
A Comprehensive Comparison between Primary Liver Cancer and Liver Metastases through scRNA-Seq Data Analysis

Abstract
:1. Introduction
2. Materials and Methods
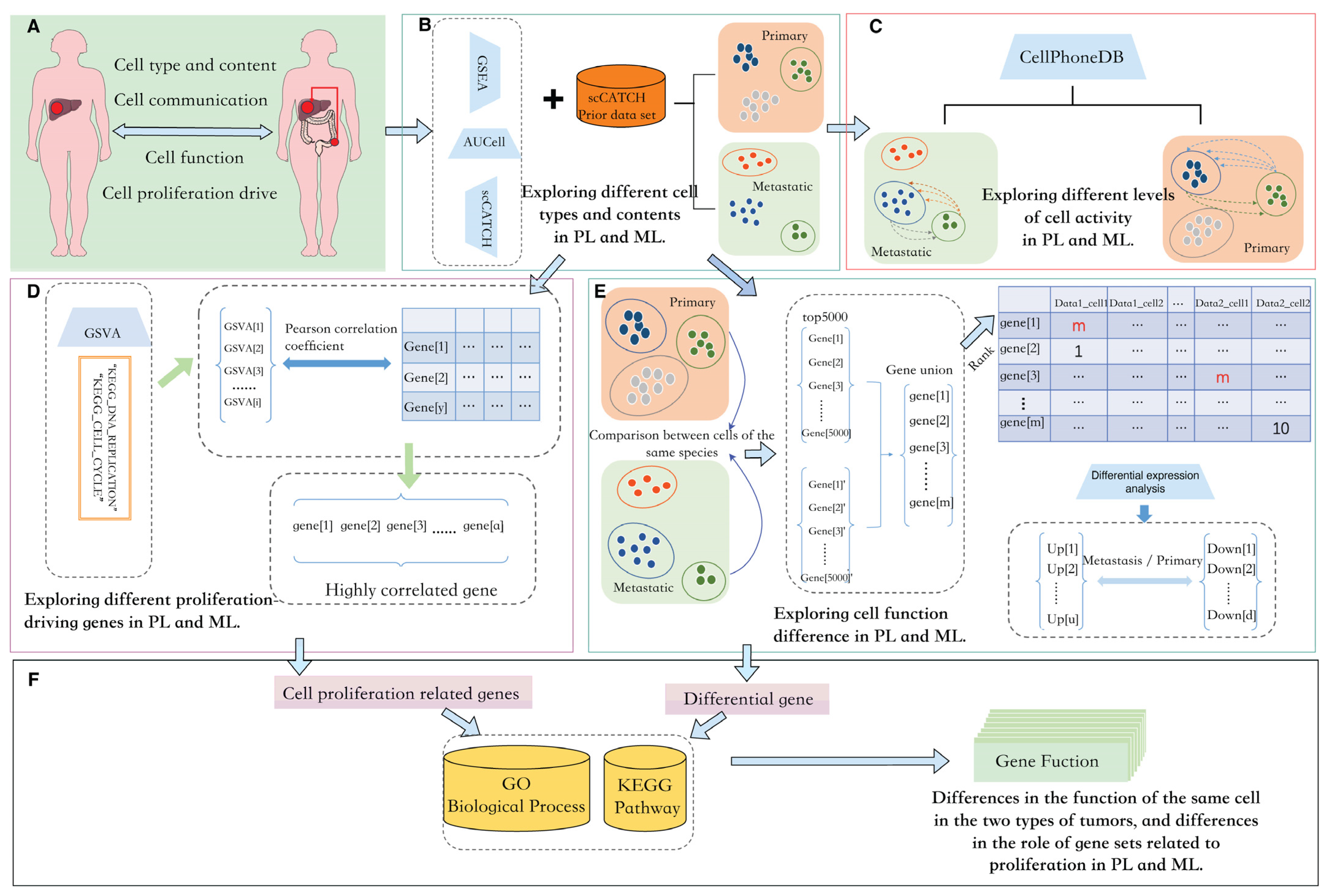
2.1. Overview
2.2. Data Collection and Processing
2.3. Dimension Reduction and Cell Type Annotation
2.4. Rank-Based Differential Expression Analysis
2.5. Enrichment Analysis
2.6. Cell Crosstalk Analysis
2.7. GSVA Enrichment and Correlation Calculation
3. Results
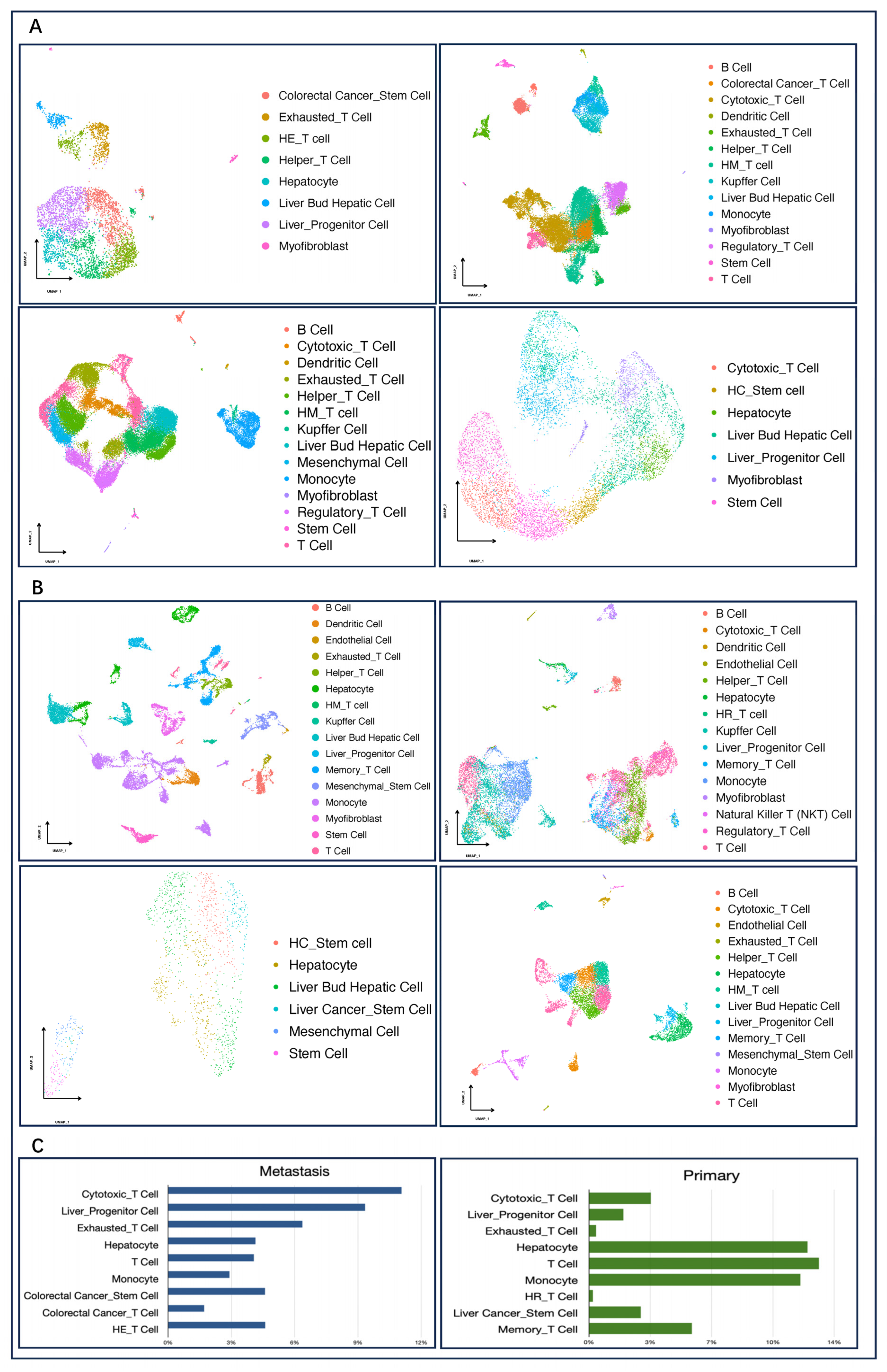
3.1. Comparison of Cell Components between Primary and Metastatic Liver Carcinoma
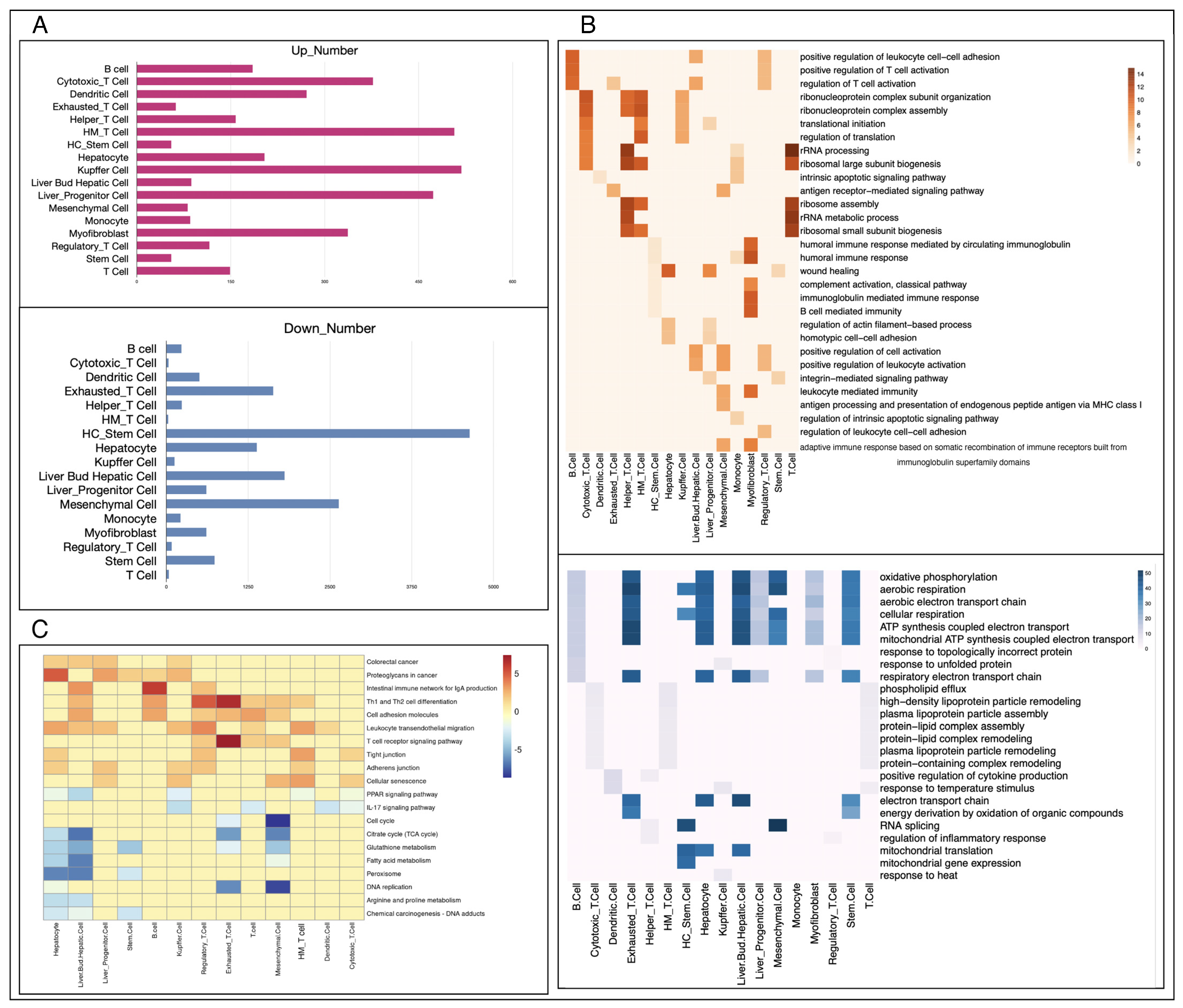
3.2. Different Functions of the Same Cell Type in Primary and Metastatic Liver Cancer
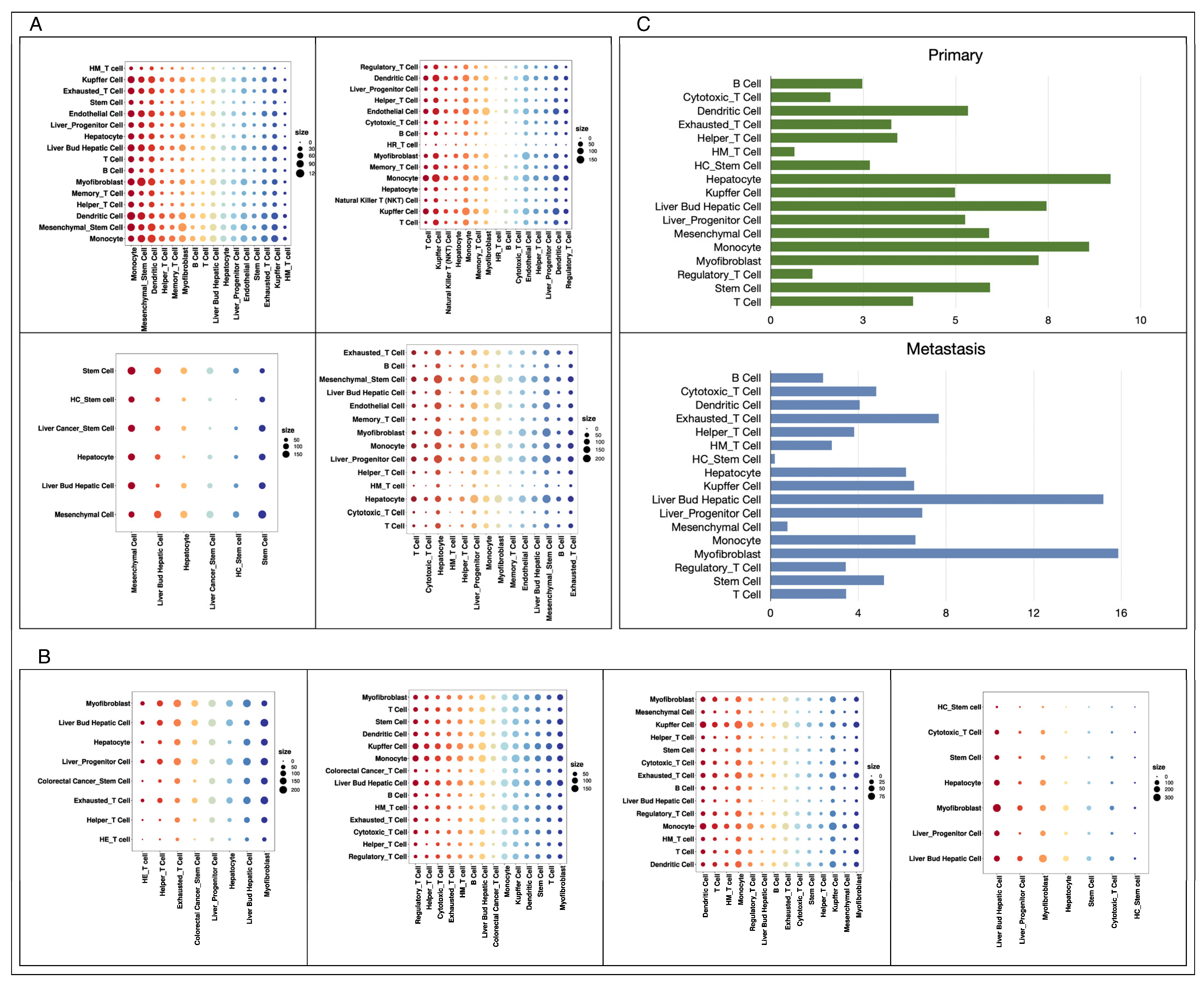
3.3. Diverse Cell–Cell Communications in Primary and Metastatic Liver Cancer
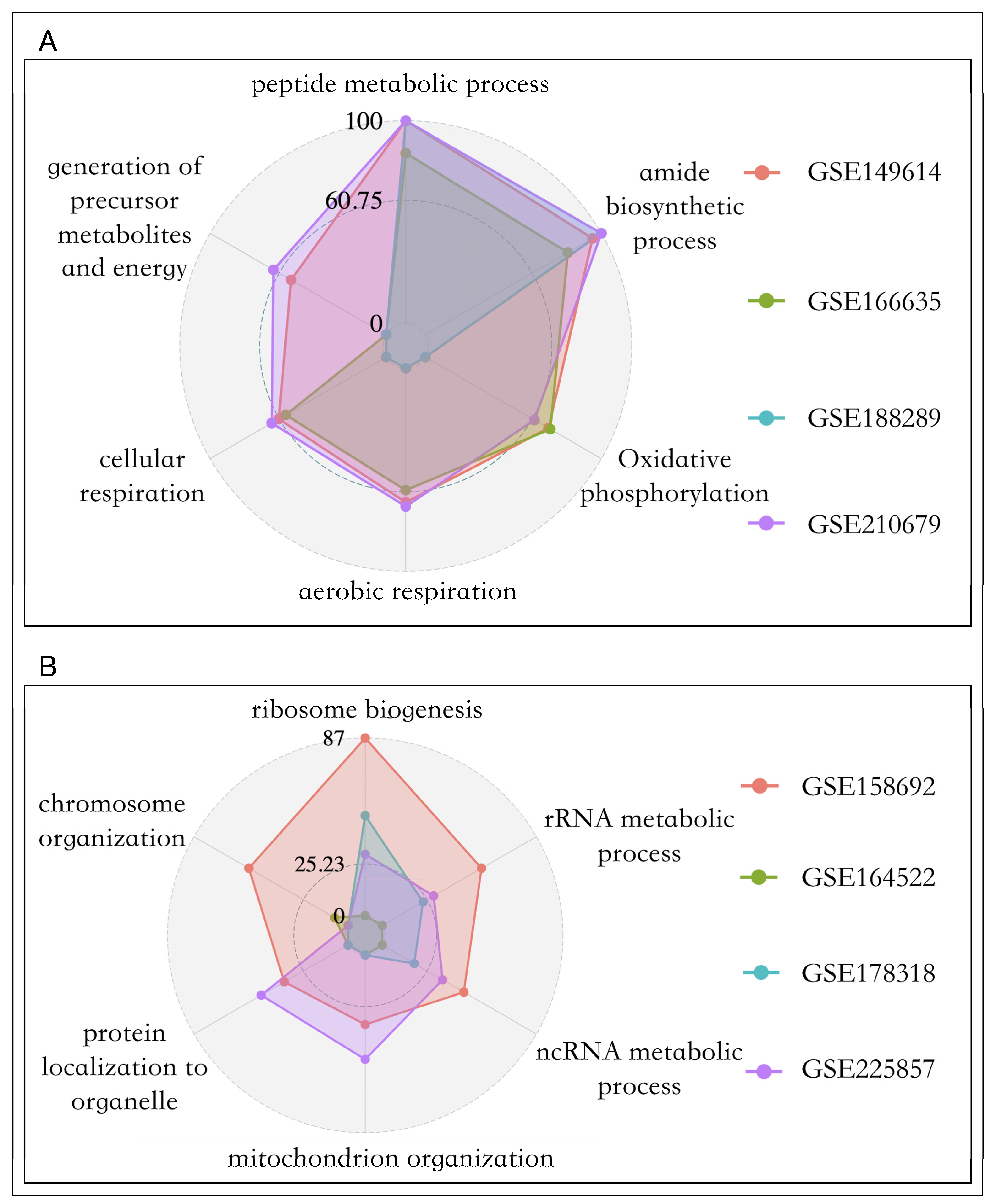
3.4. Biological Processes Related to Cell Proliferation in Primary and Metastatic Liver Cancer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Massagué, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, J.; Xu, B.; Zhang, X. Tumor metastasis: Mechanistic insights and therapeutic interventions. MedComm (2020) 2021, 2, 587–617. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Panagi, M.; Stylianopoulos, T.; Papageorgis, P. The Role of Tumor Microenvironment in Cancer Metastasis: Molecular Mechanisms and Therapeutic Opportunities. Cancers 2021, 13, 2053. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Q.; Xing, B.; Luo, N.; Gao, R.; Yu, K.; Hu, X.; Bu, Z.; Peng, J.; Ren, X.; et al. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell 2022, 40, 424–437.e5. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, J.; Webster, R.M. The hepatocellular carcinoma market. Nat. Rev. Drug Discov. 2019, 18, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhao, Z.; Zhang, X. Genetic differences between primary and metastatic cancer: A pan-cancer whole-genome comparison study. Signal Transduct. Target. Ther. 2023, 8, 363. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer. 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Swanton, C. Metastasis as an evolutionary process. Science 2016, 352, 169–175. [Google Scholar] [CrossRef]
- Gerstberger, S.; Jiang, Q.; Ganesh, K. Metastasis. Cell 2023, 186, 1564–1579. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel therapeutic targets for cancer metastasis. Expert Rev. Anticancer Ther. 2020, 20, 97–109. [Google Scholar] [CrossRef]
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 2020, 52, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Cacho-Díaz, B.; García-Botello, D.R.; Wegman-Ostrosky, T.; Reyes-Soto, G.; Ortiz-Sánchez, E.; Herrera-Montalvo, L.A. Tumor microenvironment differences between primary tumor and brain metastases. J. Transl. Med. 2020, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Rozenblit, M.; Huang, R.; Danziger, N.; Hegde, P.; Alexander, B.; Ramkissoon, S.; Blenman, K.; Ross, J.S.; Rimm, D.L.; Pusztai, L. Comparison of PD-L1 protein expression between primary tumors and metastatic lesions in triple negative breast cancers. J. Immunother. Cancer 2020, 8, e001558. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Author Correction: Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2020, 21, 65. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 2021, 13, 36. [Google Scholar] [CrossRef]
- Gohil, S.H.; Iorgulescu, J.B.; Braun, D.A.; Keskin, D.B.; Livak, K.J. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 244–256. [Google Scholar] [CrossRef]
- Zeng, Q.; Mousa, M.; Nadukkandy, A.S.; Franssens, L.; Alnaqbi, H.; Alshamsi, F.Y.; Al Safar, H.; Carmeliet, P. Understanding tumour endothelial cell heterogeneity and function from single-cell omics. Nat. Rev. Cancer 2023, 23, 544–564. [Google Scholar] [CrossRef]
- Polychronidou, M.; Hou, J.; Babu, M.M.; Liberali, P.; Amit, I.; Deplancke, B.; Lahav, G.; Itzkovitz, S.; Mann, M.; Saez-Rodriguez, J.; et al. Single-cell biology: What does the future hold? Mol. Syst. Biol. 2023, 19, e11799. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Liao, J.; Lu, X.; Xue, R.; Ai, N.; Fan, X. scCATCH: Automatic Annotation on Cell Types of Clusters from Single-Cell RNA Sequencing Data. iScience 2020, 23, 100882. [Google Scholar] [CrossRef] [PubMed]
- Van de Sande, B.; Flerin, C.; Davie, K.; De Waegeneer, M.; Hulselmans, G.; Aibar, S.; Seurinck, R.; Saelens, W.; Cannoodt, R.; Rouchon, Q.; et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020, 15, 2247–2276. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar]
- Wang, H.; Sun, Q.; Zhao, W.; Qi, L.; Gu, Y.; Li, P.; Zhang, M.; Li, Y.; Liu, S.-L.; Guo, Z. Individual-level analysis of differential expression of genes and pathways for personalized medicine. Bioinformatics 2015, 31, 62–68. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hein, D.M.; Deng, W.; Bleile, M.; Kazmi, S.A.; Rhead, B.; De La Vega, F.M.; Jones, A.L.; Kainthla, R.; Jiang, W.; Cantarel, B.; et al. Racial and Ethnic Differences in Genomic Profiling of Early Onset Colorectal Cancer. J. Natl. Cancer Inst. 2022, 114, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hao, L.; Zhang, J.; Deng, J.; Hu, X. Focus on T cell exhaustion: New advances in traditional Chinese medicine in infection and cancer. Chin. Med. 2023, 18, 76. [Google Scholar] [CrossRef]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.R.; Ho, P.C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef]
- Zhang, N.; Bai, S.; Zhang, F.; Shi, M.; Wang, L.; Wang, L.; Xu, L.; Yang, Z.; Yu, C. Molecular markers and mechanisms for stemness maintenance of liver cancer stem cells: A review. Sheng Wu Gong Cheng Xue Bao 2021, 37, 2719–2736. [Google Scholar] [PubMed]
- Yan, J.; Allen, S.; McDonald, E.; Das, I.; Mak, J.Y.W.; Liu, L.; Fairlie, D.P.; Meehan, B.S.; Chen, Z.; Corbett, A.J.; et al. MAIT Cells Promote Tumor Initiation, Growth, and Metastases via Tumor MR1. Cancer Discov. 2020, 10, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Abdeen, A.A.; Wycislo, K.L.; Fan, T.M.; Kilian, K.A. Interfacial geometry dictates cancer cell tumorigenicity. Nat. Mater. 2016, 15, 856–862. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Hou, P.-P.; Luo, L.-J.; Chen, H.-Z.; Chen, Q.-T.; Bian, X.-L.; Wu, S.-F.; Zhou, J.-X.; Zhao, W.-X.; Liu, J.-M.; Wang, X.-M.; et al. Ectosomal PKM2 Promotes HCC by Inducing Macrophage Differentiation and Remodeling the Tumor Microenvironment. Mol. Cell 2020, 78, 1192–1206.e10. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Lüscher, B.; Larsson, L.-G.; Hermeking, H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, E187–E196. [Google Scholar] [CrossRef]
- Prakash, V.; Carson, B.B.; Feenstra, J.M.; Dass, R.A.; Sekyrova, P.; Hoshino, A.; Petersen, J.; Guo, Y.; Parks, M.M.; Kurylo, C.M.; et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat. Commun. 2019, 10, 2110. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSE | Number of Samples | Number of Cells | |
|---|---|---|---|
| Metastatic | GSE178318 | 6 | 40,170 |
| GSE225857 | 2 | 8482 | |
| GSE158692 | 6 | 4587 | |
| GSE164522 | 17 | 34,995 | |
| Primary | GSE149614 | 9 | 20,762 |
| GSE166635 | 2 | 13,696 | |
| GSE188289 | 1 | 1410 | |
| GSE210679 | 1 | 9027 | |
| Total | 8 | 44 | 133,129 |
| GSE | Number of Genes | |
|---|---|---|
| Metastasis | GSE178318 | 121 |
| GSE225857 | 2849 | |
| GSE158692 | 1642 | |
| GSE164522 | 21 | |
| Primary | GSE149614 | 1909 |
| GSE166635 | 1699 | |
| GSE188289 | 1641 | |
| GSE210679 | 2911 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, S.; Chen, L.; Du, W.; Sun, H. A Comprehensive Comparison between Primary Liver Cancer and Liver Metastases through scRNA-Seq Data Analysis. Metabolites 2024, 14, 90. https://doi.org/10.3390/metabo14020090
Hao S, Chen L, Du W, Sun H. A Comprehensive Comparison between Primary Liver Cancer and Liver Metastases through scRNA-Seq Data Analysis. Metabolites. 2024; 14(2):90. https://doi.org/10.3390/metabo14020090
Chicago/Turabian StyleHao, Shuang, Liqun Chen, Wenhui Du, and Huiyan Sun. 2024. "A Comprehensive Comparison between Primary Liver Cancer and Liver Metastases through scRNA-Seq Data Analysis" Metabolites 14, no. 2: 90. https://doi.org/10.3390/metabo14020090