Abstract
Caloric restriction (CR) and its related alternatives have been shown to be the only interventions capable of extending lifespan and decreasing the risk of cancer, along with a reduction in burden in pre-clinical trials. Nevertheless, the results from clinical trials have not been as conclusive as the pre-clinical results. Recognizing the challenges associated with long-term fasting, the application of caloric restriction mimetics (CRMs), pharmacological agents that mimic the molecular effects of CR, to harness the potential benefits while overcoming the practical limitations of fasting has resulted in an interesting alternative. This review synthesizes the findings of diverse clinical trials evaluating the safety and efficacy of CR and CRMs. In dietary interventions, a fast-mimicking diet was the most tolerated to reduce tumoral growth markers and chemotherapy side effects. CRMs were well tolerated, and metformin and aspirin showed the most promising effect in reducing cancer risk in a selected group of patients. The application of CR and/or CRMs shows promising effects in anti-cancer therapy; however, there is a need for more evidence to safely include these interventions in standard-of-care therapies.
1. Introduction
Cancer is a disease caused by the uncontrollable proliferation of cells that have lost the ability to respond to normal signals and gained the ability to invade tissues and organs, affecting their functions.
In 2020, the World Health Organization (WHO) reported 19 million new cases and estimated almost 10 million deaths around the world. Even with the most recent advances in surgery, chemotherapy (CT), and radiotherapy (RT), cancer remains the second most common cause of death worldwide, indicating the need for new complementary treatments against cancer.
Metabolic shifts are one of the representative hallmarks of cancer [1]. Elevated glucose consumption is one of the most representative changes in cancer cells. It allows for the high levels of ATP needed for the metabolic processes in neoplastic cells and serves as an intermediate metabolite for the synthesis of the nucleic acids and phospholipids required for cell division and the maintenance of tumor growth [2]. Furthermore, hexose can be derived from the pentose–phosphate pathway to generate NADPH and maintain the redox state in neoplastic cells [3].
Almost a hundred years ago, Otto Warburg hypothesized a mitochondrial dysfunction in cancer cells, which makes them dependent on high glucose consumption and aerobic glycolysis, a hypothesis named the Warburg effect [4]. Further studies have demonstrated correct mitochondrial functioning, which differs from this hypothesis.
Bearing this in mind, the modulation of glucose consumption in cancer cells can be highlighted as a complementary treatment. This approach might have the potential to decrease cell division, increase oxidative stress, and ultimately lead to cell death, making it a plausible co-therapy against cancer. Many pre-clinical studies have shown the feasibility of this therapy, which can reduce tumor growth and increase the sensitivity to CT and RT [5,6], but how can the modulation of glucose consumption be applied in a clinical context?
2. Methodology for Database Analysis
A comprehensive literature search using the PubMed, Google Scholar, and ClinicalTrials.gov databases was performed to retrieve information related to caloric restriction, caloric restriction mimetics, and their effect on cancer. The selection of the dietary interventions and the mimetics was made according to the results of pre-clinical studies and their applicability in humans. The keywords used for this search included “caloric restriction”, “ketogenic diet”, “fast mimicking diet”, “caloric restriction mimetics”, “metformin”, “rapamycin”, “aspirin”, and “resveratrol” in relation to cancer. Only research studies detailing in vivo, in vitro, and clinical trials published between 2010 and 2023 were included in this review. The web application Biorender.com was used for the creation of the figures.
3. Caloric Restriction
3.1. Definition and Cancer-Associated Molecular Pathways
Caloric restriction (CR) is defined as a reduction in energy intake (<500 kCal) without incurring malnutrition [7]. The benefits of this dietary intervention in adults have been reported for years, and CR is the only reported intervention capable of extending lifespan; it is cardio-, osteo-, sarco-, and neuroprotective, and has been reported to be able to lower the incidence and progression of cancer [8,9]. However, a reduction in food consumption to achieve caloric restriction can be difficult for a healthy adult person and almost impossible for an oncologic patient, so alternative dietary regimens are necessary.
To overcome this, alternative regimens have been tested in cancer patients; among them are the ketogenic diet (KD), intermittent fasting (IF), and short-term starvation (STS).
The KD limits the consumption of carbohydrates and promotes the intake of fats; this induces ketosis through the mobilization of stored fatty acids for energy needs [10]. IF is a dietary intervention that limits the timing of caloric consumption, regardless of the content or number of calories [11]. STS is another dietary intervention that requires fasting for several days [12].
No matter the dietary intervention, all of them have been shown to be tolerable to oncologic patients and have some benefits; this effect is related to the regulation of a variety of molecular pathways (Figure 1) [13,14,15].
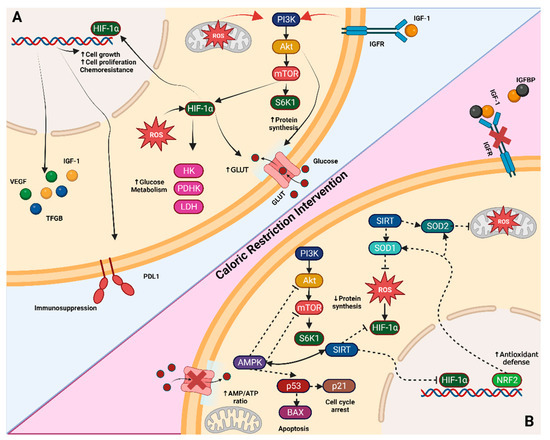
Figure 1.
Tumor-altered pathways and CR molecular targets. (A) One of the commonly altered pathways in tumor cells is the PI3K/AKT/mTOR pathway, which leads to increased protein synthesis through S6K1 activation and the upregulation of HIF1α. HIF1α further promotes glycolytic intermediaries such as hexokinase (HK), pyruvate dehydrogenase kinase (PDK), and lactate dehydrogenase (LDH). Additionally, it induces the secretion of growth factors like insulin-like growth factor (IGF-1), transforming growth factor beta (TGF-β), and vascular endothelial growth factor (VEGF). These changes collectively enhance cell growth, proliferation, and chemoresistance. (B) When a caloric-restriction intervention is applied, such as glucose starvation, the increased AMP levels activate AMPK. This activation results in the inhibition of AKT and mTOR pathways and triggers a response mediated by p53 and p21, leading to interrupted protein synthesis and cell cycle arrest or apoptosis. The energy deficit also activates the SIRT pathways, which downregulates HIF1α, and the Nrf2 pathway, which enhances antioxidant defenses (e.g., superoxide dismutase SOD1 and SOD2) and reduces reactive oxygen species (ROS). Sky blue background shows tumor-altered pathways in non-caloric restriction (normal) diet, meanwhile red background shows the pathways during caloric restriction diet. Figure created with BioRender.com. Note: Arrows indicate activation, while dashed bar lines indicate inactivation.
3.1.1. Energy Sensor Pathways: PI3K-AKT-mTOR and AMPK
The phosphoinositide 3-kinase (PI3K), AKT, mammalian target of rapamycin (mTOR) pathway (PIK3-AKT-mTOR) responds to the availability of nutrients, hormones, and growth factors, playing a crucial role in cell growth, metabolism, and proliferation [16]. AKT and mTOR can increase the transcription of glycolytic enzymes and glucose transporters (GLUTs), making them attractive targets for cancer therapy [17]. AKT and mTOR inhibitors are currently undergoing preclinical and clinical trials, underscoring the importance of regulating oncologic metabolism. As a nutrient-sensor mechanism, the PI3K-AKT-mTOR pathway can be regulated by both the depletion and increase in metabolites. CR modulates this pathway by activating AMP-activated protein kinase (AMPK), mediated by an increased AMP/ATP ratio in a glucose-limited environment [18]. AMPK activation leads to the phosphorylation of Rictor, resulting in the inactivation of the mTOR complex. Additionally, the phosphorylation of tuberous sclerosis complex 2 (TSC2) by AMPK further inhibits the mTOR complex, thereby increasing autophagy flux. This enhanced autophagy can be utilized to induce cell death in cancer cells [17,19].
3.1.2. Sirtuins
Sirtuins (SIRTs) are a family of NAD-dependent deacetylase enzymes involved in various processes such as carbohydrate metabolism, stress response, inflammatory response, lifespan regulation, and tumor formation [20]. Consequently, they have become a focal point for research into diseases such as cancer, age-related conditions, rheumatic diseases, and metabolic syndrome. SIRTs function as transcriptional regulators by deacetylating histones and modulating the chromatin structure, thereby influencing the transcription of multiple genes, including FOXO, PGC-1α, and PPARα, among others [21]. In metabolic functions, SIRTs downregulate glycolytic enzymes through deacetylation and repress the key transcriptional inducer HIF-1α [22,23].
Furthermore, SIRTs contribute to antioxidant protection by increasing superoxide dismutase 2 (SOD2) activity, an enzyme in the mitochondrial matrix, which mitigates the expression of HIF-1α in response to cellular injury [24,25]. In the context of cancer, the shift from glycolytic to oxidative metabolism and the downregulation of glutaminolysis mediated by SIRTs act as an anti-Warburg effect, reducing tumor growth. Among the sirtuins, SIRT1, SIRT3, SIRT4, and SIRT6 have been identified as significant targets for anti-tumor treatments [26]; SIRT4 has been proposed as a tumor-suppressor protein, reducing glutamine entry into mitochondria in response to DNA damage [27,28].
However, SIRT1 is a controversial protein with dual roles; it may enhance tumor growth [29,30] or prevent tumorigenesis [31,32]. Therefore, further studies incorporating controlled variables of tumor growth variables are essential to elucidate its precise role.
The most frequently reported molecular targets of CR are AMPK and SIRTs, both being dependent on each other. The exact mechanism by which CR activates SIRTs remains unclear, but it is hypothesized that the increase in NAD+ by the metabolic shift from fermentation to respiration triggers the activation of the sirtuins enzymes [33,34]. This theory implies that cells unable to undergo this metabolic shift, such as some tumoral cells, do not benefit from this pathway.
3.1.3. Nrf2
The nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that regulates the expression of antioxidant enzymes in response to oxidative damage and inflammation [35]. Additionally, Nrf2 controls the production of reducing cofactor and multi-drug resistance-associated proteins, working as a protective mechanism against cell damage. The induction and overexpression of Nrf2 have demonstrated tumor-suppressive activity in chemically induced phase 2 tumorigenesis [36,37,38,39]. Nevertheless, in well-established tumors, overexpression and mutant variants of Nrf2 and downregulation of Keap1, the main inactivator of Nrf2, have been associated with poor prognosis in cancer [40]. This pro-tumorigenic effect is mediated by protection against reactive oxygen species (ROS) generated by RT and increased resistance to CT through the production of multi-drug resistance-associated proteins [41,42].
Not well described up to date, the carcinogenic protective effect of CR has been linked to the expression of Nfr2 [43], as evidenced by studies showing that Nfr2-knockout ( mice on a CR diet develop chemically induced tumors, whereas wild-type () mice on a CR diet do not [44]. Understanding how CR activates the NRF2 pathway and elucidating the dual role of Nfr2 in tumorigenesis requires further research for its safe application in clinical trials.
3.1.4. GH/IGF-1 Axis
The growth hormone/insulin-like growth factor-1 (GH/IGF-1) is the main axis of growth in the body. Its primary function extends from development through adulthood until levels of this hormone decrease to a critical point known as “somatopause”, which leads to changes in body composition and cellular metabolism [44]. The main function of this axis is to increase the anabolic processes, such as protein synthesis and mitosis, while downregulating apoptosis. Growth hormone (GH) is secreted by the adenohypophysis under the regulation of hypothalamic growth hormone-releasing hormone (GHRH) and somatostatin. The liver is the main target, where it interacts with the growth hormone receptor (GHR), triggering IGF-1 release as the principal effector of this axis.
IGF-1 binds to its receptor (IGF1R), which is practically ubiquitous, exerting systemic body growth effects [45]. The binding of IGF-1 to IGF1R initiates the PI3K-AKT-mTOR pathway, among other important pathways such as the Ras/Raf/MEK/ERK/MAPK pathway, signaling nutrient availability of nutrients to initiate cell division.
Due to its mitogenic and anti-apoptotic effects, the GH/IGF-1 axis has garnered interest as a potential target for anti-cancer therapy. Although not considered an oncogene [46], elevated blood IGF-1 levels, and more importantly, local or peritumoral IGF-1 levels [47,48,49], have been associated with poor prognosis, increased tumor growth, and resistance to cytotoxic therapy. Consequently, antibodies and small molecules targeting IGF-1 and IGF1R have been developed and tested in pre-clinical and clinical models, showing the potential to delay tumor growth and increase sensibility to CT and RT [50,51]. CT in vivo models have demonstrated its ability to reduce IGF1 levels in rodent models, suggesting a plausible non-pharmacological intervention [52].
Nevertheless, clinical studies have not shown that a CR diet can reduce serum IGF-1 levels in patients, as observed in preclinical models [53,54]. Nonetheless, the effect of CR in reducing tumor burden persists, suggesting a possible non-IGF-1 mediated response or a reduction in tumor and peritumoral expression of IGF-1 or IGF1R.
4. Dietary Interventions and Their Anti-Tumorigenic Effect: Bench to Bedside
The anti-tumor/antiproliferative effects of caloric restriction have been widely proven in vitro, showing that multiple tumor cell lines are affected contrary to non-tumoral cell lines. The standard CR induction has been through glucose restriction (GR) in the media based on reports of dependence on the Warburg effect in different tumoral cell lines. Reducing glucose to 1 g/L and 0 g/L, compared to 4.5 g/L, has shown a decreased proliferative effect, mainly in tumor cell lines. Additionally, in prostate cancer (PC) [55] and triple-negative breast cancer (TNBC) [56] cell lines, GR has been shown to trigger an increase in ROS, leading to cell death.
Among the multiple effects of GR, disrupting the cell cycle to induce cell arrest [57,58], activating the AMPK pathway, downregulating telomerase activity [59,60], decreasing migration [58], inducing apoptosis [61], and necrosis-dependent death [62] are the principal mechanisms implicated in the antiproliferative effect. Consequently, GR has been proposed as an anti-tumoral therapy. Additionally, GR has been related to an increased sensitivity to CT [63,64], allowing for a dosage reduction in the clinical context.
In higher mammals, mouse models have demonstrated that various nutraceutical regimens can be translated to a human diet, showing similar effects to those reported in vitro. CR, fasting-mimicking diet (FMD), and low caloric intake (LCI) reduced tumor burden in a breast cancer (BC) mouse model, with CR showing the maximal effect [65]. Comparing CR against KD in a pancreatic ductal adenocarcinoma (PDCA) model, CR but not KD helped to reduce tumor growth and mortality [66]. This outcome could be explained by the increased availability of lipids in KD, which can be used as fuel for energy generation and anabolic reactions.
Other outcomes found in CR diets as a treatment against cancer include an increase in tumor-infiltrating lymphocyte (TIL) CD8+ and PD-1 expression [67], a reduction in myeloid-derived suppressor cells [68], and tumor-forming stem cells. Additionally, CR decreases poor prognosis markers such as IGF1, pAKT, and PI3K [69] and induces changes in the gut microbiome linked to anti-tumor effects [70]. Incorporating a nutraceutical regimen like CR or KD with CT has reduced tumor growth and relapse and improved the survival rate [67,71].
In a clinical context (Table 1), the applicability of a CR diet in oncologic patients is controversial due to the potential adverse effects of significant body weight loss. Therefore, most clinical trials have used a body mass index (BMI) between 18.5 and 27.5 to mitigate the risk of induced cachexia. Nevertheless, the use of CR diets has been reported as well or moderately tolerated in most cancers [13,14,15,72], except in head and neck squamous cell carcinoma (HNSCC), where not even enteral nutrition was tolerated (72). The location and compressive effects of the tumor mass can be considered the principal problem for tolerability [73].
Another topic to consider is the type of dietary intervention, with FMD being the first option, followed by KD and CR last. FMD has been considered the most cost-effective and applicable because it does not completely restrict food intake, the food is the most tolerable when compared to KD, and it has a shorter fasting period compared to CR. This makes it the most approachable nutraceutical intervention against cancer.
Even though most of these clinical trials have only reached phase 1 with a limited number of patients, the outcomes are promising as complementary therapy regimens. KD is not well tolerated, with low adherence, and there were no changes in the overall survival (OS) and partial response (PR) for the small number of patients. There was an increase in serum carbonylated proteins after a combination of CT/RT with KD [74], presumably due to increased oxidative stress and partial cell death.
The results from clinical trial outcomes have been further validated in preclinical models to minimize the concomitant variables, with findings consistently indicating that dietary restrictions render highly proliferative tumor cells more susceptible to oxidative damage [56,57]. This is due to the reduction in glucose, a precursor of NADPH, the principal co-factor for many antioxidant systems and enzymes, rendering the cells unable to control oxidative damage and more susceptible to DNA damage caused by CRT [75].
Among other beneficial effects of dietary interventions is the increased endurance of healthy cells to cytotoxic drugs and radiotherapy. For example, CR has been reported to induce cell cycle arrest in the G0/G1 phases, enabling cells to undergo DNA repair more efficiently and diminishing DNA damage by CRT [58]. In contrast, tumoral cells, which have an altered cell cycle, are unable to repair DNA, leading to cell death [76]. Additionally, this protective effect can diminish myeloid suppression and secondary tumor formation from CT/RT damage. A pilot study in patients with diffuse large B-cell lymphoma on short-term calorie reduction (SCR) showed an increase in leukocyte count after the first intervention compared to the comparison group, but no difference was observed after the second [72]. Two more extensive studies with different neoplasias, one not randomized and one randomized, evidenced that inducing CR at least one hour prior to or after a CT round reduced leukocyte DNA CT-associated damage compared to the non-fasting group [13,77].
As previously reviewed, dietary interventions not only act directly on tumor glucose metabolism but also alter other metabolic pathways not directly linked to carbohydrate metabolism. This can be seen in a report on PC patients, where CR implementation increased IGFBP3 serum levels and tended to lower insulin serum levels [15]. The insulin/IGF1 growth axis has been implicated as a poor prognostic marker, not as an oncogenic pathway, but as a proliferative additive to the unregulated tumor cell signal [47,52]. IGFBP3 is the main IGF-transporter protein in the blood, where it forms a complex that stabilizes and enhances the half-life of insulin-like growth factors. It also reduces their interaction with IGFR, thus blocking activation and reducing mitogenic effects. Decreased IGFBP3 levels have been implicated as a risk factor in multiple neoplasias, making it an interesting target in oncologic treatment [78].
Other metabolic changes associated with dietary interventions combined with conventional anti-tumoral therapies include reduced serum glucose levels and a decrease in insulin, IGF1, and leptin [15,77]. Many cancer patients receive corticosteroids as prophylactic treatment against CRT side effects, but steroids can increase glucose and insulin levels, triggering growth signals in residual tumor cells as a collateral effect. Clinical trials have shown that CR can reduce both glucose and insulin levels in patients, regardless of the use of steroids [79], highlighting the potential of nutraceutical interventions in regulating not only the tumoral but overall metabolism, potentially improving the patient’s health outcomes.
The regulation of caloric intake in pre-clinical models has shown the capability to modulate the tumor immune response, improving cytotoxic responses against the immunosuppressive tumor microenvironment [67,68]. In oncologic patients, stimulating the immune response is a primary goal of immunomodulators like anti-PDL1. Therefore, the immune-regulatory function of CR could be beneficial in enhancing the anti-tumor response with fewer side effects than immunomodulatory drugs.
A phase 2 non-randomized study that included multiple types of cancer patients on FMD demonstrated that this dietary intervention could reduce monocytic myeloid-derived suppressor cells CD14+HLA-DR- and CD14+PD-LD+, both reported as highly suppressive subsets, while upregulating CD8+PD-1+CD69+-activated T cells, cytolytic natural killer (NK) cells, and CD3+CD25+ T cells, all reported as anti-tumor effectors [80]. This immunoregulatory effect is not only attributed to the cell phenotype, as another trial showed that KD was able to decrease TNF-a and increase IL-10 [81]. IL-10, an anti-inflammatory cytokine, has been reported as a potent inducer and activator of CD8+ T cells, having anti-tumoral properties that can be targeted [82].

Table 1.
Caloric restriction and its applicability in anti-cancer trials.
Table 1.
Caloric restriction and its applicability in anti-cancer trials.
| Study Design | Type(s) of Cancer | SOC | Intervention | Outcomes | REF. |
|---|---|---|---|---|---|
| Phase 2. Randomized, controlled, observer-blind | HER- BC | CT with dexamethasone | Fast-mimicking diet | There is no difference in pCR. The toxicity was partially reduced after FMD without the use of dexamethasone. Increase in the MP. Reduction in glucose, insulin, IGF1, and DNA leukocyte damage. | [77] |
| Phase 1, non-randomized | Advanced NSCLC and PaCa | CRT | Ketogenic diet | No tolerability of the intervention. Not enough patients to analyze overall survival nor partial response. Increase in carbonylated proteins in serum as an indicator of increased oxidative stress. | [74] |
| Phase 1, non-randomized | HNSCC | CRT | Ketogenic diet | Poor tolerability of the diet. Not enough patients to analyze overall survival nor partial response. No evident changes in carbonylated proteins nor GSH as indicators of increased redox stress. | [73] |
| Phase 1, non-randomized | Multiple | CT | Short-term fasting | The intervention was well tolerated by all participants.Reduction in blood leukocyte damage after 24 h of fasting. | [13] |
| Pilot study, randomized, controlled | PC | Sx or active surveillance | Caloric restriction | The restriction diet was well tolerated. Increase in serum IGFBP-3 levels, along with insulin and C-peptide. | [15] |
| Phase 1, single arm | Multiple | SOC | Fast-mimicking diet | FMD along with the SOC decreased glucose, insulin, and IGF1 levels in serum. There was also a reduction in myeloid-derived suppressor cells and PD+ cells. In tumor and blood, there was an increase in CD8+, NK, and macrophage infiltration. | [80] |
| Phase 1, randomized, not blind | BC | CT | Ketogenic diet | Dietetic intervention reduced serum TNF-a and insulin levels. Also, CT plus KD reduced tumor size and TNM compared to control. | [81] |
| Pilot study, randomized, blind | DLBCL | CT + biologic | Short-term caloric reduction | The intervention was safe and feasible. The intervention improved hematological parameters. | [72] |
| Pilot study, randomized, cross-over | BC and OC | CT | Short-term fasting | Short-term fasting is feasible and reduces chemotherapy side effects, improving quality of life. | [83] |
| Phase 1, randomized, controlled | BC | SOC | Ketogenic diet | Ketogenic diet improved quality of life, but not in biomarkers after 12 weeks of intervention. | [84] |
BC: breast cancer, BTC: biliary tract cancer, CC: Cervix Cancer, CRC: colorectal cancer, CRT: chemoradiotherapy, CT: chemotherapy, DLBCL: Diffuse Large B Cell Lymphoma, EC: endometrial cancer, FMD: fast-mimicking diet, FTC: follicular thyroid cancer, GC: Gastric Cancer, GSH: Glutathione, IGF: insulin-like growth factor, IGFBP-3: IGF Binding Protein 3, MP: Miller–Payne, NSCLC: non-small cell lung cancer, OC: ovarian cancer, OS: overall survival, PaCa: Pancreatic Cancer, PC: prostate cancer, pCR: pathological complete response, PR: partial response, SOC: standard of care therapy, Sx: surgery, TNBC: triple-negative breast cancer.
5. Caloric Restriction Mimetics (CRM)
5.1. Definition
Considering the difficulties oncologic patients face in maintaining a CR diet, pharmacological therapies have been developed to mimic the effects of CR interventions. Caloric restriction mimetics (CRM) are a variety of compounds that replicate the biochemical and signaling pathways triggered by CR, such as autophagy induction, AMPK activation, and SIRT expression, among other pathways. Examples of the most studied CRM and their anti-cancer effects include metformin, rapamycin, aspirin, and resveratrol and its by-products [8] (Figure 2).
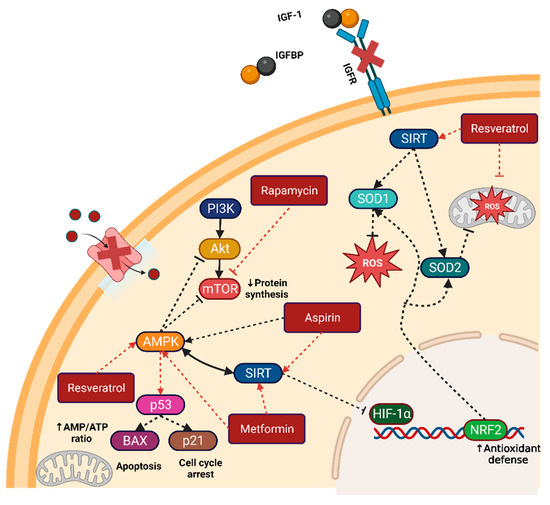
Figure 2.
Caloric restriction mimetics and their targets. Metformin, aspirin, resveratrol, and rapamycin are the most common CRMs used in preclinical and clinical research. Their principal target is the direct or indirect activation of the master regulators AMPK and SIRT. Activation of these master regulators can lead to the inactivation of key growth and division pathways such as PI3K/AKT/mTOR and HIF1α. Figure created with BioRender.com. Note: arrows indicate activation, while dashed bar lines indicate inactivation.
5.1.1. Metformin
Metformin is a biguanide molecule extensively used as an antidiabetic drug, primarily known as an insulin-sensitizer drug; it exerts multiple effects on most cells and their organelles [85]. The main effect on the mitochondria is that it inhibits complex I electron transfer, reducing NADH oxidation and thereby preventing ATP production [86]. The changes in the AMP/ATP ratio lead to activation of the AMPK pathway [59]. In tumor cells, AMPK activation can repress the PI3K/AKT/mTOR pathway, thereby inhibiting cell growth and tumorigenesis.
In preclinical studies, metformin has demonstrated the ability to induce cell cycle arrest and promote cell senescence [87]. Additionally, intrinsic apoptosis and autophagy-induced cell death have also been reported as anti-tumoral mechanisms of this drug [88]. Metformin also increases the activation of CD8+ lymphocytes and decreases T-reg levels [89], enhancing the anti-tumor immune response. Consequently, metformin has become a target of interest in oncology (Table 2).
A 2014 retrospective cohort study of 65,754 patients in Taiwan compared the effects of metformin therapy to other antidiabetic drugs used either alone or in combination. The analysis revealed that metformin reduced cancer risk in a dose-dependent manner, regardless of whether used alone or in combination, highlighting the growing interest in its anti-tumorigenic effects [90].
In non-diabetic women with operable stage I/II breast cancer, short-term neoadjuvant treatment with metformin, although not affecting tumor size, led to decreased Ki67 and phosphodiesterase 3 (PDE3B) staining in postoperative samples. It also resulted in reductions in weight, BMI, and glucose levels [91,92]. Metformin usage additionally increased transcriptomic signatures of TNFR1 pathway genes and TUNEL-positive cells [92].
In non-diabetic endometrial cancer patients, neoadjuvant use of metformin likewise decreased Ki67 staining along with insulin, IGF1, IGFBP7, and the surrogate marker of mTOR activation S6 ribosomal protein (Ps6) [93,94,95]. Also, they identified an increased activation of AMPK and p27, with subsequent reduction in cyclin D1 expression and cell cycle arrest [93].
For other cancer types, such as colorectal cancer (CRC), the addition of metformin to classical chemotherapy increased overall survival (OS) up to 7 months in obese patients without metabolic complications [94]. In non-small cell lung cancer (NSCLC) patients randomized to receive standard therapy with or without metformin, the combination of both therapies increased 1-year progression-free survival (PFS) to 45% compared to 15% in other trials without metformin, demonstrating the benefit of adding metformin to standard therapy [96].
Nevertheless, other studies have not shown benefits from adding metformin to the standard of care CT (SOC). These include studies on metastatic pancreatic [97] and breast cancer [98,99], advanced NSCLC [100,101], and castration-resistant prostate cancer [102]. This raises questions about whether metformin’s effects are primarily beneficial in the early stages of tumorigenesis, where glucose and growth signals are crucial for initial establishment and exponential growth.
Table 2.
Metformin and its application in anti-cancer therapies.
Table 2.
Metformin and its application in anti-cancer therapies.
| Study Design | Type(s) of Cancer | SOC | CRM | Outcomes | REF. |
|---|---|---|---|---|---|
| Pilot study, randomized, non-blinded | BC | Sx | Metformin 1 g/12 h | Preoperative use of metformin showed no decrease in tumor size but a significant reduction of Ki-67 and PDE3B cells. | [91] |
| Population-based retrospective cohort study | None | None | Metformin | The usage of metformin monotherapy or in combination with other antidiabetic drugs reduced the incidence of cancer in type 2 diabetic patients. | [90] |
| Pilot study, single arm | EC | Sx | Metformin 2 g/d | Preoperative use of metformin decreased Ki-67 and Ps6 staining and reduced serum levels of glucose and IGF1. It decreased proliferative markers ERK1/2 and Cyclin D1. | [93] |
| Phase 2, randomized | NSCLC | CT | Metformin 2 g/d | Concomitant use of metformin increased progression-free survival by 32% at one year compared to control. | [96] |
| Phase 2, randomized | CC | CRT | Metformin | Metformin increased 2-year disease-free survival by up to 67% compared to 33% in the controls. | [103] |
| Phase 2, randomized, double-blind, placebo-controlled | BC | Sx | Metformin 850 mg/12 h | Overall, no difference in proliferation rates between groups. In subgroup analysis, metformin showed a heterogeneous effect dependent on insulin resistance. | [104] |
| Phase 2, randomized, placebo-controlled | BC | CR | Metformin 850 mg/12 h | There is no difference in progression-free survival nor overall response. | [99] |
| Phase 2, randomized | NSCLC | CR | Metformin 1000 mg/12 h | Overall, there was no difference in risk of progression or death compared to control. In high fluorodeoxyglucose-uptake cancers, the addition of metformin to CT decreased the risk of progression and death. | [105] |
BC: breast cancer, BTC: biliary tract cancer, CC: Cervix Cancer, CRC: colorectal cancer, CRT: chemoradiotherapy, CT: chemotherapy, DLBCL: Diffuse Large B Cell Lymphoma, EC: endometrial cancer, FMD: fast-mimicking diet, FTC: follicular thyroid cancer, GC: Gastric Cancer, GSH: Glutathione, IGF: insulin-like growth factor, IGFBP-3: IGF Binding Protein 3, MP: Miller–Payne, NSCLC: non-small cell lung cancer, OC: ovarian cancer, OS: overall survival, PaCa: Pancreatic Cancer, PC: prostate cancer, pCR: pathological complete response, PR: partial response, SOC: standard of care therapy, Sx: surgery, TNBC: triple-negative breast cancer.
5.1.2. Rapamycin and Its Analogs
Rapamycin, also known as Sirolimus, is the most studied CRM. It is a macrolide compound used as an immunosuppressant in solid organ transplants. The main function of rapamycin is the inhibition of the mTOR complex through its binding to the FK-binding protein 12 (FKBP12) and subsequently binding to mTOR Complex 1 (mTORC1), inhibiting its nutritional and proliferative effects [106]. Due to adverse effects such as edema, nausea, hypertriglyceridemia, anemia, and diabetes-like symptoms, the use of rapamycin is somewhat limited. Nevertheless, Temsirolimus and Everolimus, both rapamycin analogs (rapalogs), tend to have fewer adverse effects compared to rapamycin.
In clinical settings, Everolimus is FDA-approved as a complementary therapy for treating postmenopausal women with ER+HER- locally advanced or secondary BC (Table 3). The addition of rapalogs to aromatase inhibitors therapy, or their use as monotherapy, has resulted in varying increases in PFS, with reported durations of 22 months [107], 8.4 months [108], and 6.4 months [109]. The significant difference in PFS between studies must be carefully analyzed, as the discrepancy may be attributed to variations in the patient’s clinical history. The Everolimus dosage and experimental design could explain these variations and guide the effective use of rapalogs.
In follicular thyroid cancer, the addition of Everolimus led to better disease management and fewer adverse reactions compared to other treatments, such as Tyrosine Kinase Inhibitors (TKI). Notwithstanding, this was not observed in anaplastic thyroid cancer patients, highlighting the variability in cancer subtypes and their response to treatment [110]. In advanced biliary tract cancer (ABTC) patients, first-line therapy with Everolimus showed a median PFS of up to 5.5 months compared to the historical SOC platinum-gemcitabine of 8 months. This suggests the potential feasibility of adding mTOR inhibitors to increase PFS and OS [111].
In metastatic colorectal cancer, the addition of rapalogs to CT and Bevacizumab modestly increased PFS by 20% up to 6 months. Interestingly, the best response was observed in patients with mutations in PTEN and PIK3CA proteins [112]. Similarly, in HNSCC, a beneficial effect in a subgroup of PTEN mutation was observed after adding rapalogs [113], making plausible the selection of patients who might show the best response to mTOR inhibitors.
Nonetheless, clinical trials in patients with triple-negative breast cancer (TNBC) [114], HER2-BC [115,116], and prostate cancer [117,118] have not demonstrated additional benefits from incorporating Everolimus into standard therapy. This underscores how specific groups of patients with particular tumor characteristics may benefit from using rapalogs as a complementary regimen.
Table 3.
Rapamycin and rapalogs and their application in anti-cancer therapies.
Table 3.
Rapamycin and rapalogs and their application in anti-cancer therapies.
| Study Design | Type(s) of Cancer | SOC | CRM | Outcomes | REF. |
|---|---|---|---|---|---|
| Phase 2, non-randomized | FTC | None | Everolimus 10 mg/d | Increased stable disease and overall survival compared to other interventions with fewer side effects. | [110] |
| Phase 2, single arm | BTC | None | Everolimus 10 mg/d | Median progression-free survival of 5.5 months and median OS of 9.5 months | [111] |
| Phase 2 | CRC | CT+ biologic | Everolimus | The addition of Everolimus to standard therapy increased progression-free survival by 20% at 6 months. The subgroup of patients with PTEN mutations might show a better response. | [112] |
| Phase 2, single arm | BC | Aromatase inhibitor | Everolimus 10 mg/d | Everolimus and aromatase inhibitor (Exemestane) bitherapy increased progression-free survival by up to 6 months compared to aromatase inhibitor monotherapy. | [109] |
| Phase 2, randomized, double-blind, placebo-controlled | TNBC | CT | Everolimus 5 mg/d | Neoadjuvant addition of Everolimus to CT did not increase pathological complete response compared to placebo and added toxicity. | [114] |
| Phase 2, randomized | PC | Sx | Everolimus 5 mg/d vs. 10 mg/d | No difference in mTOR markers, nor PSA pre- and post-operative with the intervention. | [117] |
| Phase 2, randomized | HER2-BC | CT | Everolimus 5 mg/d | No difference in progression-free survival and overall survival compared to monotherapy alone. | [115] |
| Phase 2, non-randomized | FTC | None | Everolimus 10 mg/d | Increased stable disease and overall survival compared to other interventions with fewer side effects. | [110] |
BC: breast cancer, BTC: biliary tract cancer, CC: Cervix Cancer, CRC: colorectal cancer, CRT: chemoradiotherapy, CT: chemotherapy, DLBCL: Diffuse Large B Cell Lymphoma, EC: endometrial cancer, FMD: fast-mimicking diet, FTC: follicular thyroid cancer, GC: Gastric Cancer, GSH: Glutathione, IGF: insulin-like growth factor, IGFBP-3: IGF Binding Protein 3, MP: Miller–Payne, NSCLC: non-small cell lung cancer, OC: ovarian cancer, OS: overall survival, PaCa: Pancreatic Cancer, PC: prostate cancer, pCR: pathological complete response, PR: partial response, SOC: standard of care therapy, Sx: surgery, TNBC: triple-negative breast cancer.
5.1.3. Aspirin
Aspirin is a nonsteroidal anti-inflammatory drug (NSAID) whose main effect is the inhibition of cyclooxygenases (COX), suppressing the production of prostaglandins and thromboxanes, thereby dampening the inflammatory response. Besides its anti-inflammatory effects, aspirin and its salicylate derivatives are increasingly being considered as CRMs [119]. Further experiments have shown that aspirin inhibits the dephosphorylation of AMPK, maintaining high levels of this molecule and dampening the activation of anabolic pathways such as PI3K-AKT-mTOR [120].
The anti-carcinogenic effects of aspirin in the development of CRC have already been well-documented and are related to its COX inhibitor properties (Table 4). As such, aspirin has been associated with reduced CRC risk in Lynch syndrome patients [121] but not in non-colorectal cancers [122]. A retrospective cohort study also showed that aspirin reduces CRC risk in healthy patients when used before the age of 70 and for a duration of at least 5 years [123]. In a pilot study of healthy patients who consumed aspirin, no difference in the expression and catabolism of prostaglandins was observed, but there was a reduction in the expression of pS6 protein, the main target of the mTOR pathway, implicating that the protective effect of aspirin could be related to its CRM properties [124].
In non-CRC cancers, aspirin has shown various effects worth mentioning. In PC retrospective studies, aspirin use has been associated with a decrease in prostatic-specific antigen (PSA) levels in serum and a possible reduction in PC development risk [125]. A randomized follow-up study found that pre-diagnostic aspirin use reduced the risk of lethal PC, and post-diagnostic use improved survival [126].
Meanwhile, in other hormone-dependent cancers, aspirin has not shown relevant results. In BC, there was no apparent effect in decreasing cancer risk or improving survival [127,128]. In ovarian cancer, no association was found between aspirin use and reduced cancer risk, even though a trend toward a protective effect was found with low-dose aspirin [129].
Table 4.
Aspirin and resveratrol and their application in anti-cancer therapies.
Table 4.
Aspirin and resveratrol and their application in anti-cancer therapies.
| Study Design | Type(s) of Cancer | SOC | CRM | Outcomes | REF. |
|---|---|---|---|---|---|
| Multicenter, double-blind, placebo-controlled | None | None | Aspirin and/or NSAIDs | The use of aspirin and NSAIDs reduces serum levels of prostate-specific antigen. Aspirin consumption reduced the risk of prostate cancer development. | [125] |
| Multicenter, double-blind, randomized, placebo-controlled | None | None | Aspirin 600 mg/d | Obesity-related risk of developing colorectal cancer is reduced after aspirin consumption. | [130] |
| Randomized, placebo-controlled | None | None | Aspirin 235 mg | Pre-diagnostic use of aspirin lowered the risk of lethal prostate cancer. Post-diagnostic use improved survival compared to placebo. | [126] |
| Pilot study | None | None | Resveratrol 500 mg | Doses up to 2.5 g of resveratrol were well tolerated in healthy patients and were able to reduce IGF-1 and IGFBP-3 levels. | [131] |
| Phase 1, non-randomized | CRC | Sx | Resveratrol 0.5 or 1.0 g | Use of resveratrol reduced cell proliferation. | [132] |
| Phase 1, randomized, double-blind, placebo-controlled | CRC | None | Resveratrol 5 g (SRT501) | The micronized formulation of resveratrol increases apoptosis rate in CRC. No difference in IGF-1, Ki67, AKT, GSK, ERK, JNK, or beta-catenin. | [133] |
BC: breast cancer, BTC: biliary tract cancer, CC: Cervix Cancer, CRC: colorectal cancer, CRT: chemoradiotherapy, CT: chemotherapy, DLBCL: Diffuse Large B Cell Lymphoma, EC: endometrial cancer, FMD: fast-mimicking diet, FTC: follicular thyroid cancer, GC: Gastric Cancer, GSH: Glutathione, IGF: insulin-like growth factor, IGFBP-3: IGF Binding Protein 3, MP: Miller–Payne, NSCLC: non-small cell lung cancer, OC: ovarian cancer, OS: overall survival, PaCa: Pancreatic Cancer, PC: prostate cancer, pCR: pathological complete response, PR: partial response, SOC: standard of care therapy, Sx: surgery, TNBC: triple-negative breast cancer.
5.1.4. Resveratrol
Among the natural products related to anti-cancer effects, resveratrol is one of the most studied and the only one considered a CRM. Resveratrol is a stilbene belonging to the polyphenols group, found mostly in grape skin, berries, and in small amounts in red wine and other foods. Its primary activity is as an antioxidant, which can be direct or indirect, scavenging ROS and through the modulation of cellular antioxidant pathways [134,135]. In the anti-cancer effects, resveratrol has shown properties both in vitro and in vivo to inhibit the carcinogenesis stages [136] and as a chemotherapeutic agent [137,138] potentially linked to its pro-apoptotic and anti-proliferative actions.
Even though its promising potential is shown in pre-clinical studies, resveratrol has only been tested in clinical studies for CRC (Table 4). In a phase 1 study of stage 1 CRC, the consumption of 0.5 g or 1 g of resveratrol daily for eight days significantly reduced Ki-67-positive cancer cells [131], illustrating the potential neoadjuvant effect of this CRM. In another phase 1 clinical trial of metastatic CRC, the consumption of 5 g of resveratrol daily increased activated caspase-3 activity by up to 40% without affecting the mTOR and WNT/b-catenin pathways [133].
In healthy volunteers, the consumption of resveratrol reduced IGF1 and IGFBP-3 levels [131], but this effect was not observed in oncologic patients, indicating the need for more trials to characterize the effect of resveratrol on the GH/IGF-1 axis [133].
The principal challenge in using natural products like resveratrol is its poor solubility in water and the low bioavailability after intestinal and liver metabolism, reducing the plasmatic and intra-tumoral levels and, thereby, lowering its efficacy. Luckily, recently, advances have been made in the formulation of delivery systems based on nanoparticles that can enhance the target delivery and bioavailability of resveratrol and its derivates [139].
Once an efficient delivery system for resveratrol is established, the dosage must be determined that provides the best effect with minimal side effects, considering its high intestinal and liver metabolism [140,141] and the potential interactions with other SOC therapies or complementary drugs. More extensive clinical trials are needed to determine the effects of resveratrol in a clinical context. Additionally, more comprehensive pre-clinical and clinical studies on metabolites of resveratrol could address the main challenges of delivery and bioavailability of its parental compound [141,142].
6. Discussion
Despite the most recent advances in surgery, chemotherapy, radiotherapy, and biological targeting, cancer remains one of the leading causes of morbidity and mortality. This underscores the necessity for research into complementary therapies that are able to increase disease-free survival, improve quality of life, and potentially lead to complete healing.
Among the most promising complementary therapies, caloric restriction has shown beneficial results in pre-clinical models, including increased lifespan in both non-mammal and mammal models, reduced risk of metabolic diseases, delayed disease progression, and significant anti-carcinogenic and anti-tumoral effects. However, translating these promising preclinical findings to clinical settings has proven challenging.
Firstly, the controlled environment and syngeneic background used in pre-clinical studies reduce the heterogeneity seen in clinical trials. Unlike clinical studies, where patients can be affected by multiple health issues simultaneously, the controlled conditions in preclinical models do not account for the complex interplay of variabilities present in human subjects, potentially rendering CR interventions less effective.
Additionally, the efficacy of CR depends on maintaining a specific caloric intake and fasting duration, which may be challenging for oncologic patients [13,72,74]. The use of SOC therapy, prescribed or non-prescribed drugs, must also be considered when evaluating the potential benefits of a CR intervention. Even though the ketogenic diet may be the most famous and commonly applied CR intervention, its efficacy in an oncologic context remains questionable. This is due to the difficulty in adapting to ketogenic foods, potential early side effects of carbohydrate restriction, and the challenge of achieving and maintaining ketone blood levels that complicate its application [73,74,84]. Hence, all previous factors may interfere with the outcomes, which could explain the lack of conclusive clinical evidence regarding the addition of KD in oncologic patients.
Given these complexities, it is crucial to interpret these results with caution. Most studies reviewed here are phase I/II trials with very limited sample sizes and often involve highly specific cancer types, which may result in heterogeneous responses when applied to other cancer types and comorbidities. Nonetheless, CR interventions might effectively reduce side effects and potentially enhance anti-tumor responses.
To overcome the challenges of implementing a full CR diet in non-compliant patients, whether due to intolerance or difficulty maintaining fasting periods, the use of CRMs might be the most promising intervention alternative. Preclinical models have demonstrated that CRM can replicate most of the molecular and metabolic effects of CR without the need for a restrictive diet. Among CRM, metformin and aspirin are the most well-known, widely used, and have minimal side effects. They are also inexpensive and over-the-counter drugs [90,123,125,143].
After carefully reviewing the current knowledge, the use of CRMs might be recommended when a particular pathway, such as PI3K/AKT/mTOR or IGF1/IGFBP, is upregulated in the tumor and can be modulated by the intervention. However, more evidence is necessary to determine the most appropriate scenarios for the correct CRM.
As a preventive measure, aspirin is the only CRM with sufficient evidence as a chemopreventive agent in patients with an average cancer risk, particularly for CRC. This may be related to its COX-inhibition effects or its role as a CRM. More studies are needed to elucidate its principal mechanism and potential benefits against other cancer types and to determine the optimal timing for its use.
Metformin also shows potential as a chemopreventive agent. Retrospective studies have shown a reduced cancer risk in patients with Type 2 Diabetes Mellitus who use metformin or combine it with other antidiabetic drugs [92,103,143]. Nonetheless, more studies are required to confirm these findings in both diabetic and healthy populations. Furthermore, there is a need to standardize metformin dosage in oncologic studies, as variability in dosing complicates comparisons between trials and expected outcomes.
7. Conclusions
In conclusion, caloric restriction interventions and the use of caloric restriction mimetics may serve as beneficial complementary therapies in oncology. Their potential to modulate tumor metabolism, growth, and immune response could enhance the efficacy of standard-of-care treatments. Additionally, these complementary therapies’ protective effects on healthy cells may render them more resistant to cytotoxic therapies, potentially decreasing the side effects of CRT. Thus, integrating these approaches into standard oncologic treatment regimens could provide significant benefits to patients.
Author Contributions
U.E.D.-L.-C., writing—original draft, editing, and conceptualization; J.J.P.-T. and S.A.V.-C., writing—review and editing; R.M.-d.-O.-L., supervision; M.d.J.L.-A., A.G.-G. and O.S.-C., conceptualization; A.G.M.-P. and C.R.M.-d.-O.-S., feedback on the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Council for Humanities Science and Technology (CONAHCYT) grant No. CF-2023-I-1251.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
For the National Council for Humanities, Science and Technology (Consejo Nacional de Humanidades, Ciencia y Tecnología, CONAHCYT: CF-2023-I-1251). U.E.D-C received a scholarship (No. CVU: 1077619) from CONAHCYT. All figures were created with Biorender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
ABTC: advanced biliary tract cancer, AMPK: AMP-activated protein kinase, BC: breast cancer, CT: chemotherapy, CR: caloric restriction, CRM: caloric restriction mimetic, CRC: colorectal cancer, GH: growth hormone, GR: glucose restriction, HNSS: Head and Neck Squamous Cancer, IF: intermittent gasting, IGF1: insulin-like growth factor 1, LCI: low caloric intake, mTOR: Mammalian Target of Rapamycin, NSCLC: non-small cell lung cancer, NRF2: nuclear factor erythroid 2-related factor 2, NSAID: nonsteroidal anti-inflammatory drug, OS: overall survival, PDCA: pancreatic ductal adenocarcinoma, PR: partial response, PDE3B: phosphodiesterase 3, PI3KS: phosphoinositide 3-kinase, PFS: progression-free survival, PC: prostate cancer, PSA: prostatic-specific antigen, RT: radiotherapy, rapalogs: rapamycin analogs, ROS: reactive oxygen species, STS: short-term starvation, SIRT: sirtuins, TNBC: triple-negative breast cancer, TIL: tumor-infiltrating lymphocyte.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S.H.; Guo, H. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front. Oncol. 2020, 10, 523226. [Google Scholar] [CrossRef]
- Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating Glycolysis to Improve Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2606. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Ravussin, E. Caloric Restriction in Humans: Impact on Physiological, Psychological, and Behavioral Outcomes. Antioxid. Redox Signal. 2011, 14, 275–287. [Google Scholar] [CrossRef]
- Gillespie, Z.E.; Pickering, J.; Eskiw, C.H. Better Living through Chemistry: Caloric Restriction (CR) and CR Mimetics Alter Genome Function to Promote Increased Health and Lifespan. Front. Genet. 2016, 7, 142. [Google Scholar] [CrossRef]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef]
- Paoli, A.; Bianco, A.; Moro, T.; Mota, J.F.; Coelho-Ravagnani, C.F. The Effects of Ketogenic Diet on Insulin Sensitivity and Weight Loss, Which Came First: The Chicken or the Egg? Nutrients 2023, 15, 3120. [Google Scholar] [CrossRef]
- Pascual, P.E.; Rolands, M.R.; Eldridge, A.L.; Kassis, A.; Mainardi, F.; Lê, K.; Karagounis, L.G.; Gut, P.; Varady, K.A. A meta-analysis comparing the effectiveness of alternate day fasting, the 5:2 diet, and time-restricted eating for weight loss. Obesity 2023, 31 (Suppl. S1), 9–21. [Google Scholar] [CrossRef] [PubMed]
- Zauner, C.; Schneeweiss, B.; Kranz, A.; Madl, C.; Ratheiser, K.; Kramer, L.; Roth, E.; Schneider, B.; Lenz, K. Resting energy expenditure in short-term starvation is increased as a result of an increase in serum norepinephrine. Am. J. Clin. Nutr. 2000, 71, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Dorff, T.B.; Groshen, S.; Garcia, A.; Shah, M.; Tsao-Wei, D.; Pham, H.; Cheng, C.-W.; Brandhorst, S.; Cohen, P.; Wei, M.; et al. Safety and feasibility of fasting in combination with platinum-based chemotherapy. BMC Cancer 2016, 16, 360. [Google Scholar] [CrossRef] [PubMed]
- Valdemarin, F.; Caffa, I.; Persia, A.; Cremonini, A.L.; Ferrando, L.; Tagliafico, L.; Tagliafico, A.; Guijarro, A.; Carbone, F.; Ministrini, S.; et al. Safety and feasibility of fasting-mimicking diet and effects on nutritional status and circulating metabolic and inflammatory factors in cancer patients undergoing active treatment. Cancers 2021, 13, 4013. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.L.; Plymate, S.; D’Oria-Cameron, A.; Bain, C.; Haugk, K.; Xiao, L.; Lin, D.W.; Stanford, J.L.; McTiernan, A. A study of caloric restriction versus standard diet in overweight men with newly diagnosed prostate cancer: A randomized controlled trial. Prostate 2013, 73, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Garza-Lombó, C.; Schroder, A.; Reyes-Reyes, E.M.; Franco, R. mTOR/AMPK signaling in the brain: Cell metabolism, proteostasis and survival. Curr. Opin. Toxicol. 2018, 8, 102–110. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, F. Coordination of the AMPK, Akt, mTOR, and p53 Pathways under Glucose Starvation. Int. J. Mol. Sci. 2022, 23, 14945. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, R.-Y.; Song, J.; Guan, Y.-F.; Xu, T.-Y.; Du, H.; Viollet, B.; Miao, C.-Y. Loss of AMP-activated protein kinase-α2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes 2012, 61, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. Targeting sirtuin 1 to improve metabolism: All you need is NAD+? Pharmacol. Rev. 2012, 64, 166–187. [Google Scholar] [CrossRef]
- Chen, Y.R.; Fang, S.R.; Fu, Y.C.; Zhou, X.H.; Xu, M.Y.; Xu, W.C. Calorie restriction on insulin resistance and expression of SIRT1 and SIRT4 in rats. Biochem. Cell Biol. 2010, 88, 715–722. [Google Scholar]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Lu, C.; Zhao, H.; Liu, Y.; Yang, Z.; Yao, H.; Liu, T.; Gou, T.; Wang, L.; Zhang, J.; Tian, Y.; et al. Novel Role of the SIRT1 in Endocrine and Metabolic Diseases. Int. J. Biol. Sci. 2023, 19, 484–501. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Guo, Z.; Feng, R.; Wu, N.; Zhong, X.; Fang, Z.; Hu, Y.; Yu, X.; Zhao, S.; Zhao, G.; et al. Multi-omics analysis reveals the regulation of SIRT6 on protein processing of endoplasmic reticulum to alleviate oxidative stress in endothelial cells. Clin. Transl. Med. 2022, 12, e1039. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yu, X.; Zhao, S.; Zhong, X.; Huang, D.; Feng, R.; Li, P.; Fang, Z.; Hu, Y.; Zhang, Z.; et al. SIRT6 deficiency in endothelial cells exacerbates oxidative stress by enhancing HIF1α accumulation and H3K9 acetylation at the Ero1α promoter. Clin. Transl. Med. 2023, 13, e1377. [Google Scholar] [CrossRef]
- de Oliveira, M.V.M.; Andrade, J.M.O.; Paraíso, A.F.; Santos, S.H.S. Sirtuins and cancer: New insights and cell signaling. Cancer Investig. 2013, 31, 645–653. [Google Scholar] [CrossRef]
- Li, J.; Zhan, H.; Ren, Y.; Feng, M.; Wang, Q.; Jiao, Q.; Wang, Y.; Liu, X.; Zhang, S.; Du, L.; et al. Sirtuin 4 activates autophagy and inhibits tumorigenesis by upregulating the p53 signaling pathway. Cell Death Differ. 2023, 30, 313–326. [Google Scholar] [CrossRef]
- Cai, G.; Ge, Z.; Xu, Y.; Cai, L.; Sun, P.; Huang, G. SIRT4 functions as a tumor suppressor during prostate cancer by inducing apoptosis and inhibiting glutamine metabolism. Sci. Rep. 2022, 12, 12208. [Google Scholar] [CrossRef]
- Huffman, D.M.; Grizzle, W.E.; Bamman, M.M.; Kim, J.-S.; Eltoum, I.A.; Elgavish, A.; Nagy, T.R. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007, 67, 6612–6618. [Google Scholar] [CrossRef]
- Peck, B.; Chen, C.Y.; Ho, K.K.; Di Fruscia, P.; Myatt, S.S.; Coombes, R.C.; Fuchter, M.J.; Hsiao, C.D.; Lam, E.W.F. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 2010, 9, 844–855. [Google Scholar] [CrossRef]
- Herranz, D.; Muñoz-Martin, M.; Cañamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Zheng, Y.; Kim, H.-S.; Xu, X.; Cao, L.; Lahusen, T.; Lee, M.-H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-Associated Tumorigenesis. Mol. Cell 2008, 32, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol. Metab. 2009, 20, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, D.A.; McCabe, M.T.; Arnold, R.S.; Day, M.L. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 2008, 27, 4353–4362. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Huang, M.-T.; Shen, G.; Yuan, X.; Lin, W.; Khor, T.O.; Conney, A.H.; Kong, A.-N.T. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2–related factor 2. Cancer Res. 2006, 66, 8293–8296. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.-K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef]
- Shibata, T.; Saito, S.; Kokubu, A.; Suzuki, T.; Yamamoto, M.; Hirohashi, S. Global downstream pathway analysis reveals a dependence of oncogenic NF-E2–related factor 2 mutation on the mTOR growth signaling pathway. Cancer Res. 2010, 70, 9095–9105. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.-X.; Singh, G. Expression of xCT and activity of system xc− are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Kim, S.; Kim, E.-J.; Park, J.-H.; Yang, S.-H.; Jeong, E.-T.; Park, C.; Youn, M.-J.; So, H.-S.; Park, R. Suppression of Nrf2-driven heme oxygenase-1 enhances the chemosensitivity of lung cancer A549 cells toward cisplatin. Lung Cancer 2008, 60, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Martín-Montalvo, A.; Villalba, J.M.; Navas, P.; de Cabo, R. NRF2, cancer and calorie restriction. Oncogene 2011, 30, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.J.; Lewis, K.N.; Price, N.L.; Chang, J.W.; Perez, E.; Cascajo, M.V.; Tamashiro, K.L.; Poosala, S.; Csiszar, A.; Ungvari, Z.; et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc. Natl. Acad. Sci. USA 2008, 105, 2325–2330. [Google Scholar] [CrossRef]
- Blum, W.F.; Alherbish, A.; Alsagheir, A.; El Awwa, A.; Kaplan, W.; Koledova, E.; O Savage, M. The growth hormone–insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocr. Connect. 2018, 7, R212–R222. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Laron, Z. Role of the GH-IGF1 system in progression of cancer. Mol. Cell. Endocrinol. 2020, 518, 111003. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; You, M.-L.; Chong, Q.-Y.; Pandey, V.; Zhuang, Q.-S.; Liu, D.-X.; Ma, L.; Zhu, T.; Lobie, P.E. Autocrine Human Growth Hormone Promotes Invasive and Cancer Stem Cell-Like Behavior of Hepatocellular Carcinoma Cells by STAT3 Dependent Inhibition of CLAUDIN-1 Expression. Int. J. Mol. Sci. 2017, 18, 1274. [Google Scholar] [CrossRef] [PubMed]
- de Ostrovich, K.K.; Lambertz, I.; Colby, J.K.; Tian, J.; Rundhaug, J.E.; Johnston, D.; Conti, C.J.; DiGiovanni, J.; Fuchs-Young, R. Paracrine Overexpression of Insulin-Like Growth Factor-1 Enhances Mammary Tumorigenesis in Vivo. Am. J. Pathol. 2008, 173, 824–834. [Google Scholar] [CrossRef]
- Brunet-Dunand, S.E.; Vouyovitch, C.; Araneda, S.; Pandey, V.; Vidal, L.J.-P.; Print, C.; Mertani, H.C.; Lobie, P.E.; Perry, J.K. Autocrine Human Growth Hormone Promotes Tumor Angiogenesis in Mammary Carcinoma. Endocrinology 2009, 150, 1341–1352. [Google Scholar] [CrossRef]
- Basu, R.; Baumgaertel, N.; Wu, S.; Kopchick, J.J. Growth Hormone Receptor Knockdown Sensitizes Human Melanoma Cells to Chemotherapy by Attenuating Expression of ABC Drug Efflux Pumps. Horm. Cancer 2017, 8, 143–156. [Google Scholar] [CrossRef]
- Basu, R.; Kopchick, J.J. GH and IGF1 in cancer therapy resistance. Endocr.-Relat. Cancer 2023, 30, e220414. [Google Scholar] [CrossRef]
- Patel, S.A.; Chaudhari, A.; Gupta, R.; Velingkaar, N.; Kondratov, R.V. Circadian clocks govern calorie restriction-mediated life span extension through BMAL1- and IGF-1-dependent mechanisms. FASEB J. 2016, 30, 1634–1642. [Google Scholar] [CrossRef]
- Hołowko, J.; Michalczyk, M.M.; Zając, A.; Czerwińska-Rogowska, M.; Ryterska, K.; Banaszczak, M.; Jakubczyk, K.; Stachowska, E. Six Weeks of Calorie Restriction Improves Body Composition and Lipid Profile in Obese and Overweight Former Athletes. Nutrients 2019, 11, 1461. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Villareal, D.T.; Das, S.K.; Smith, S.R.; Meydani, S.N.; Pittas, A.G.; Klein, S.; Bhapkar, M.; Rochon, J.; Ravussin, E.; et al. Effects of 2-year calorie restriction on circulating levels of IGF-1, IGF-binding proteins and cortisol in nonobese men and women: A randomized clinical trial. Aging Cell 2016, 15, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Menendez, P.; Hevia, D.; Alonso-Arias, R.; Álvarez-Artime, A.; Rodriguez-Garcia, A.; Kinet, S.; Gonzalez-Pola, I.; Taylor, N.; Mayo, J.C.; Sainz, R.M. GLUT1 protects prostate cancer cells from glucose deprivation-induced oxidative stress. Redox Biol. 2018, 17, 112–127. [Google Scholar] [CrossRef]
- Raut, G.K.; Chakrabarti, M.; Pamarthy, D.; Bhadra, M.P. Glucose starvation-induced oxidative stress causes mitochondrial dysfunction and apoptosis via Prohibitin 1 upregulation in human breast cancer cells. Free Radic. Biol. Med. 2019, 145, 428–441. [Google Scholar] [CrossRef]
- Han, J.; Zhang, L.; Guo, H.; Wysham, W.Z.; Roque, D.R.; Willson, A.K.; Sheng, X.; Zhou, C.; Bae-Jump, V.L. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol. Oncol. 2015, 138, 668–675. [Google Scholar] [CrossRef]
- Ferretti, A.C.; Hidalgo, F.; Tonucci, F.M.; Almada, E.; Pariani, A.; Larocca, M.C.; Favre, C. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: Targeting AMPK activation to control migration. Sci. Rep. 2019, 9, 2815. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Tollefsbol, T.O. Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression. FASEB J. 2010, 24, 1442–1453. [Google Scholar] [CrossRef]
- Wardi, L.; Alaaeddine, N.; Raad, I.; Sarkis, R.; Serhal, R.; Khalil, C.; Hilal, G. Glucose restriction decreases telomerase activity and enhances its inhibitor response on breast cancer cells: Possible extra-telomerase role of BIBR 1532. Cancer Cell Int. 2014, 14, 60. [Google Scholar] [CrossRef]
- Peng, Y.; Xing, S.-N.; Tang, H.-Y.; Wang, C.-D.; Yi, F.-P.; Liu, G.-L.; Wu, X.-M. Influence of glucose transporter 1 activity inhibition on neuroblastoma in vitro. Gene 2019, 689, 11–17. [Google Scholar] [CrossRef]
- Lee, H.Y.; Itahana, Y.; Schuechner, S.; Fukuda, M.; Je, H.S.; Ogris, E.; Virshup, D.M.; Itahana, K. Ca2+-dependent demethylation of phosphatase PP2Ac promotes glucose deprivation–induced cell death independently of inhibiting glycolysis. Sci. Signal. 2018, 11, 7893. [Google Scholar] [CrossRef] [PubMed]
- Schroll, M.M.; Ludwig, K.R.; LaBonia, G.J.; Herring, E.L.; Hummon, A.B. Combined Short-Term Glucose Starvation and Chemotherapy in 3D Colorectal Cancer Cell Culture Decreases 14-3-3 Family Protein Expression and Phenotypic Response to Therapy. J. Am. Soc. Mass Spectrom. 2018, 29, 2012–2022. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Huang, C.-Y.; Pai, Y.-C.; Lin, B.-R.; Lee, T.-C.; Liang, P.-H.; Yu, L.C.-H. Glucose Metabolites Exert Opposing Roles in Tumor Chemoresistance. Front. Oncol. 2019, 9, 1282. [Google Scholar] [CrossRef]
- Pomatto-Watson, L.C.D.; Bodogai, M.; Bosompra, O.; Kato, J.; Wong, S.; Carpenter, M.; Duregon, E.; Chowdhury, D.; Krishna, P.; Ng, S.; et al. Daily caloric restriction limits tumor growth more effectively than caloric cycling regardless of dietary composition. Nat. Commun. 2021, 12, 12. [Google Scholar] [CrossRef]
- Lien, E.C.; Westermark, A.M.; Zhang, Y.; Yuan, C.; Li, Z.; Lau, A.N.; Sapp, K.M.; Wolpin, B.M.; Heiden, M.G.V. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 2021, 599, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Manukian, G.; Kivolowitz, C.; DeAngelis, T.; Shastri, A.A.; Savage, J.E.; Camphausen, K.; Rodeck, U.; Zarif, J.C.; Simone, N.L. Caloric Restriction Impairs Regulatory T cells within the Tumor Microenvironment After Radiation and Primes Effector T cells. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 1341–1349. [Google Scholar] [CrossRef]
- Pomatto-Watson, L.C.D.; Bodogai, M.; Carpenter, M.; Chowdhury, D.; Krishna, P.; Ng, S.; Bosompra, O.; Kato, J.; Wong, S.; Reyes-Sepulveda, C.; et al. Replenishment of myeloid-derived suppressor cells (MDSCs) overrides CR-mediated protection against tumor growth in a murine model of triple-negative breast cancer. GeroScience 2022, 44, 2471–2490. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Parris, A.B.; Howard, E.W.; Shi, Y.; Yang, S.; Jiang, Y.; Kong, L.; Yang, X. Caloric restriction inhibits mammary tumorigenesis in MMTV-ErbB2 transgenic mice through the suppression of ER and ErbB2 pathways and inhibition of epithelial cell stemness in premalignant mammary tissues. Carcinogenesis 2018, 39, 1264–1273. [Google Scholar] [CrossRef]
- Mao, Y.-Q.; Huang, J.-T.; Zhang, S.-L.; Li, Z.-M.; Jing, H.; Chen, H.-L.; Kong, C.-Y.; Huang, S.-H.; Cai, P.-R.; Han, B.; et al. The antitumour effects of caloric restriction are mediated by the gut microbiome. Nat. Metab. 2023, 5, 96–110. [Google Scholar] [CrossRef]
- Simone, B.A.; Palagani, A.; Strickland, K.; Ko, K.; Jin, L.; Lim, M.K.; Dan, T.D.; Sarich, M.; Monti, D.A.; Cristofanilli, M.; et al. Caloric restriction counteracts chemotherapy-induced inflammation and increases response to therapy in a triple negative breast cancer model. Cell Cycle 2018, 17, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-C.; Huang, T.-C.; Tien, F.-M.; Lin, J.-M.; Yeh, Y.-C.; Lee, C.-Y. Safety, Feasibility, and Effects of Short-Term Calorie Reduction during Induction Chemotherapy in Patients with Diffuse Large B-Cell Lymphoma: A Pilot Study. Nutrients 2021, 13, 3268. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.C.; Anderson, C.M.; Rodman, S.N.; Buranasudja, V.; McCormick, M.L.; Davis, A.; Loth, E.; Bodeker, K.L.; Ahmann, L.; Parkhurst, J.R.; et al. Ketogenic Diet with Concurrent Chemoradiation in Head and Neck Squamous Cell Carcinoma: Preclinical and Phase 1 Trial Results. Radiat. Res. 2021, 196, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Zahra, A.; Fath, M.A.; Opat, E.; Mapuskar, K.A.; Bhatia, S.K.; Ma, D.C.; Rodman, S.N., III; Snyders, T.P.; Chenard, C.A.; Eichenberger-Gilmore, J.M.; et al. Consuming a Ketogenic Diet while Receiving Radiation and Chemotherapy for Locally Advanced Lung Cancer and Pancreatic Cancer: The University of Iowa Experience of Two Phase 1 Clinical Trials. Radiat. Res. 2017, 187, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Firsanov, D.; Spencer, B.; Seluanov, A.; Gorbunova, V. Short-term calorie restriction enhances DNA repair by non-homologous end joining in mice. npj Aging Mech. Dis. 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef] [PubMed]
- de Groot, S.; Lugtenberg, R.T.; Cohen, D.; Welters, M.J.; Ehsan, I.; Vreeswijk, M.P.; Smit, V.T.; de Graaf, H.; Heijns, J.B.; Portielje, J.E.; et al. Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun. 2020, 11, 3083. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Dozmorov, M.; Oh, Y. IGFBP-3/IGFBP-3 Receptor System as an Anti-Tumor and Anti-Metastatic Signaling in Cancer. Cells 2020, 9, 1261. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Pfetzer, N.; Schwab, M.; Strauss, I.; Kämmerer, U. Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: A pilot trial. Nutr. Metab. 2011, 8, 54. [Google Scholar] [CrossRef]
- Vernieri, C.; Fucà, G.; Ligorio, F.; Huber, V.; Vingiani, A.; Iannelli, F.; Raimondi, A.; Rinchai, D.; Frigè, G.; Belfiore, A.; et al. Fasting-Mimicking Diet Is Safe and Reshapes Metabolism and Antitumor Immunity in Patients with Cancer. Cancer Discov. 2022, 12, 90–107. [Google Scholar] [CrossRef]
- Khodabakhshi, A.; Akbari, M.E.; Mirzaei, H.R.; Seyfried, T.N.; Kalamian, M.; Davoodi, S.H. Effects of Ketogenic metabolic therapy on patients with breast cancer: A randomized controlled clinical trial. Clin. Nutr. 2021, 40, 751–758. [Google Scholar] [CrossRef]
- Sheikhpour, E.; Noorbakhsh, P.; Foroughi, E.; Farahnak, S.; Nasiri, R.; Neamatzadeh, H. A Survey on the Role of Interleukin-10 in Breast Cancer: A Narrative. Rep. Biochem. Mol. Biol. 2018, 7, 30–37. [Google Scholar]
- Bauersfeld, S.P.; Kessler, C.S.; Wischnewsky, M.; Jaensch, A.; Steckhan, N.; Stange, R.; Kunz, B.; Brückner, B.; Sehouli, J.; Michalsen, A. The effects of short-term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: A randomized cross-over pilot study. BMC Cancer 2018, 18, 476. [Google Scholar] [CrossRef] [PubMed]
- Khodabakhshi, A.; Seyfried, T.N.; Kalamian, M.; Beheshti, M.; Davoodi, S.H. Does a ketogenic diet have beneficial effects on quality of life, physical activity or biomarkers in patients with breast cancer: A randomized controlled clinical trial. Nutr. J. 2020, 19, 87. [Google Scholar] [CrossRef]
- Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; Loera-Arias, M.J.; Montes-De-Oca-Luna, R.; Rodriguez-Rocha, H.; Garcia-Garcia, A. Metformin and Trehalose-Modulated Autophagy Exerts a Neurotherapeutic Effect on Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 7253–7273. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef]
- Zimmermann, M.; Arachchige-Don, A.P.S.; Donaldson, M.S.; Patriarchi, T.; Horne, M.C. Cyclin G2 promotes cell cycle arrest in breast cancer cells responding to fulvestrant and metformin and correlates with patient survival. Cell Cycle 2016, 15, 3278–3295. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Qi, L.; Chen, K.; Li, R.; Song, S.; Zhou, C.; Zhai, W. Metformin induces TPC-1 cell apoptosis through endoplasmic reticulum stress-associated pathways in vitro and in vivo. Int. J. Oncol. 2019, 55, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.; Richa, T.; Mollaee, M.; Zhan, T.; Tassone, P.; Johnson, J.; Luginbuhl, A.; Cognetti, D.; Martinez-Outschoorn, U.; Stapp, R.; et al. Metformin Effects on FOXP3+ and CD8+ T Cell Infiltrates of Head and Neck Squamous Cell Carcinoma. Laryngoscope 2020, 130, E490–E498. [Google Scholar] [CrossRef]
- Lin, H.-C.; Kachingwe, B.H.; Cheng, H.W.; Uang, Y.-S.; Wang, L.-H. Effects of metformin dose on cancer risk reduction in patients with type 2 diabetes mellitus: A 6-year follow-up study. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2014, 34, 36–45. [Google Scholar] [CrossRef]
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, D.G.; Pusztai, L.; et al. Evidence for biological effects of metformin in operable breast cancer: A pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 2011, 128, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Niraula, S.; Dowling, R.J.O.; Ennis, M.; Chang, M.C.; Done, S.J.; Hood, N.; Escallon, J.; Leong, W.L.; McCready, D.R.; Reedijk, M.; et al. Metformin in early breast cancer: A prospective window of opportunity neoadjuvant study. Breast Cancer Res. Treat. 2012, 135, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, A.; Kiyokawa, T.; Sato, Y.; Shozu, M. Effects of metformin on endometrial cancer cell growth in vivo: A preoperative prospective trial. Cancer 2014, 120, 2986–2995. [Google Scholar] [CrossRef] [PubMed]
- Petchsila, K.; Prueksaritanond, N.; Insin, P.; Yanaranop, M.; Chotikawichean, N. Effect of metformin for decreasing proliferative marker in women with endometrial cancer: A randomized double-blind placebo-controlled trial. Asian Pac. J. Cancer Prev. 2020, 21, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Laskov, I.; Drudi, L.; Beauchamp, M.-C.; Yasmeen, A.; Ferenczy, A.; Pollak, M.; Gotlieb, W.H. Anti-diabetic doses of metformin decrease proliferation markers in tumors of patients with endometrial cancer. Gynecol. Oncol. 2014, 134, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Marrone, K.A.; Zhou, X.; Forde, P.M.; Purtell, M.; Brahmer, J.R.; Hann, C.L.; Kelly, R.J.; Coleman, B.; Gabrielson, E.; Rosner, G.L.; et al. A Randomized Phase II Study of Metformin plus Paclitaxel/Carboplatin/Bevacizumab in Patients with Chemotherapy-Naïve Advanced or Metastatic Nonsquamous Non-Small Cell Lung Cancer. Oncologist 2018, 23, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L.; et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res. Treat. 2019, 174, 433–442. [Google Scholar] [CrossRef]
- Parikh, A.B.; Kozuch, P.; Rohs, N.; Becker, D.J.; Levy, B.P. Metformin as a repurposed therapy in advanced non-small cell lung cancer (NSCLC): Results of a phase II trial. Investig. New Drugs 2017, 35, 813–819. [Google Scholar] [CrossRef]
- Skinner, H.; Hu, C.; Tsakiridis, T.; Santana-Davila, R.; Lu, B.; Erasmus, J.J.; Doemer, A.J.; Videtic, G.M.M.; Coster, J.; Yang, A.X.; et al. Addition of Metformin to Concurrent Chemoradiation in Patients with Locally Advanced Non–Small Cell Lung Cancer: The NRG-LU001 Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Borchiellini, D.; Thamphya, B.; Guillot, A.; Paoli, J.-B.; Besson, D.; Hilgers, W.; Priou, F.; El Kouri, C.; Hoch, B.; et al. TAXOMET: A French Prospective Multicentric Randomized Phase II Study of Docetaxel Plus Metformin Versus Docetaxel Plus Placebo in Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2021, 19, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Fyles, A.; Shek, T.; Croke, J.; Dhani, N.; D’Souza, D.; Lee, T.-Y.; Chaudary, N.; Bruce, J.; Pintilie, M.; et al. A Phase II Randomized Trial of Chemoradiation with or without Metformin in Locally Advanced Cervical Cancer. Clin. Cancer Res. 2022, 28, 5263–5271. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, B.; Puntoni, M.; Cazzaniga, M.; Pruneri, G.; Serrano, D.; Guerrieri-Gonzaga, A.; Gennari, A.; Trabacca, M.S.; Galimberti, V.; Veronesi, P.; et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J. Clin. Oncol. 2012, 30, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Joo, J.; Lee, Y.J.; Lee, E.K.; Park, S.; Kim, T.-S.; Lee, S.-H.; Kim, S.Y.; Wie, G.-A.; Park, M.; et al. Randomized phase II study of platinum-based chemotherapy plus controlled diet with or without metformin in patients with advanced non-small cell lung cancer. Lung Cancer 2021, 151, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Royce, M.; Bachelot, T.; Villanueva, C.; Özgüroglu, M.; Azevedo, S.J.; Cruz, F.M.; Debled, M.; Hegg, R.; Toyama, T.; Falkson, C.; et al. Everolimus Plus Endocrine Therapy for Postmenopausal Women with Estrogen Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: A Clinical Trial. JAMA Oncol. 2018, 4, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, G.; de Boer, R.H.; Hurvitz, S.; Yardley, D.A.; Kovalenko, E.; Ejlertsen, B.; Blau, S.; Özgüroglu, M.; Landherr, L.; Ewertz, M.; et al. Everolimus Plus Exemestane vs Everolimus or Capecitabine Monotherapy for Estrogen Receptor-Positive, HER2-Negative Advanced Breast Cancer The BOLERO-6 Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1367–1374. [Google Scholar] [CrossRef]
- Yardley, D.A.; Liggett, W.; Mainwaring, M.; Castrellon, A.; Blakely, L.; Hemphill, B.; Anz, B.; Young, R.R.; Shastry, M.; DeBusk, L.M.; et al. A Phase II Open Label Study of Everolimus in Combination with Endocrine Therapy in Resistant Hormone Receptor-Positive HER2-Negative Advanced Breast Cancer. Clin. Breast Cancer 2020, 20, 89–97. [Google Scholar] [CrossRef]
- Schneider, T.C.; De Wit, D.; Links, T.P.; Van Erp, N.P.; Van Der Hoeven, J.J.M.; Gelderblom, H.; Roozen, I.C.F.M.; Bos, M.; Corver, W.E.; Van Wezel, T.; et al. Everolimus in Patients with Advanced Follicular-Derived Thyroid Cancer; Results of a Phase II Clinical Trial. J. Clin. Endocrinol. Metab. 2017, 102, 698–707. [Google Scholar] [CrossRef]
- Lau, D.K.; Tay, R.Y.; Yeung, Y.H.; Chionh, F.; Mooi, J.; Murone, C.; Skrinos, E.; Price, T.J.; Mariadason, J.M.; Tebbutt, N.C. Phase II study of everolimus (RAD001) monotherapy as first-line treatment in advanced biliary tract cancer with biomarker exploration: The RADiChol Study. Br. J. Cancer 2018, 118, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Gilcrease, G.W.; Stenehjem, D.D.; Wade, M.L.; Weis, J.; McGregor, K.; Whisenant, J.; Boucher, K.M.; Thorne, K.; Orgain, N.; Garrido-Laguna, I.; et al. Phase I/II study of everolimus combined with mFOLFOX-6 and bevacizumab for first–line treatment of metastatic colorectal cancer. Investig. New Drugs 2019, 37, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.-A.O.; Hayes, D.N.; Karrison, T.; Harismendy, O.; Flores, J.M.; Moore-Medlin, T.; Vokes, E.E.; Gutkind, J.S.; Neupane, P.; Mills, G.; et al. A Randomized Multi-institutional Phase II Trial of Everolimus as Adjuvant Therapy in Patients with Locally Advanced Squamous Cell Cancer of the Head and Neck. Clin. Cancer Res. 2022, 28, 5040–5048. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel with or without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Marschner, N.; Muendlein, A.; Welt, A.; Hagen, V.; Rauh, J.; Schröder, H.; Jaehnig, P.; Potthoff, K.; Lerchenmüller, C. VicTORia: A randomised phase II study to compare vinorelbine in combination with the mTOR inhibitor everolimus versus vinorelbine monotherapy for second-line chemotherapy in advanced HER2-negative breast cancer. Breast Cancer Res. Treat. 2019, 176, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Cottu, P.; Chabaud, S.; Dalenc, F.; Allouache, D.; Delaloge, S.; Jacquin, J.-P.; Grenier, J.; Bouvet, L.V.; Jegannathen, A.; et al. Everolimus Added to Adjuvant Endocrine Therapy in Patients with High-Risk Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Primary Breast Cancer. J. Clin. Oncol. 2022, 40, 3699–3708. [Google Scholar] [CrossRef]
- Koshkin, V.S.; Mir, M.C.; Barata, P.; Gul, A.; Gupta, R.; Stephenson, A.J.; Kaouk, J.; Berglund, R.; Magi-Galluzzi, C.; Klein, E.A.; et al. Randomized phase II trial of neoadjuvant everolimus in patients with high-risk localized prostate cancer. Investig. New Drugs 2019, 37, 559–566. [Google Scholar] [CrossRef] [PubMed]
- George, D.J.; Halabi, S.; Healy, P.; Jonasch, D.; Anand, M.; Rasmussen, J.; Wood, S.Y.; Spritzer, C.; Madden, J.F.; Armstrong, A.J. Phase 2 clinical trial of TORC1 inhibition with everolimus in men with metastatic castration-resistant prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 79.e15–79.e22. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Castoldi, F.; Markaki, M.; Lachkar, S.; Chen, G.; Enot, D.P.; Durand, S.; Bossut, N.; Tong, M.; Malik, S.A.; et al. Aspirin Recapitulates Features of Caloric Restriction. Cell Rep. 2018, 22, 2395–2407. [Google Scholar] [CrossRef]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin Inhibits mTOR Signaling, Activates AMP-Activated Protein Kinase, and Induces Autophagy in Colorectal Cancer Cells. Gastroenterology 2012, 142, 1504–1515.e3. [Google Scholar] [CrossRef]
- Burn, J.; Gerdes, A.-M.; Macrae, F.; Mecklin, J.-P.; Moeslein, G.; Olschwang, S.; Eccles, D.M.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.-P.; Möslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Guo, C.-G.; Ma, W.; Drew, D.A.; Cao, Y.; Nguyen, L.H.; Joshi, A.D.; Ng, K.; Ogino, S.; Meyerhardt, J.A.; Song, M.; et al. Aspirin Use and Risk of Colorectal Cancer among Older Adults. JAMA Oncol. 2021, 7, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Sacco, A.; Sostres, C.; Bruno, A.; Dovizio, M.; Piazuelo, E.; Di Francesco, L.; Contursi, A.; Zucchelli, M.; Schiavone, S.; et al. Low-Dose Aspirin Acetylates Cyclooxygenase-1 in Human Colorectal Mucosa: Implications for the Chemoprevention of Colorectal Cancer. Clin. Pharmacol. Ther. 2017, 102, 52–61. [Google Scholar] [CrossRef]
- Vidal, A.C.; Howard, L.E.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freedland, S.J. Aspirin, NSAIDs, and risk of prostate cancer: Results from the REDUCE study. Clin. Cancer Res. 2015, 21, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Downer, M.K.; Allard, C.B.; Preston, M.A.; Gaziano, J.M.; Stampfer, M.J.; Mucci, L.A.; Batista, J.L. Regular Aspirin Use and the Risk of Lethal Prostate Cancer in the Physicians’ Health Study. Eur. Urol. 2017, 72, 821–827. [Google Scholar] [CrossRef]
- Zhang, S.M.; Cook, N.R.; E Manson, J.; Lee, I.-M.; E Buring, J. Low-dose aspirin and breast cancer risk: Results by tumour characteristics from a randomised trial. Br. J. Cancer 2008, 98, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.D.; Olsson, H.; Pawitan, Y.; Holm, J.; Lundholm, C.; Andersson, T.M.-L.; Adami, H.-O.; Askling, J.; Smedby, K.E. Aspirin intake and breast cancer survival—A nation-wide study using prospectively recorded data in Sweden. BMC Cancer 2014, 14, 391. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, L.M.; Pinsky, P.F.; Huang, W.-Y.; Freedman, N.D.; Trabert, B. Aspirin use and ovarian cancer risk using extended follow-up of the PLCO Cancer Screening Trial. Gynecol. Oncol. 2020, 159, 522–526. [Google Scholar] [CrossRef]
- Movahedi, M.; Bishop, D.T.; Macrae, F.; Mecklin, J.-P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Obesity, Aspirin, and Risk of Colorectal Cancer in Carriers of Hereditary Colorectal Cancer: A Prospective Investigation in the CAPP2 Study. J. Clin. Oncol. 2015, 33, 3591–3597. [Google Scholar] [CrossRef]
- Brown, V.A.; Patel, K.R.; Viskaduraki, M.; Crowell, J.A.; Perloff, M.; Booth, T.D.; Vasilinin, G.; Sen, A.; Schinas, A.M.; Piccirilli, G.; et al. Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: Safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res. 2010, 70, 9003–9011. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Brown, V.A.; Jones, D.J.; Britton, R.G.; Hemingway, D.; Miller, A.S.; West, K.P.; Booth, T.D.; Perloff, M.; Crowell, J.A.; et al. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 2010, 70, 7392–7399. [Google Scholar] [CrossRef]
- Howells, L.M.; Berry, D.P.; Elliott, P.J.; Jacobson, E.W.; Hoffmann, E.; Hegarty, B.; Brown, K.; Steward, W.P.; Gescher, A.J. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases—Safety, pharmacokinetics, and pharmacodynamics. Cancer Prev. Res. 2011, 4, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Park, S.; Kim, M.; Yang, W.K.; Im, D.U.; Yang, K.R.; Hong, J.; Choe, W.; Kang, I.; Kim, S.S.; et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J. 2014, 281, 4421–4438. [Google Scholar] [CrossRef] [PubMed]
- Inglés, M.; Gambini, J.; Miguel, M.G.; Bonet-Costa, V.; Abdelaziz, K.M.; El Alami, M.; Viña, J.; Borrás, C. PTEN Mediates the Antioxidant Effect of Resveratrol at Nutritionally Relevant Concentrations. BioMed Res. Int. 2014, 2014, 580852. [Google Scholar] [CrossRef] [PubMed]
- Saud, S.M.; Li, W.; Morris, N.L.; Matter, M.S.; Colburn, N.H.; Kim, Y.S.; Young, M.R. Resveratrol prevents tumorigenesis in mouse model of Kras activated sporadic colorectal cancer by suppressing oncogenic Kras expression. Carcinogenesis 2014, 35, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Alkhalaf, M.; El-Mowafy, A.; Renno, W.; Rachid, O.; Ali, A.; Al-Attyiah, R. Resveratrol-induced apoptosis in human breast cancer cells is mediated primarily through the caspase-3-dependent pathway. Arch. Med. Res. 2008, 39, 162–168. [Google Scholar] [CrossRef]
- van Ginkel, P.R.; Sareen, D.; Subramanian, L.; Walker, Q.; Darjatmoko, S.R.; Lindstrom, M.J.; Kulkarni, A.; Albert, D.M.; Polans, A.S. Resveratrol inhibits tumor growth of human neuroblastoma and mediates apoptosis by directly targeting mitochondria. Clin. Cancer Res. 2007, 13, 5162–5169. [Google Scholar] [CrossRef]
- Li, C.; Wang, Z.; Lei, H.; Zhang, D. Recent progress in nanotechnology-based drug carriers for resveratrol delivery. Drug Deliv. 2023, 30, 2174206. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Springer, M.; Moco, S. Resveratrol and Its Human Metabolites—Effects on Metabolic Health and Obesity. Nutrients 2019, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Andreadi, C.; Britton, R.G.; Horner-Glister, E.; Karmokar, A.; Sale, S.; Brown, V.A.; Brenner, D.E.; Singh, R.; Steward, W.P.; et al. Sulfate metabolites provide an intracellular pool for resveratrol generation and induce autophagy with senescence. Sci. Transl. Med. 2013, 5, 205ra133. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, Y.; Guo, Z.; Zeng, Y.; Zhang, W.; Wang, H. Metformin: Current clinical applications in nondiabetic patients with cancer. Aging 2020, 12, 3993–4009. [Google Scholar] [CrossRef] [PubMed]
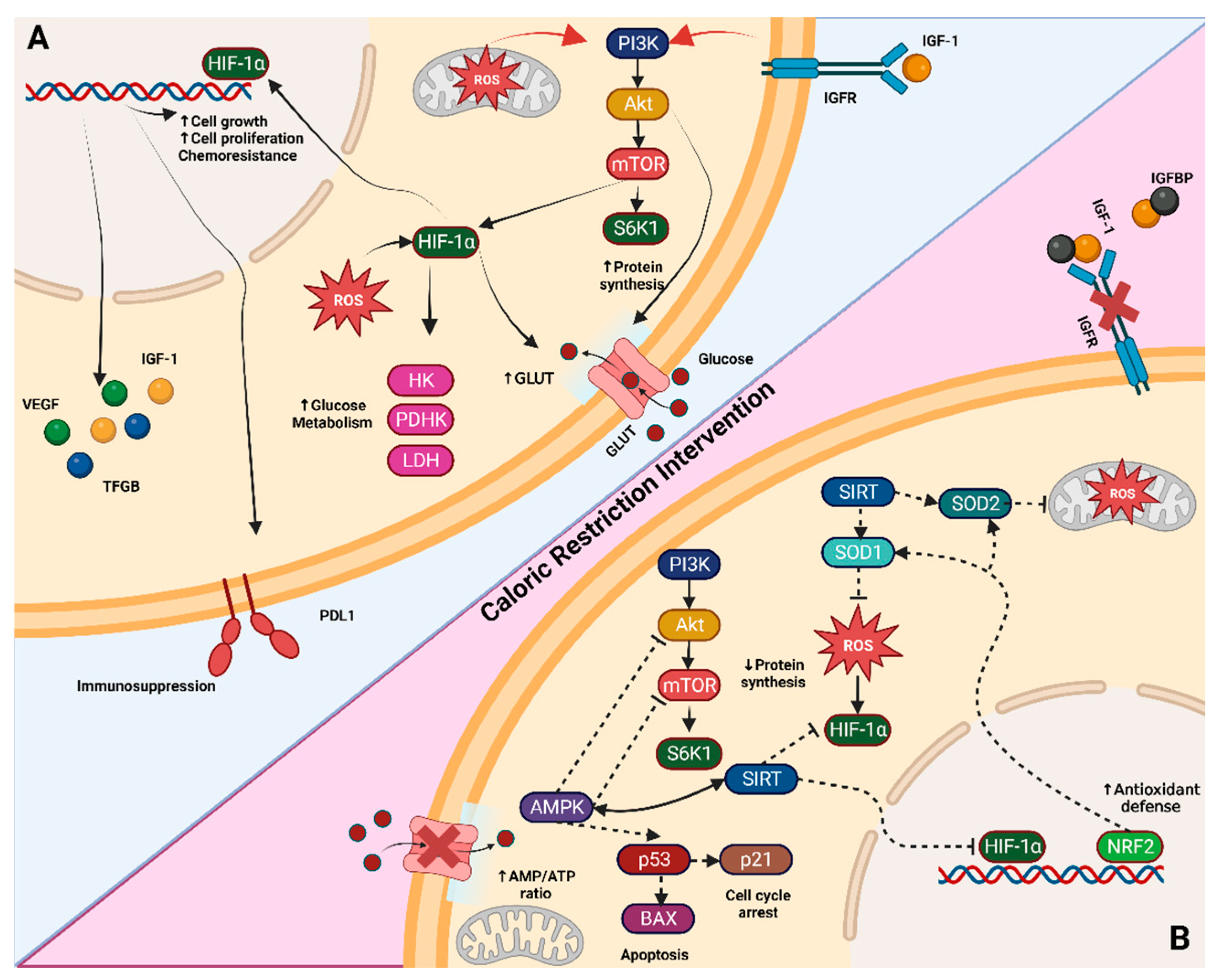
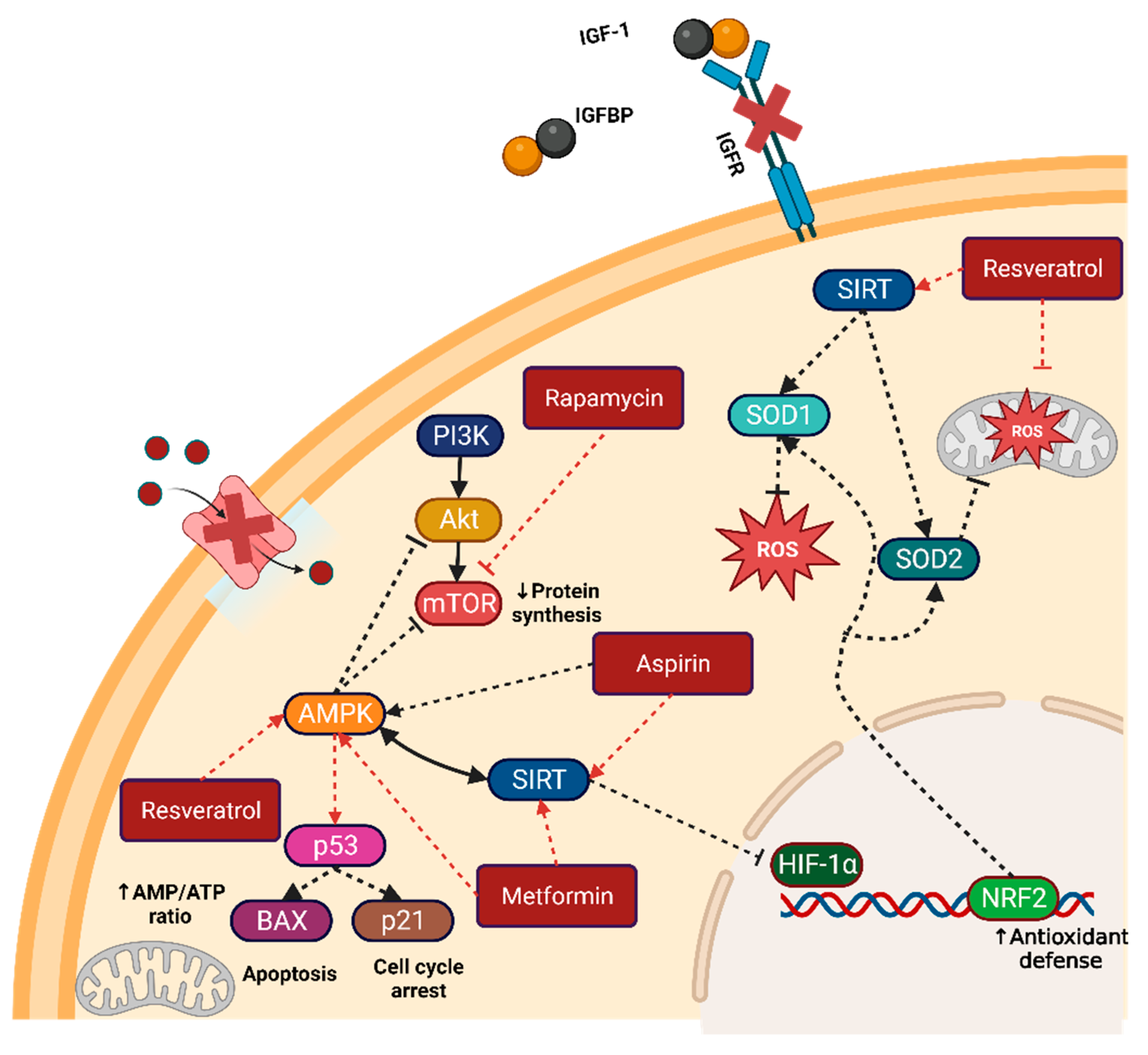
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).