

Current State, Challenges, and Opportunities in Genome-Scale Resource Allocation Models: A Mathematical Perspective

 , and
, and
Abstract

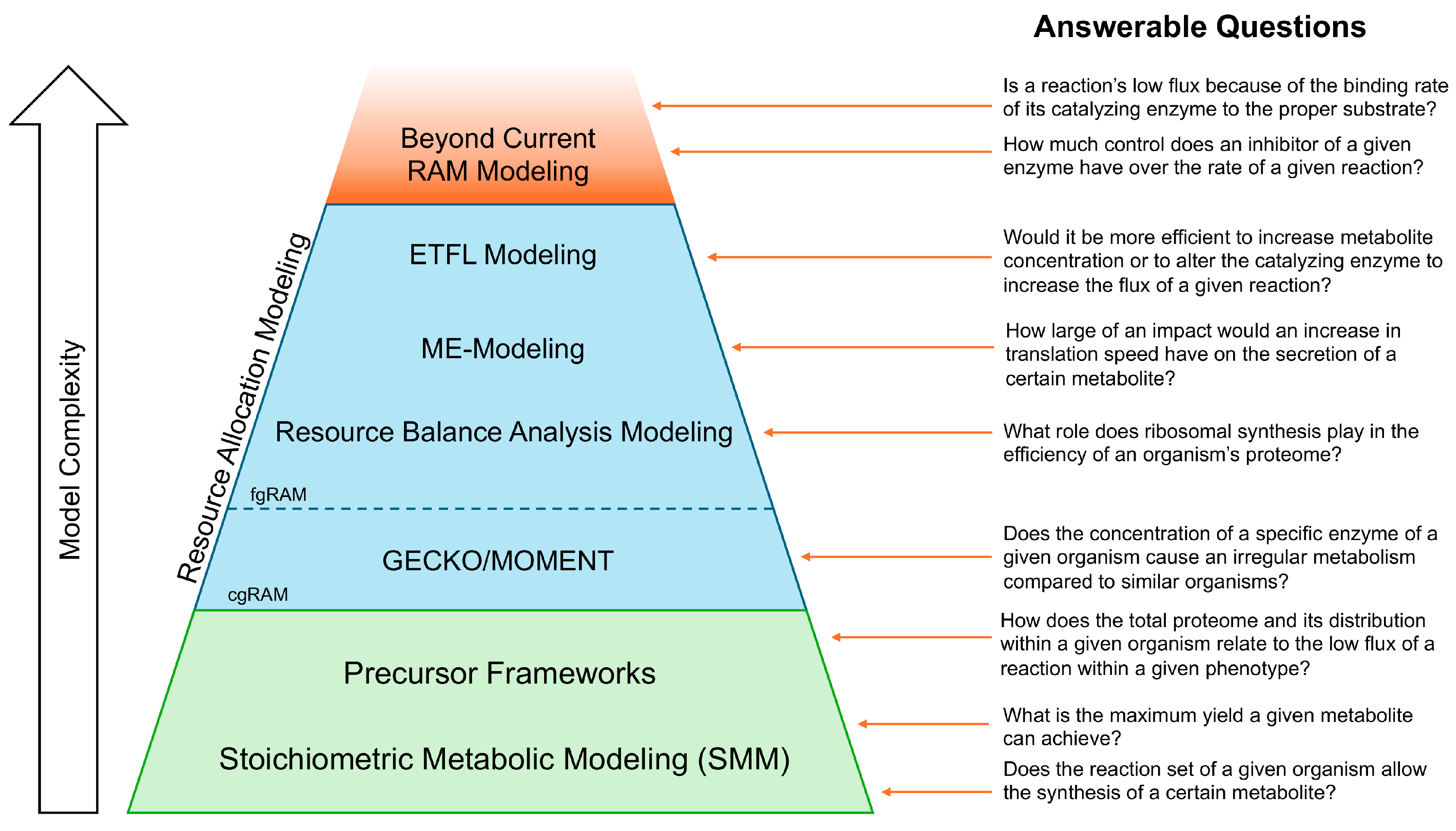
1. Introduction
2. Stoichiometric Models of Metabolism (SMMs) and Flux Balance Analysis (FBA)
3. Precursor Frameworks
3.1. Flux Balance Analysis with Molecular Crowding (FBAwMC)
3.2. FBA with Solvent Capacity Constraints (FBAwSCC)
4. Resource Allocation Model (RAM) Frameworks
4.1. Coarse-Grained RAMs (cgRAMs)
4.1.1. Metabolic Modeling with Enzyme Kinetics (MOMENT) Framework and Successors
4.1.2. GEM with Enzymatic Constraints Using Kinetics and Omics (GECKO) Framework and Its Progeny
4.1.3. Automated Reconstruction of MOMENT and GECKO Models
4.2. Fine-Grained RAMs (fgRAMs)
4.2.1. Resource Balance Analysis (RBA)
4.2.2. Model of Metabolism and Macromolecular Expression (ME-Models)
4.2.3. Expression and Thermodynamics Flux (ETFL) Framework
5. Discussion and Conclusions
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Durot, M.; Bourguignon, P.Y.; Schachter, V. Genome-scale models of bacterial metabolism: Reconstruction and applications. FEMS Microbiol. Rev. 2009, 33, 164–190. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.G.; Bi, X.Y.; Liu, Y.F.; Lv, X.Q.; Li, J.H.; Du, G.C.; Liu, L. In silico cell factory design driven by comprehensive genome-scale metabolic models: Development and challenges. Syst. Microbiol. Biomanufact. 2023, 3, 207–222. [Google Scholar] [CrossRef]
- Simeonidis, E.; Price, N.D. Genome-scale modeling for metabolic engineering. J. Ind. Microbiol. Biotechnol. 2015, 42, 327–338. [Google Scholar] [CrossRef]
- Santos, F.; Boele, J.; Teusink, B. A Practical Guide to Genome-Scale Metabolic Models and Their Analysis. Methods Enzymol. 2011, 500, 509–532. [Google Scholar] [CrossRef]
- Schroeder, W.L.; Kuil, T.; van Maris, A.J.A.; Lynd, L.R.; Maranas, C.D. A detailed genome-scale metabolic model of Clostridium thermocellum investigates sources of pyrophosphate for driving glycolysis. Metab. Eng. 2023, 77, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Nogales, J.; Mueller, J.; Gudmundsson, S.; Canalejo, F.J.; Duque, E.; Monk, J.; Feist, A.M.; Ramos, J.L.; Niu, W.; Palsson, B.O. High-quality genome-scale metabolic modelling of Pseudomonas putida highlights its broad metaboliccapabilities. Environ. Microbiol. 2020, 22, 255–269. [Google Scholar] [CrossRef]
- Heinken, A.; Basile, A.; Hertel, J.; Thinnes, C.; Thiele, I. Genome-Scale Metabolic Modeling of the Human Microbiome in the Era of Personalized Medicine. Annu. Rev. Microbiol. 2021, 75, 199–222. [Google Scholar] [CrossRef]
- Abdel-Haleem, A.M.; Hefzi, H.; Mineta, K.; Gao, X.; Gojobori, T.; Palsson, B.O.; Lewis, N.E.; Jamshidi, N. Functional interrogation of Plasmodium genus metabolism identifies species- and stage-specific differences in nutrient essentiality and drug targeting. PLoS Comput. Biol. 2018, 14, e1005895. [Google Scholar] [CrossRef]
- Folger, O.; Jerby, L.; Frezza, C.; Gottlieb, E.; Ruppin, E.; Shlomi, T. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 2011, 7, 501. [Google Scholar] [CrossRef]
- Gu, C.D.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Reimers, A.M.; Knoop, H.; Bockmayr, A.; Steuer, R. Cellular trade-offs and optimal resource allocation during cyanobacterial diurnal growth. Proc. Natl. Acad. Sci. USA 2017, 114, E6457–E6465. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Cluett, W.R.; Mahadevan, R. Constructing kinetic models of metabolism at genome-scales: A review. Biotechnol. J. 2015, 10, 1345–1359. [Google Scholar] [CrossRef]
- Dahal, S.; Zhao, J.; Yang, L. Recent advances in genome-scale modeling of proteome allocation. Curr. Opin. Syst. Biol. 2021, 26, 39–45. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Dash, S.; Maranas, C. K-FIT: An accelerated kinetic parameterization algorithm using steady-state fluxomic data. Metab. Eng. 2020, 61, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Dinh, H.V.; Maranas, C.D. Evaluating proteome allocation of Saccharomyces cerevisiae phenotypes with resource balance analysis. Metab. Eng. 2023, 77, 242–255. [Google Scholar] [CrossRef]
- Strain, B.; Morrissey, J.; Antonakoudis, A.; Kontoravdi, C. Genome-scale models as a vehicle for knowledge transfer from microbial to mammalian cell systems. Comput. Struct. Biotechnol. J. 2023, 21, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Reimers, A.M.; Lindhorst, H.; Waldherr, S. A Protocol for Generating and Exchanging (Genome-Scale) Metabolic Resource Allocation Models. Metabolites 2017, 7, 47. [Google Scholar] [CrossRef]
- Chen, Y.; Nielsen, J. Mathematical modeling of proteome constraints within metabolism. Curr. Opin. Syst. Biol. 2021, 25, 50–56. [Google Scholar] [CrossRef]
- O’Brien, E.J.; Lerman, J.A.; Chang, R.L.; Hyduke, D.R.; Palsson, B.O. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 2013, 9, 693. [Google Scholar] [CrossRef]
- Salvy, P.; Hatzimanikatis, V. The ETFL formulation allows multi-omics integration in thermodynamics-compliant metabolism and expression models. Nat. Commun. 2020, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Hwa, T.; Martin, O.C.; De Martino, A.; Marinari, E. Constrained Allocation Flux Balance Analysis. PLoS Comput. Biol. 2016, 12, e1004913. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Yang, L.; Lloyd, C.J.; Fang, X.; Palsson, B.O. Genome-scale model of metabolism and gene expression provides a multi-scale description of acid stress responses in. PLoS Comput. Biol. 2019, 15, e1007525. [Google Scholar] [CrossRef] [PubMed]
- De Becker, K.; Totis, N.; Bernaerts, K.; Waldherr, S. Using resource constraints derived from genomic and proteomic data in metabolic network models. Curr. Opin. Syst. Biol. 2022, 29, 100400. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; ColladoVides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Palsson, B.O. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. USA 2000, 97, 5528–5533. [Google Scholar] [CrossRef]
- Heinken, A.; Hertel, J.; Acharya, G.; Ravcheev, D.A.; Nyga, M.; Okpala, O.E.; Hogan, M.; Magnúsdóttir, S.; Martinelli, F.; Nap, B.; et al. Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat. Biotechnol. 2023, 41, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lloyd, C.J.; Palsson, B.O. Reconstructing organisms in silico: Genome-scale models and their emerging applications. Nat. Rev. Microbiol. 2020, 18, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Beg, Q.K.; Vazquez, A.; Ernst, J.; de Menezes, M.A.; Bar-Joseph, Z.; Barabási, A.L.; Oltvai, Z.N. Intracellular crowding defines the mode and sequence of substrate uptake by and constrains its metabolic activity. Proc. Natl. Acad. Sci. USA 2007, 104, 12663–12668. [Google Scholar] [CrossRef]
- Shlomi, T.; Benyamini, T.; Gottlieb, E.; Sharan, R.; Ruppin, E. Genome-Scale Metabolic Modeling Elucidates the Role of Proliferative Adaptation in Causing the Warburg Effect. PLoS Comput. Biol. 2011, 7, e1002018. [Google Scholar] [CrossRef] [PubMed]
- De, R.K.; Das, M.; Mukhopadhyay, S. Incorporation of enzyme concentrations into FBA and identification of optimal metabolic pathways. BMC Syst. Biol. 2008, 2, 65. [Google Scholar] [CrossRef] [PubMed]
- Van Hoek, M.J.A.; Merks, R.M.H. Redox balance is key to explaining full vs. partial switching to low-yield metabolism. BMC Syst. Biol. 2012, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Beg, Q.K.; deMenezes, M.A.; Ernst, J.; Bar-Joseph, Z.; Barabási, A.L.; Boros, L.G.; Oltvai, Z.N. Impact of the solvent capacity constraint on E. coli metabolism. BMC Syst. Biol. 2008, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res 2021, 49, D498–D508. [Google Scholar] [CrossRef] [PubMed]
- Kerkhoven, E.J. Advances in constraint-based models: Methods for improved predictive power based on resource allocation constraints. Curr. Opin. Microbiol. 2022, 68, 102168. [Google Scholar] [CrossRef]
- Wang, Q.L.; Chen, J.H.; He, N.Y.; Guo, F.Q. Metabolic Reprogramming in Chloroplasts under Heat Stress in Plants. Int. J. Mol. Sci. 2018, 19, 849. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.J.; Kerkhoven, E.J.; Nielsen, J. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 2017, 13, 935. [Google Scholar] [CrossRef]
- Ferreira, M.A.D.; Silveira, W.B.D.; Nikoloski, Z. PARROT: Prediction of enzyme abundances using protein-constrained metabolic models. PLoS Comput. Biol. 2023, 19, e1011549. [Google Scholar] [CrossRef]
- Adadi, R.; Volkmer, B.; Milo, R.; Heinemann, M.; Shlomi, T. Prediction of Microbial Growth Rate versus Biomass Yield by a Metabolic Network with Kinetic Parameters. PLoS Comput. Biol. 2012, 8, e1002575. [Google Scholar] [CrossRef]
- Wittig, U.; Rey, M.; Weidemann, A.; Kania, R.; Müller, W. SABIO-RK: An updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 2018, 46, D656–D660. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Cukura, A.; Denny, P.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Bekiaris, P.S.; Klamt, S. Automatic construction of metabolic models with enzyme constraints. BMC Bioinform. 2020, 21, 19. [Google Scholar] [CrossRef]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A.; et al. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef]
- Hu, X.P.; Schroeder, S.; Lercher, M.J. Proteome efficiency of metabolic pathways in Escherichia coli increases along the nutrient flow. mSystems 2023, 8, e0076023. [Google Scholar] [CrossRef]
- Chen, Y.; Gustafsson, J.; Rangel, A.T.; Anton, M.; Domenzain, I.; Kittikunapong, C.; Li, F.R.; Yuan, L.; Nielsen, J.; Kerkhoven, E.J. Reconstruction, simulation and analysis of enzyme-constrained metabolic models using GECKO Toolbox 3.0. Nat. Protoc. 2024, 19, 629–667. [Google Scholar] [CrossRef]
- Massaiu, I.; Pasotti, L.; Sonnenschein, N.; Rama, E.; Cavaletti, M.; Magni, P.; Calvio, C.; Herrgård, M.J. Integration of enzymatic data in genome-scale metabolic model improves phenotype predictions and enables in silico design of poly-gamma-glutamic acid production strains. Microb. Cell Fact. 2019, 18, 3. [Google Scholar] [CrossRef]
- Domenzain, I.; Sanchez, B.; Anton, M.; Kerkhoven, E.J.; Millan-Oropeza, A.; Henry, C.; Siewers, V.; Morrissey, J.P.; Sonnenschein, N.; Nielsen, J. Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0. Nat. Commun. 2022, 13, 3766. [Google Scholar] [CrossRef]
- Gustafsson, J.; Roshanzamir, F.; Hagnestål, A.; Patel, S.M.; Daudu, O.I.; Becker, D.F.; Robinson, J.L.; Nielsen, J. Metabolic collaboration between cells in the tumor microenvironment has a negligible effect on tumor growth. Innovation 2024, 5, 100583. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.T.; Zhao, X.; Yang, X.; Zhang, P.J.; Du, J.W.; Yuan, Q.Q.; Ma, H.W. ECMpy, a Simplified Workflow for Constructing Enzymatic Constrained Metabolic Network Model. Biomolecules 2022, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Abedpour, N.; Kollmann, M. Resource constrained flux balance analysis predicts selective pressure on the global structure of metabolic networks. BMC Syst. Biol. 2015, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Jabarivelisdeh, B.; Waldherr, S. Optimization of bioprocess productivity based on metabolic-genetic network models with bilevel dynamic programming. Biotechnol. Bioeng. 2018, 115, 1829–1841. [Google Scholar] [CrossRef]
- Goelzer, A.; Fromion, V.; Scorletti, G. Cell design in bacteria as a convex optimization problem. Automatica 2011, 47, 1210–1218. [Google Scholar] [CrossRef]
- Goelzer, A.; Muntel, J.; Chubukov, V.; Jules, M.; Prestel, E.; Nölker, R.; Mariadassou, M.; Aymerich, S.; Hecker, M.; Noirot, P.; et al. Quantitative prediction of genome-wide resource allocation in bacteria. Metab. Eng. 2015, 32, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Phillips, R. Cell Biology by the Numbers, 1st ed.; Garland Science: Boca Raton, FL, USA, 2015. [Google Scholar]
- Bulovic, A.; Fischer, S.; Dinh, M.; Golib, F.; Liebermeister, W.; Poirier, C.; Tournier, L.; Klipp, E.; Fromion, V.; Goelzer, A. Automated generation of bacterial resource allocation models. Metab. Eng. 2019, 55, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Bodeit, O.; Ben Samir, I.; Karr, J.R.; Goelzer, A.; Liebermeister, W. RBAtools: A programming interface for Resource Balance Analysis models. Bioinform. Adv. 2023, 3, vbad056. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Steuer, R. Modelling microbial communities using biochemical resource allocation analysis. J. R. Soc. Interface 2019, 16, 20190474. [Google Scholar] [CrossRef]
- Thiele, I.; Fleming, R.M.T.; Que, R.; Bordbar, A.; Diep, D.; Palsson, B.O. Multiscale Modeling of Metabolism and Macromolecular Synthesis in E. coli and Its Application to the Evolution of Codon Usage. PLoS ONE 2012, 7, e45635. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, K.; Palsson, B.O.; Yang, L.R.C. StressME: Unified computing framework of metabolism, gene expression, and stress responses. PLoS Comput. Biol. 2024, 20, e1011865. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Gao, Y.; Mih, N.; O’Brien, E.J.; Yang, L.; Palsson, B.O. Thermosensitivity of growth is determined by chaperone-mediated proteome reallocation. Proc. Natl. Acad. Sci. USA 2017, 114, 11548–11553. [Google Scholar] [CrossRef]
- Yang, L.; Mih, N.; Anand, A.; Park, J.H.; Tan, J.; Yurkovich, J.T.; Monk, J.M.; Lloyd, C.J.; Sandberg, T.E.; Seo, S.W.; et al. Cellular responses to reactive oxygen species are predicted from molecular mechanisms. Proc. Natl. Acad. Sci. USA 2019, 116, 14368–14373. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.J.; Ebrahim, A.; Yang, L.; King, Z.A.; Catoiu, E.; O’Brien, E.J.; Liu, J.K.; Polsson, B.O. COBRAme: A computational framework for genome-scale models of metabolism and gene expression. PLoS Comput. Biol. 2018, 14, e1006302. [Google Scholar] [CrossRef]
- Lerman, J.A.; Hyduke, D.R.; Latif, H.; Portnoy, V.A.; Lewis, N.E.; Orth, J.D.; Schrimpe-Rutledge, A.C.; Smith, R.D.; Adkins, J.N.; Zengler, K.; et al. In silico method for modelling metabolism and gene product expression at genome scale. Nat. Commun. 2012, 3, 929. [Google Scholar] [CrossRef]
- Yang, L.; Ma, D.; Ebrahim, A.; Lloyd, C.J.; Saunders, M.A.; Palsson, B.O. solveME: Fast and reliable solution of nonlinear ME models. BMC Bioinform. 2016, 17, 391. [Google Scholar] [CrossRef]
- Ma, D.; Yang, L.; Fleming, R.M.T.; Thiele, I.; Palsson, B.O.; Saunders, M.A. Reliable and efficient solution of genome-scale models of Metabolism and macromolecular Expression. Sci. Rep. 2017, 7, 40863. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamics-based metabolic flux analysis. Biophys. J. 2007, 92, 1792–1805. [Google Scholar] [CrossRef]
- Jankowski, M.D.; Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 2008, 95, 1487–1499. [Google Scholar] [CrossRef]
- Oftadeh, O.; Salvy, P.; Masid, M.; Curvat, M.; Miskovic, L.; Hatzimanikatis, V. A genome-scale metabolic model of Saccharomyces cerevisiae that integrates expression constraints and reaction thermodynamics. Nat. Commun. 2021, 12, 4790. [Google Scholar] [CrossRef]
- Lynd, L.R.; Beckham, G.T.; Guss, A.M.; Jayakody, L.N.; Karp, E.M.; Maranas, C.; McCormick, R.L.; Amador-Noguez, D.; Bomble, Y.J.; Davison, B.H.; et al. Toward low-cost biological and hybrid biological/catalytic conversion of cellulosic biomass to fuels. Energy Environ. Sci. 2022, 15, 938–990. [Google Scholar] [CrossRef]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. OptKnock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Maranas, C.D. Designing overall stoichiometric conversions and intervening metabolic reactions. Sci. Rep. 2015, 5, 16009. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Maranas, C.D. OptCom: A Multi-Level Optimization Framework for the Metabolic Modeling and Analysis of Microbial Communities. PLoS Comput. Biol. 2012, 8, e1002363. [Google Scholar] [CrossRef] [PubMed]
- Boorla, V.S.; Maranas, C.D. CatPred: A comprehensive framework for deep learning in vitro enzyme kinetic parameters kcat, Kmand Ki. bioRxiv 2024. [Google Scholar] [CrossRef]
- Chen, Y.; Nielsen, J. In vitro turnover numbers do not reflect in vivo activities of yeast enzymes. Proc. Natl. Acad. Sci. USA 2021, 118, e2108391118. [Google Scholar] [CrossRef] [PubMed]
- Davidi, D.; Noor, E.; Liebermeister, W.; Bar-Even, A.; Flamholz, A.; Tummler, K.; Barenholz, U.; Goldenfeld, M.; Shlomi, T.; Milo, R. Global characterization of in vivo enzyme catalytic rates and their correspondence to in vitro measurements. Proc. Natl. Acad. Sci. USA 2016, 113, 3401–3406. [Google Scholar] [CrossRef] [PubMed]
- Küken, A.; Gennermann, K.; Nikoloski, Z. Characterization of maximal enzyme catalytic rates in central metabolism of. Plant J. 2020, 103, 2168–2177. [Google Scholar] [CrossRef]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef]
- Mao, Z.; Niu, J.; Zhao, J.; Huang, Y.; Wu, K.; Yun, L.; Guan, J.; Yuan, Q.; Liao, X.; Wang, Z.; et al. ECMpy 2.0: A Python package for automated construction and analysis of enzyme-constrained models. Synth. Syst. Biotechnol. 2024, 9, 494–502. [Google Scholar] [CrossRef]
- Sarkar, D.; Maranas, C.D. SNPeffect: Identifying functional roles of SNPs using metabolic networks. Plant J. 2020, 103, 512–531. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.; Boorla, V.S.; Dash, S.; Gopalakrishnan, S.; Jacobson, T.B.; Olson, D.G.; Amador-Noguez, D.; Lynd, L.R.; Maranas, C.D. Assessing the impact of substrate-level enzyme regulations limiting ethanol titer in vivo using a core kinetic model. Metab. Eng. 2022, 69, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Olson, D.G.; Chan, S.H.J.; Amador-Noguez, D.; Lynd, L.R.; Maranas, C.D. Thermodynamic analysis of the pathway for ethanol production from cellobiose in. Metab. Eng. 2019, 55, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Crafts-Brandner, S.J.; Salvucci, M.E. Sensitivity of photosynthesis in a C4 plant, maize, to heat stress. Plant Physiol. 2002, 129, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Marcisauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, H.S. The RNA world hypothesis: The worst theory of the early evolution of life (except for all the others)a. Biol. Direct 2012, 7, 23. [Google Scholar] [CrossRef]
- Oftadeh, O.; Hatzimanikatis, V. Genome-scale models of metabolism and expression predict the metabolic burden of recombinant protein expression. Metab. Eng. 2024, 84, 109–116. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Framework Category | Constraints | |||||||
|---|---|---|---|---|---|---|---|---|
| Precursor | cgRAM | fgRAM | ||||||
| FBAwMC | FBAwSCC | GECKO | MOMENT | RBA | ME Model | ETFL | Conceptual Description | Eqn. No. |
| × | × | × | × | × | × | × | Objective function | (1) |
| × | × | × | × | × | × | × | Mass balance | (2) |
| × | × | × | × | × | × | × | Flux bounds | (3) |
| × | Molecular crowding | (4) | ||||||
| × | Solute capacity | (5) | ||||||
| × | × | × | × | × | Linear enzyme kinetics limitation | (6) | ||
| × | × | × | × | × | Enzyme capacity | (9) | ||
| × | Enzyme pool determination | (10) | ||||||
| × | Enzyme pool limit | (11) | ||||||
| × | × | × | Macromolecule mass balance (pseudosteady-state) | (14) | ||||
| × | × | rRNA capacity constraint | (17) | |||||
| × | Protein-ribosome coupling constraint | (18) | ||||||
| × | Transcription capacity constraint | (19) | ||||||
| × | Macromolecular machinery capacity constraint | (20) | ||||||
| × | Thermodynamic constraints on reaction direction | (21)–(26) | ||||||
| × | Petersen linearization of growth-driven dilution | (29)–(31) | ||||||
| LP | LP | LP | LP | Iterative LP | Iterative LP or NLP | MILP | Type of problem | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schroeder, W.L.; Suthers, P.F.; Willis, T.C.; Mooney, E.J.; Maranas, C.D. Current State, Challenges, and Opportunities in Genome-Scale Resource Allocation Models: A Mathematical Perspective. Metabolites 2024, 14, 365. https://doi.org/10.3390/metabo14070365
Schroeder WL, Suthers PF, Willis TC, Mooney EJ, Maranas CD. Current State, Challenges, and Opportunities in Genome-Scale Resource Allocation Models: A Mathematical Perspective. Metabolites. 2024; 14(7):365. https://doi.org/10.3390/metabo14070365
Chicago/Turabian StyleSchroeder, Wheaton L., Patrick F. Suthers, Thomas C. Willis, Eric J. Mooney, and Costas D. Maranas. 2024. "Current State, Challenges, and Opportunities in Genome-Scale Resource Allocation Models: A Mathematical Perspective" Metabolites 14, no. 7: 365. https://doi.org/10.3390/metabo14070365
APA StyleSchroeder, W. L., Suthers, P. F., Willis, T. C., Mooney, E. J., & Maranas, C. D. (2024). Current State, Challenges, and Opportunities in Genome-Scale Resource Allocation Models: A Mathematical Perspective. Metabolites, 14(7), 365. https://doi.org/10.3390/metabo14070365