; et al. Directed Differentiation and Functional Maturation of Cortical Interneurons from Human Embryonic Stem Cells. Cell Stem Cell 2013, 12, 559–572. [Google Scholar] [CrossRef]
- Efthymiou, A.; Shaltouki, A.; Steiner, J.P.; Jha, B.; Heman-Ackah, S.M.; Swistowski, A.; Zeng, X.; Rao, M.S.; Malik, N. Functional Screening Assays with Neurons Generated from Pluripotent Stem Cell–Derived Neural Stem Cells. J. Biomol. Screen. 2014, 19, 32–43. [Google Scholar] [CrossRef]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 2013, 56, 355–364. [Google Scholar] [CrossRef]
- Fields, R.D.; Stevens-Graham, B. New Insights into Neuron-Glia Communication. Science 2002, 298, 556–562. [Google Scholar] [CrossRef]
- Barres, B.A. The Mystery and Magic of Glia: A Perspective on Their Roles in Health and Disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, F.; He, Z.; Ma, Y.; Uchiyama, K.; Lin, J.-M. A novel approach for precisely controlled multiple cell patterning in microfluidic chips by inkjet printing and the detection of drug metabolism and diffusion. Analyst 2016, 141, 2940–2947. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Lian, Q.; Xu, F. Perspective: Fabrication of integrated organ-on-a-chip via bioprinting. Biomicrofluidics 2017, 11, 031301. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Hockaday, L.A.; Kang, K.H.; Butcher, J.T. 3D Bioprinting of heterogeneous aortic valve conduits with alginate/gelatin hydrogels. J. Biomed. Mater. Res. Part A 2013, 101, 1255–1264. [Google Scholar] [CrossRef]
- Fang, L.; Liu, Y.; Qiu, J.; Wan, W. Bioprinting and its Use in Tumor-On-A-Chip Technology for Cancer Drug Screening: A Review. Int. J. Bioprinting 2022, 8, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Bräuninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; A Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef]
- Glass, M.R.; Waxman, E.A.; Yamashita, S.; Lafferty, M.; Beltran, A.; Farah, T.; Patel, N.K.; Matoba, N.; Ahmed, S.; Stein, J.L.; et al. Cross-site reproducibility of human cortical organoids reveals consistent cell type composition and architecture. bioRxiv 2023. [Google Scholar] [CrossRef]
- Nuciforo, S.; Heim, M.H. Organoids to model liver disease. JHEP Rep. 2021, 3, 100198. [Google Scholar] [CrossRef]
- Vollmar, B.; Menger, M.D. The Hepatic Microcirculation: Mechanistic Contributions and Therapeutic Targets in Liver Injury and Repair. Physiol. Rev. 2009, 89, 1269–1339. [Google Scholar] [CrossRef]
- Prior, N.; Inacio, P.; Huch, M. Liver organoids: From basic research to therapeutic applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef] [PubMed]
- Shinozawa, T.; Yoshikawa, H.Y.; Takebe, T. Reverse engineering liver buds through self-driven condensation and organization towards medical application. Dev. Biol. 2016, 420, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Zalyte, R.; Urnavicius, L.; Carter, A.P.; Ge, J.; Li, W.; Chen, M. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Montes-Olivas, S.; Marucci, L.; Homer, M. Mathematical Models of Organoid Cultures. Front. Genet. 2019, 10, 873. [Google Scholar] [CrossRef] [PubMed]
- Koenig, L.; Ramme, A.P.; Faust, D.; Mayer, M.; Flötke, T.; Gerhartl, A.; Brachner, A.; Neuhaus, W.; Appelt-Menzel, A.; Metzger, M.; et al. A Human Stem Cell-Derived Brain-Liver Chip for Assessing Blood-Brain-Barrier Permeation of Pharmaceutical Drugs. Cells 2022, 11, 3295. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Valderrama, A.; Messina, A.; Mitjavila-Garcia, M.T.; Guenou, H. The endothelium, a key actor in organ development and hPSC-derived organoid vascularization. J. Biomed. Sci. 2020, 27, 67. [Google Scholar] [CrossRef]
- Turinetto, V.; Orlando, L.; Giachino, C. Induced Pluripotent Stem Cells: Advances in the Quest for Genetic Stability during Reprogramming Process. Int. J. Mol. Sci. 2017, 18, 1952. [Google Scholar] [CrossRef]
- Wang, K.; Guzman, A.K.; Yan, Z.; Zhang, S.; Hu, M.Y.; Hamaneh, M.B.; Yu, Y.-K.; Tolu, S.; Zhang, J.; Kanavy, H.E.; et al. Ultra-High-Frequency Reprogramming of Individual Long-Term Hematopoietic Stem Cells Yields Low Somatic Variant Induced Pluripotent Stem Cells. Cell Rep. 2019, 26, 2580–2592.e7. [Google Scholar] [CrossRef]
- Ruiz, S.; Gore, A.; Li, Z.; Panopoulos, A.D.; Montserrat, N.; Fung, H.-L.; Giorgetti, A.; Bilic, J.; Batchelder, E.M.; Zaehres, H.; et al. Analysis of protein-coding mutations in hiPSCs and their possible role during somatic cell reprogramming. Nat. Commun. 2013, 4, 1382. [Google Scholar] [CrossRef]
- Perrera, V.; Martello, G. How Does Reprogramming to Pluripotency Affect Genomic Imprinting? Front. Cell Dev. Biol. 2019, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Buckberry, S.; Liu, X.; Poppe, D.; Tan, J.P.; Sun, G.; Chen, J.; Nguyen, T.V.; de Mendoza, A.; Pflueger, J.; Frazer, T.; et al. Transient naive reprogramming corrects hiPS cells functionally and epigenetically. Nature 2023, 620, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, Z.; Ortega, J.A.; Sato, K.; Sasselli, I.R.; Kolberg-Edelbrock, A.N.; Qiu, R.; Marshall, K.A.; Nguyen, T.P.; Smith, C.S.; Quinlan, K.A.; et al. Artificial extracellular matrix scaffolds of mobile molecules enhance maturation of human stem cell-derived neurons. Cell Stem Cell 2023, 30, 219–238.e14. [Google Scholar] [CrossRef] [PubMed]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis. Model Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Bashore, F.M.; Marquez, A.B.; Chaikuad, A.; Howell, S.; Dunn, A.S.; Beltran, A.A.; Smith, J.L.; Drewry, D.H.; Beltran, A.S.; Axtman, A.D. Modulation of tau tubulin kinases (TTBK1 and TTBK2) impacts ciliogenesis. Sci. Rep. 2023, 13, 6118. [Google Scholar] [CrossRef] [PubMed]
- Necarsulmer, J.C.; Simon, J.M.; Evangelista, B.A.; Chen, Y.; Tian, X.; Nafees, S.; Marquez, A.B.; Jiang, H.; Wang, P.; Cohen, T.J.; et al. RNA-binding deficient TDP-43 drives cognitive decline in a mouse model of TDP-43 proteinopathy. eLife 2023, 12, RP85921. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Wen, Y.; Dong, Z.; Liu, J.; Axerio-Cilies, P.; Du, Y.; Li, J.; Chen, l.; Zhang, L.; Lu, J.; Wang, Y.T. Glutamate and GABA(A) receptor crosstalk mediates homeostatic regulation of neuronal excitation in the mammalian brain. Signal Transduct. Target Ther. 2022, 7, 340. [Google Scholar] [CrossRef]
- Feng, Y.; Wei, Z.-H.; Liu, C.; Li, G.-Y.; Qiao, X.-Z.; Gan, Y.-J.; Zhang, C.-C.; Deng, Y.-C. Genetic variations in GABA metabolism and epilepsy. Seizure 2022, 101, 22–29. [Google Scholar] [CrossRef]
- Kopel, J.J.; Bhutia, Y.D.; Sivaprakasam, S.; Ganapathy, V. Consequences of NaCT/SLC13A5/mINDY deficiency: Good versus evil, separated only by the blood-brain barrier. Biochem. J. 2021, 478, 463–486. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Sriram, K. Neuron-astrocyte omnidirectional signaling in neurological health and disease. Front. Mol. Neurosci. 2023, 16, 1169320. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Willenbockel, H.F.; Cordes, T. Mapping the Metabolic Niche of Citrate Metabolism and SLC13A5. Metabolites 2023, 13, 331. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beltran, A.S. Novel Approaches to Studying SLC13A5 Disease. Metabolites 2024, 14, 84. https://doi.org/10.3390/metabo14020084
Beltran AS. Novel Approaches to Studying SLC13A5 Disease. Metabolites. 2024; 14(2):84. https://doi.org/10.3390/metabo14020084
Chicago/Turabian StyleBeltran, Adriana S. 2024. "Novel Approaches to Studying SLC13A5 Disease" Metabolites 14, no. 2: 84. https://doi.org/10.3390/metabo14020084
APA StyleBeltran, A. S. (2024). Novel Approaches to Studying SLC13A5 Disease. Metabolites, 14(2), 84. https://doi.org/10.3390/metabo14020084