
Impact of Phenylketonuria on the Serum Metabolome and Plasma Lipidome: A Study in Early-Treated Patients
 , , , , and
, , , , and
Abstract
:1. Introduction
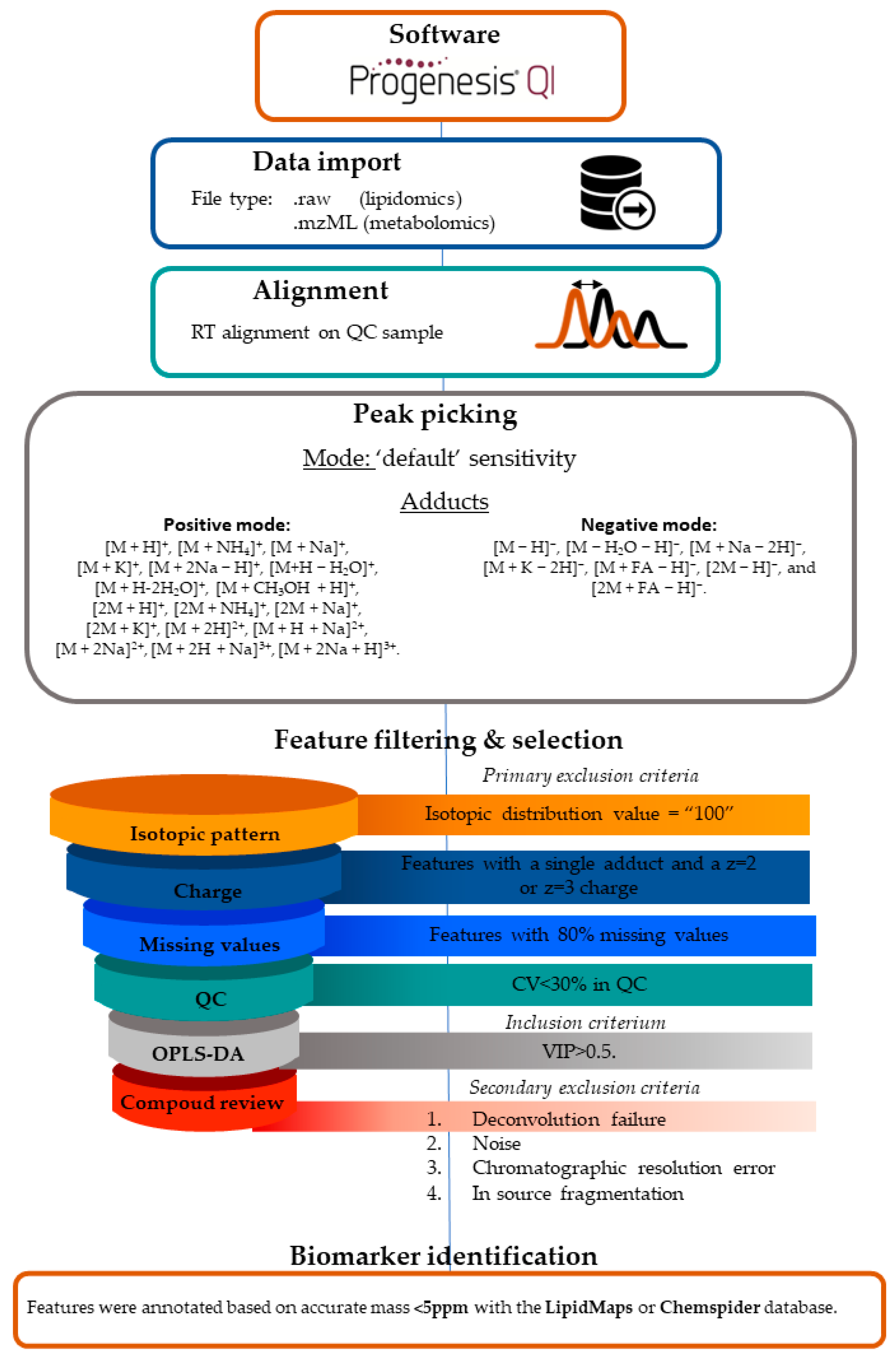
2. Materials and Methods
Lipidomics LC-MS Workflow and Data Processing
3. Results
3.1. Population Characteristics
3.2. Data Processing
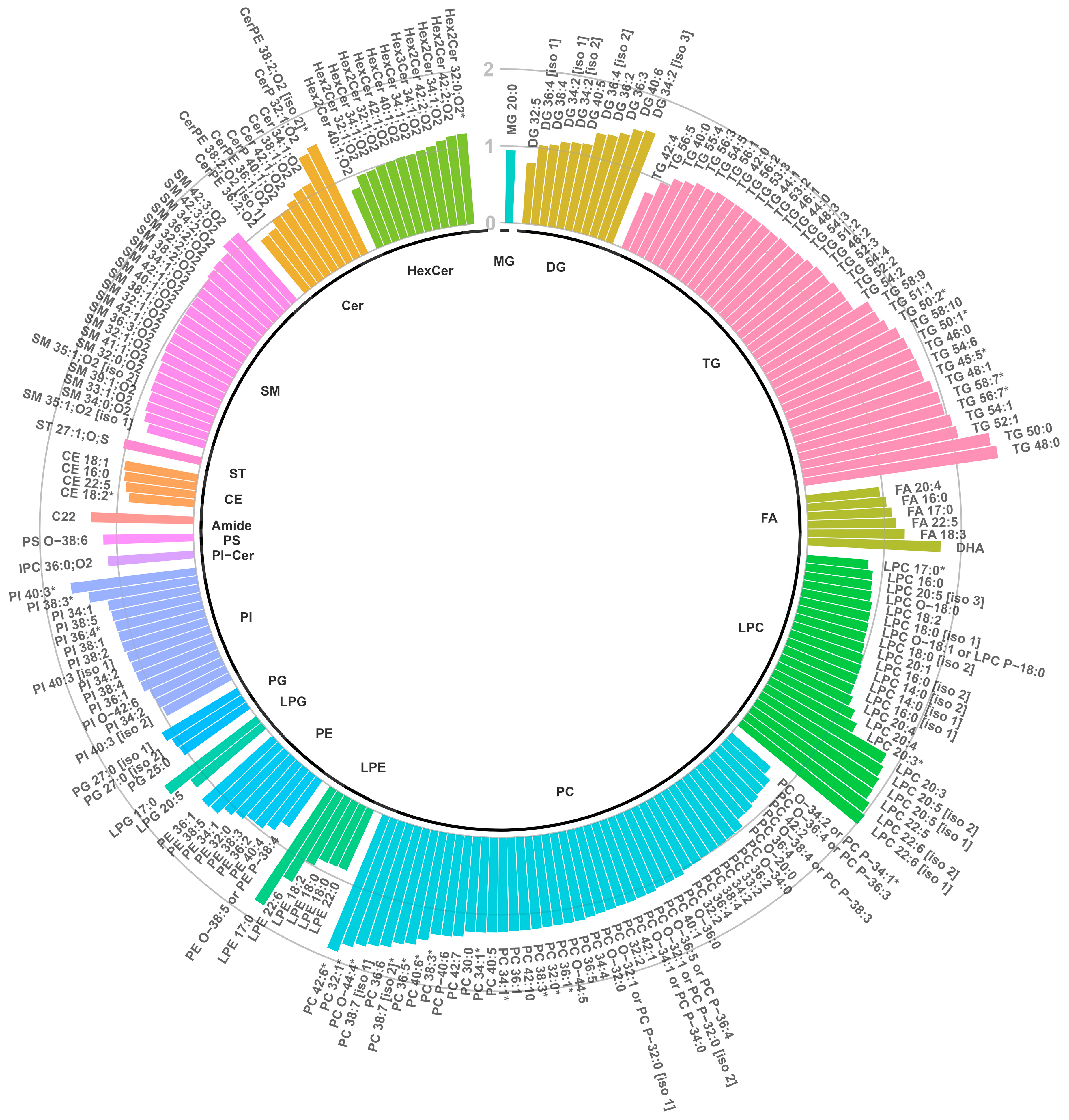
3.3. Lipidomics
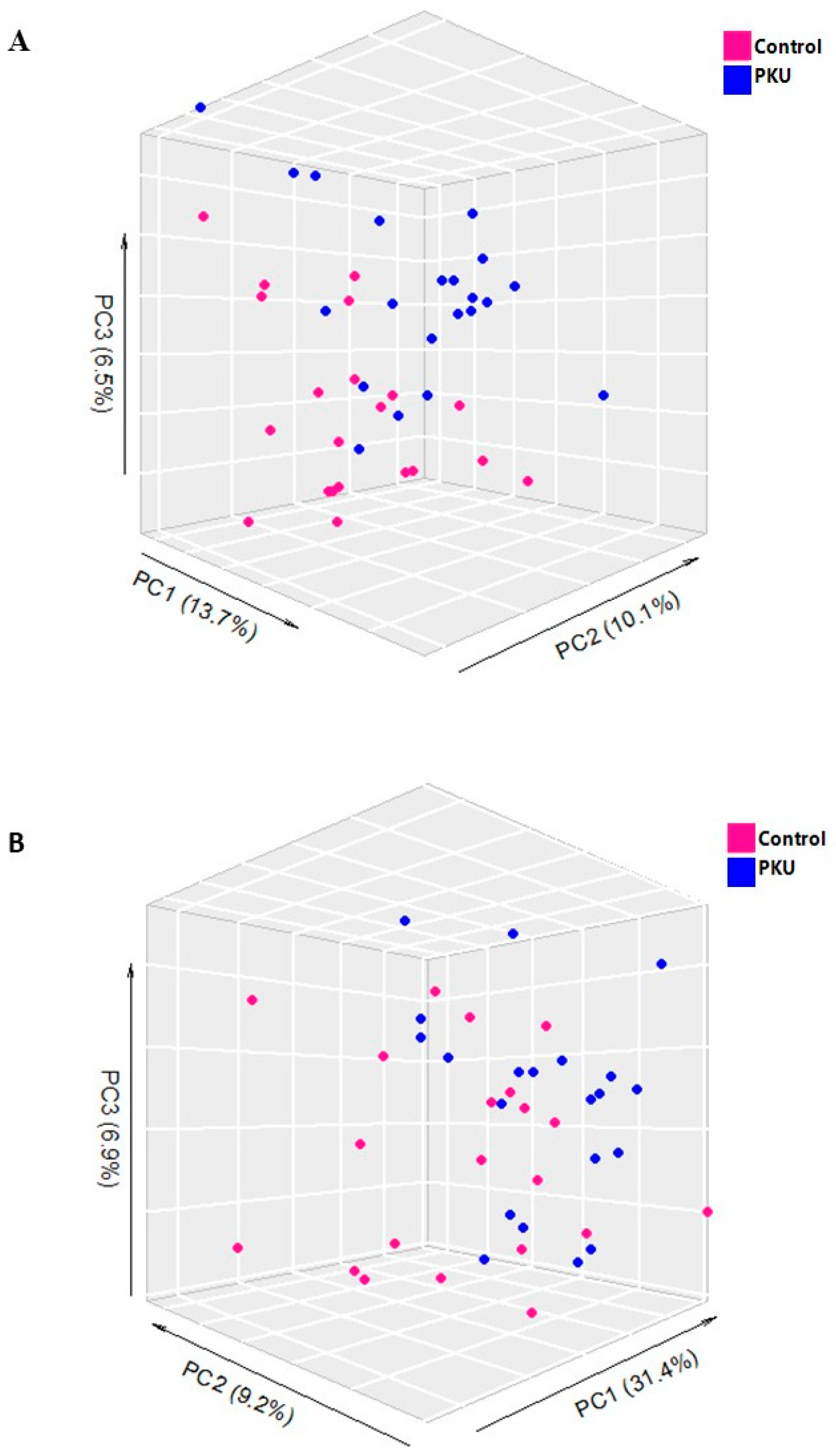
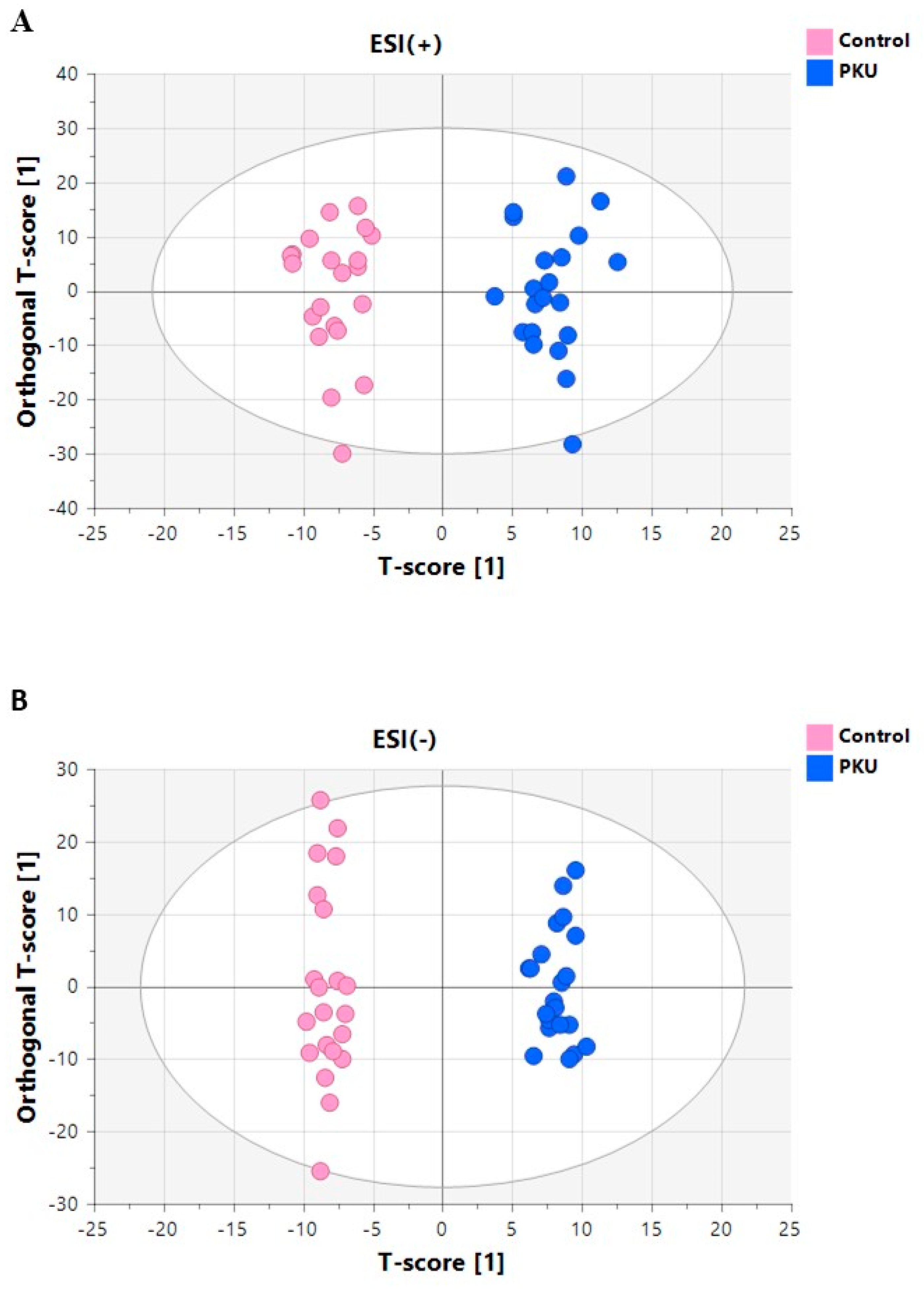
3.3.1. Multivariate Analyses of Plasma Lipidomic Data
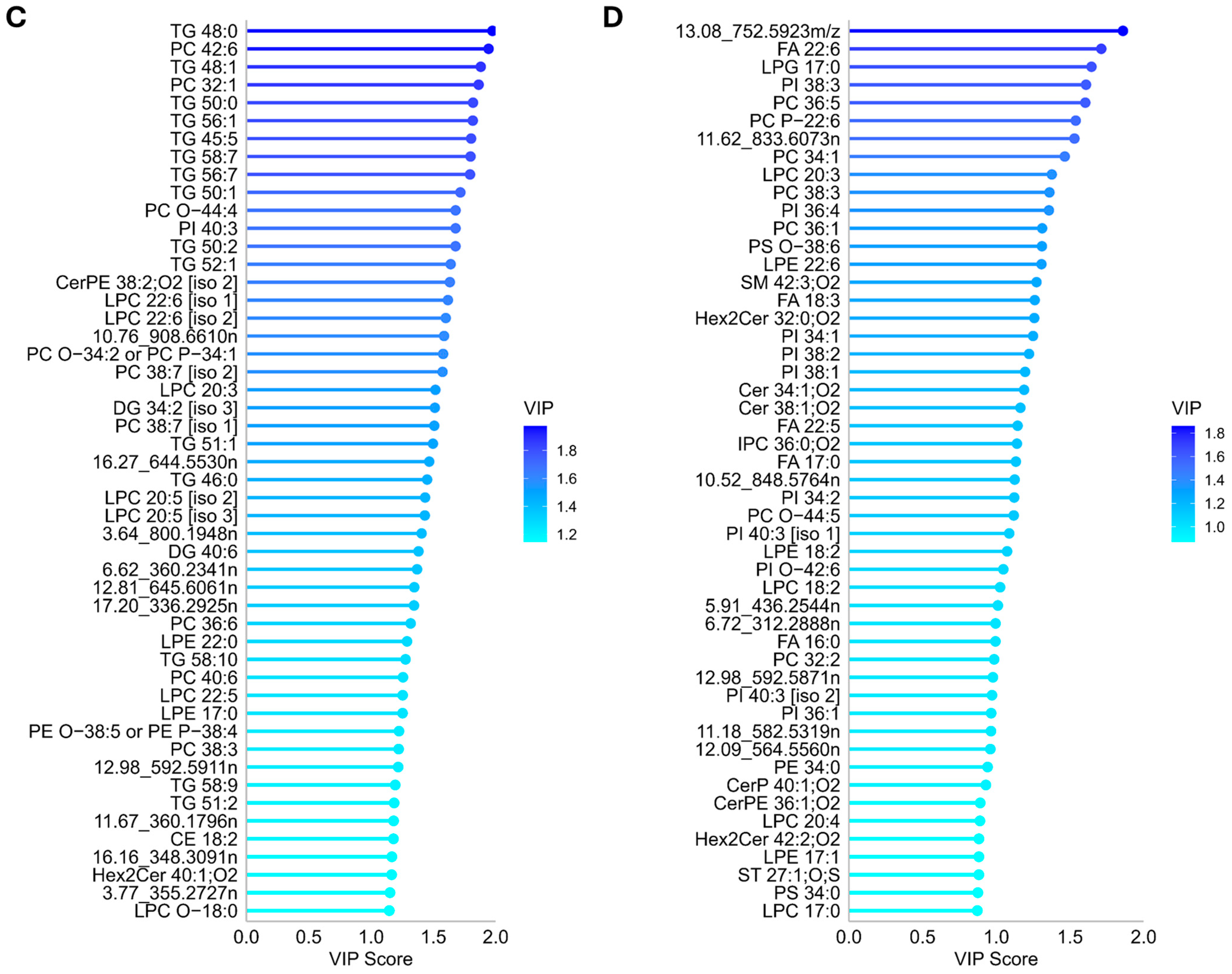
3.3.2. Annotation and Univariate Significance of Discriminating Lipid Features
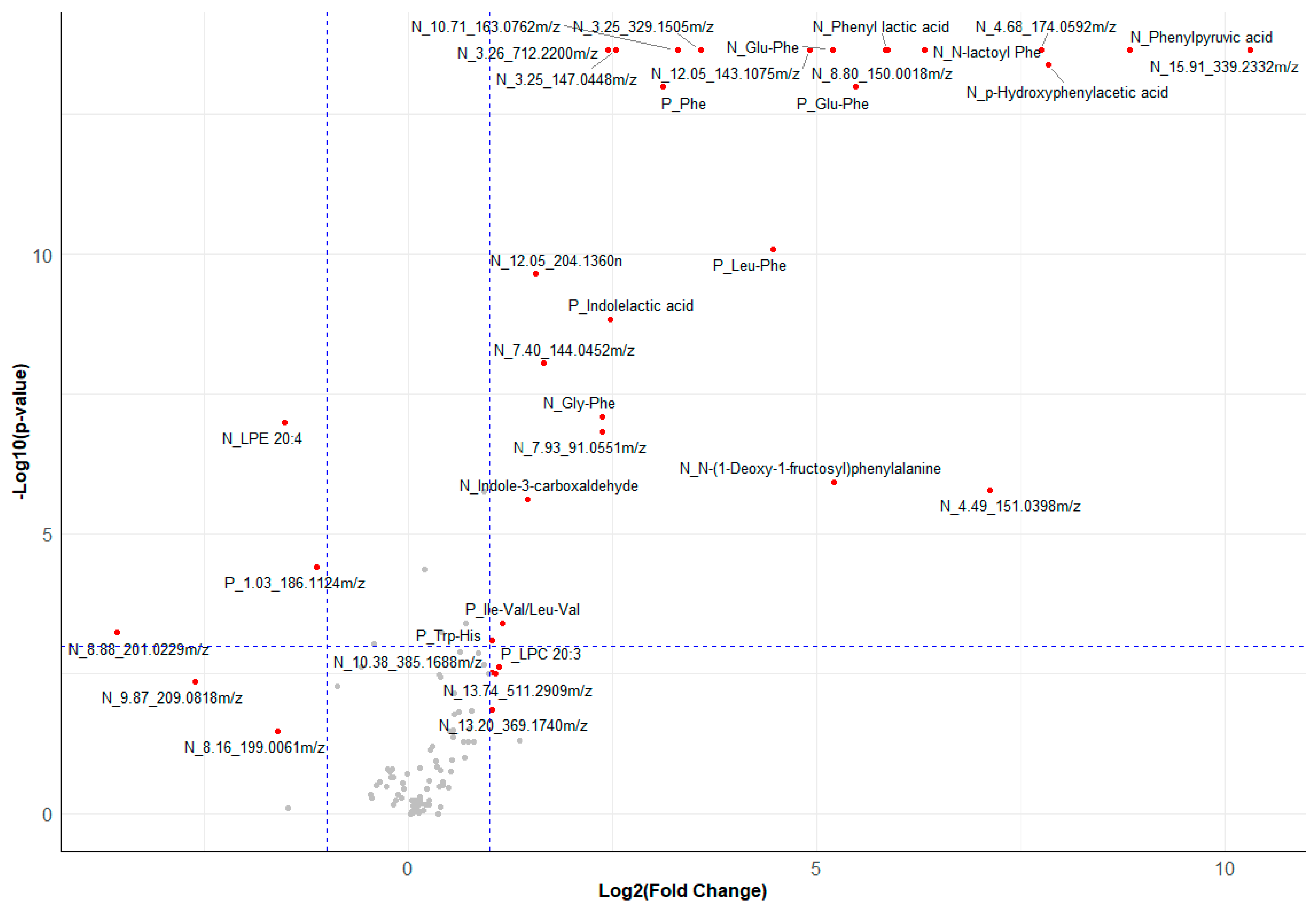
3.4. Metabolomics
3.4.1. Multivariate Analyses of Serum Metabolomics Data
3.4.2. Annotation and Univariate Significance of Discriminating Metabolite Features
3.5. Correlation between Concurrent Dried Blood Spot Phe and Potential Biomarkers
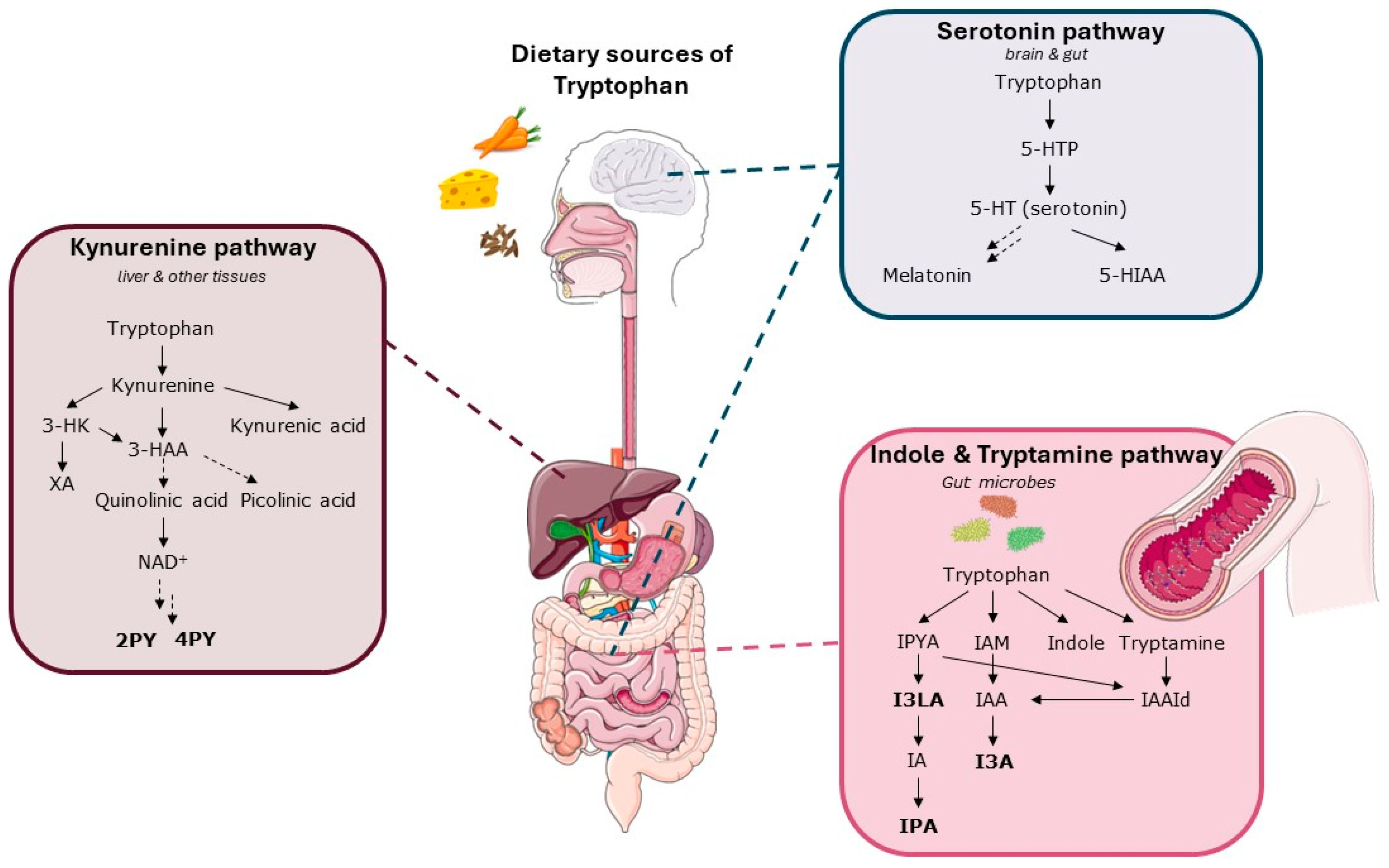
3.6. Metabolic Pathway Enrichment Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef]
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Anikster, Y.; Haack, T.B.; Vilboux, T.; Pode-Shakked, B.; Thöny, B.; Shen, N.; Guarani, V.; Meissner, T.; Mayatepek, E.; Trefz, F.K.; et al. Biallelic Mutations in DNAJC12 Cause Hyperphenylalaninemia, Dystonia, and Intellectual Disability. Am. J. Hum. Genet. 2017, 100, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.A.; Walter, J.H.; Wraith, J.E.; Jenkins, J.P.; Alani, S.M.; Tyler, K.; Whittle, D. Magnetic resonance imaging of the brain in phenylketonuria. Lancet 1994, 344, 87–90. [Google Scholar] [CrossRef]
- Fölling, A. Über Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität. Biol. Chem. 1934, 227, 169–181. [Google Scholar] [CrossRef]
- Følling, I. The discovery of phenylketonuria. Acta Paediatr. Suppl. 1994, 407, 4–10. [Google Scholar] [CrossRef]
- Leuzzi, V.; Trasimeni, G.; Gualdi, G.F.; Antonozzi, I. Biochemical, clinical and neuroradiological (MRI) correlations in late-detected PKU patients. J. Inherit. Metab. Dis. 1995, 18, 624–634. [Google Scholar] [CrossRef]
- Penrose, L.; Quastel, J.H. Metabolic studies in phenylketonuria. Biochem. J. 1937, 31, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Pietz, J.; Kreis, R.; Rupp, A.; Mayatepek, E.; Rating, D.; Boesch, C.; Bremer, H.J. Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. J. Clin. Investig. 1999, 103, 1169–1178. [Google Scholar] [CrossRef]
- Smith, I.; Beasley, M.G.; Ades, A.E. Intelligence and quality of dietary treatment in phenylketonuria. Arch. Dis. Child. 1990, 65, 472–478. [Google Scholar] [CrossRef]
- Thompson, A.J.; Tillotson, S.; Smith, I.; Kendall, B.; Moore, S.G.; Brenton, D.P. Brain MRI changes in phenylketonuria. Associations with dietary status. Brain 1993, 116 Pt 4, 811–821. [Google Scholar] [CrossRef] [PubMed]
- VanZutphen, K.; Packman, W.; Sporri, L.; Needham, M.; Morgan, C.; Weisiger, K.; Packman, S. Executive functioning in children and adolescents with phenylketonuria. Clin. Genet. 2007, 72, 13–18. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; van Wegberg, A.M.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017, 5, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.; Verduci, E.; Salvatici, E.; Paci, S.; Riva, E. Phenylketonuria: Nutritional advances and challenges. Nutr. Metab. 2012, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef]
- Burlina, A.; Biasucci, G.; Carbone, M.T.; Cazzorla, C.; Paci, S.; Pochiero, F.; Spada, M.; Tummolo, A.; Zuvadelli, J.; Leuzzi, V. Italian national consensus statement on management and pharmacological treatment of phenylketonuria. Orphanet J. Rare Dis. 2021, 16, 476. [Google Scholar] [CrossRef]
- Harding, C.O.; Longo, N.; Northrup, H.; Sacharow, S.; Singh, R.; Thomas, J.A.; Vockley, J.; Zori, R.T.; Bulloch Whitehall, K.; Lilienstein, J.; et al. Pegvaliase for the treatment of phenylketonuria: Final results of a long-term phase 3 clinical trial program. Mol. Genet. Metab. Rep. 2024, 39, 101084. [Google Scholar] [CrossRef]
- Krämer, J.; Baerwald, C.; Heimbold, C.; Kamrath, C.; Parhofer, K.G.; Reichert, A.; Rutsch, F.; Stolz, S.; Weinhold, N.; Muntau, A.C. Two years of pegvaliase in Germany: Experiences and best practice recommendations. Mol. Genet. Metab. 2023, 139, 107564. [Google Scholar] [CrossRef]
- Dijkstra, A.M.; van Vliet, N.; van Vliet, D.; Romani, C.; Huijbregts, S.C.J.; van der Goot, E.; Hovens, I.B.; van der Zee, E.A.; Kema, I.P.; Heiner-Fokkema, M.R.; et al. Correlations of blood and brain biochemistry in phenylketonuria: Results from the Pah-enu2 PKU mouse. Mol. Genet. Metab. 2021, 134, 250–256. [Google Scholar] [CrossRef]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.; Keularts, I.; et al. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef] [PubMed]
- van Outersterp, R.E.; Moons, S.J.; Engelke, U.F.H.; Bentlage, H.; Peters, T.M.A.; van Rooij, A.; Huigen, M.; de Boer, S.; van der Heeft, E.; Kluijtmans, L.A.J.; et al. Amadori rearrangement products as potential biomarkers for inborn errors of amino-acid metabolism. Commun. Biol. 2021, 4, 367. [Google Scholar] [CrossRef]
- Guerra, I.M.S.; Ferreira, H.B.; Neves, B.; Melo, T.; Diogo, L.M.; Domingues, M.R.; Moreira, A.S.P. Lipids and phenylketonuria: Current evidences pointed the need for lipidomics studies. Arch. Biochem. Biophys. 2020, 688, 108431. [Google Scholar] [CrossRef] [PubMed]
- Jahja, R.; van Spronsen, F.J.; de Sonneville, L.M.J.; van der Meere, J.J.; Bosch, A.M.; Hollak, C.E.M.; Rubio-Gozalbo, M.E.; Brouwers, M.; Hofstede, F.C.; de Vries, M.C.; et al. Long-Term Follow-Up of Cognition and Mental Health in Adult Phenylketonuria: A PKU-COBESO Study. Behav. Genet. 2017, 47, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Jahja, R.; van Spronsen, F.J.; de Sonneville, L.M.J.; van der Meere, J.J.; Bosch, A.M.; Hollak, C.E.M.; Rubio-Gozalbo, M.E.; Brouwers, M.; Hofstede, F.C.; de Vries, M.C.; et al. Social-cognitive functioning and social skills in patients with early treated phenylketonuria: A PKU-COBESO study. J. Inherit. Metab. Dis. 2016, 39, 355–362. [Google Scholar] [CrossRef]
- Jahja, R.; Huijbregts, S.C.J.; de Sonneville, L.M.J.; van der Meere, J.J.; Legemaat, A.M.; Bosch, A.M.; Hollak, C.E.M.; Rubio-Gozalbo, M.E.; Brouwers, M.; Hofstede, F.C.; et al. Cognitive profile and mental health in adult phenylketonuria: A PKU-COBESO study. Neuropsychology 2017, 31, 437–447. [Google Scholar] [CrossRef]
- Jahja, R.; Huijbregts, S.C.; de Sonneville, L.M.; van der Meere, J.J.; Bosch, A.M.; Hollak, C.E.; Rubio-Gozalbo, M.E.; Brouwers, M.C.; Hofstede, F.C.; de Vries, M.C.; et al. Mental health and social functioning in early treated Phenylketonuria: The PKU-COBESO study. Mol. Genet. Metab. 2013, 110, S57–S61. [Google Scholar] [CrossRef]
- Eisenga, M.F.; Gomes-Neto, A.W.; van Londen, M.; Ziengs, A.L.; Douwes, R.M.; Stam, S.P.; Osté, M.C.J.; Knobbe, T.J.; Hessels, N.R.; Buunk, A.M.; et al. Rationale and design of TransplantLines: A prospective cohort study and biobank of solid organ transplant recipients. BMJ Open 2018, 8, e024502. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Zhang, W.; Wolters, J.C.; Permentier, H.; Boer, T.; Horvatovich, P.; Heiner-Fokkema, M.R.; Reijngoud, D.J.; Bischoff, R. One- vs. two-phase extraction: Re-evaluation of sample preparation procedures for untargeted lipidomics in plasma samples. Anal. Bioanal. Chem. 2018, 410, 5859–5870. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Forest, A.; Ruiz, M.; Bouchard, B.; Boucher, G.; Gingras, O.; Daneault, C.; Robillard Frayne, I.; Rhainds, D.; Tardif, J.C.; Rioux, J.D.; et al. Comprehensive and Reproducible Untargeted Lipidomic Workflow Using LC-QTOF Validated for Human Plasma Analysis. J. Proteome Res. 2018, 17, 3657–3670. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Royal Society of Chemistry ChemSpider|Search and Share Chemistry. Available online: https://www.chemspider.com/ (accessed on 16 April 2024).
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [PubMed]
- t’Kindt, R.; Telenga, E.D.; Jorge, L.; Van Oosterhout, A.J.; Sandra, P.; Ten Hacken, N.H.; Sandra, K. Profiling over 1500 lipids in induced lung sputum and the implications in studying lung diseases. Anal. Chem. 2015, 87, 4957–4964. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Gao, K.; Mu, C.L.; Farzi, A.; Zhu, W.Y. Tryptophan Metabolism: A Link between the Gut Microbiota and Brain. Adv. Nutr. 2020, 11, 709–723. [Google Scholar] [CrossRef]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef] [PubMed]
- Kałużna-Czaplińska, J.; Gątarek, P.; Chirumbolo, S.; Chartrand, M.S.; Bjørklund, G. How important is tryptophan in human health? Crit. Rev. Food Sci. Nutr. 2019, 59, 72–88. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.J.; Bader, M. A unique central tryptophan hydroxylase isoform. Biochem. Pharmacol. 2003, 66, 1673–1680. [Google Scholar] [CrossRef]
- van Wegberg, A.M.J.; van der Weerd, J.C.; Engelke, U.F.H.; Coene, K.L.M.; Jahja, R.; Bakker, S.J.L.; Huijbregts, S.C.J.; Wevers, R.A.; Heiner-Fokkema, M.R.; van Spronsen, F.J. The clinical relevance of novel biomarkers as outcome parameter in adults with phenylketonuria. J. Inherit. Metab. Dis. 2024, 47, 524–635. [Google Scholar] [CrossRef]
- Andrade, F.; Cano, A.; Unceta Suarez, M.; Arza, A.; Vinuesa, A.; Ceberio, L.; López-Oslé, N.; de Frutos, G.; López-Oceja, R.; Aznal, E.; et al. Urine Phenylacetylglutamine Determination in Patients with Hyperphenylalaninemia. J. Clin. Med. 2021, 10, 3674. [Google Scholar] [CrossRef]
- Wild, J.; Shanmuganathan, M.; Hayashi, M.; Potter, M.; Britz-McKibbin, P. Metabolomics for improved treatment monitoring of phenylketonuria: Urinary biomarkers for non-invasive assessment of dietary adherence and nutritional deficiencies. Analyst 2019, 144, 6595–6608. [Google Scholar] [CrossRef]
- Xiong, X.; Sheng, X.; Liu, D.; Zeng, T.; Peng, Y.; Wang, Y. A GC/MS-based metabolomic approach for reliable diagnosis of phenylketonuria. Anal. Bioanal. Chem. 2015, 407, 8825–8833. [Google Scholar] [CrossRef]
- Berguig, G.Y.; Martin, N.T.; Creer, A.Y.; Xie, L.; Zhang, L.; Murphy, R.; Pacheco, G.; Bullens, S.; Olbertz, J.; Weng, H.H. Of mice and men: Plasma phenylalanine reduction in PKU corrects neurotransmitter pathways in the brain. Mol. Genet. Metab. 2019, 128, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Stroup, B.M.; Ney, D.M.; Murali, S.G.; Rohr, F.; Gleason, S.T.; van Calcar, S.C.; Levy, H.L. Metabolomic Insights into the Nutritional Status of Adults and Adolescents with Phenylketonuria Consuming a Low-Phenylalanine Diet in Combination with Amino Acid and Glycomacropeptide Medical Foods. J. Nutr. Metab. 2017, 2017, 6859820. [Google Scholar] [CrossRef] [PubMed]
- Bokayeva, K.; Jamka, M.; Walkowiak, D.; Duś-Żuchowska, M.; Herzig, K.H.; Walkowiak, J. Vitamin Status in Patients with Phenylketonuria: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2024, 25, 5065. [Google Scholar] [CrossRef] [PubMed]
- Cannet, C.; Bayat, A.; Frauendienst-Egger, G.; Freisinger, P.; Spraul, M.; Himmelreich, N.; Kockaya, M.; Ahring, K.; Godejohann, M.; MacDonald, A.; et al. Phenylketonuria (PKU) Urinary Metabolomic Phenotype Is Defined by Genotype and Metabolite Imbalance: Results in 51 Early Treated Patients Using Ex Vivo (1)H-NMR Analysis. Molecules 2023, 28, 4916. [Google Scholar] [CrossRef]
- Rutkowski, B.; Rutkowski, P.; Słomińska, E.; Smolenski, R.T.; Swierczyński, J. Cellular toxicity of nicotinamide metabolites. J. Ren. Nutr. 2012, 22, 95–97. [Google Scholar] [CrossRef]
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-methyl-2-pyridone-5-carboxamide: A novel uremic toxin? Kidney Int. 2003, 63, S19–S21. [Google Scholar] [CrossRef]
- Slominska, E.M.; Kowalik, K.; Smolenski, R.T.; Szolkiewicz, M.; Rutkowski, P.; Rutkowski, B.; Swierczynski, J. Accumulation of poly(ADP-ribose) polymerase inhibitors in children with chronic renal failure. Pediatr. Nephrol. 2006, 21, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Synesiou, E.; Fairbanks, L.D.; Simmonds, H.A.; Slominska, E.M.; Smolenski, R.T.; Carrey, E.A. 4-Pyridone-3-carboxamide-1-β-D-ribonucleoside triphosphate (4PyTP), a novel NAD metabolite accumulating in erythrocytes of uremic children: A biomarker for a toxic NAD analogue in other tissues? Toxins 2011, 3, 520–537. [Google Scholar] [CrossRef]
- Mancilla, V.J.; Mann, A.E.; Zhang, Y.; Allen, M.S. The Adult Phenylketonuria (PKU) Gut Microbiome. Microorganisms 2021, 9, 530. [Google Scholar] [CrossRef]
- Verduci, E.; Carbone, M.T.; Borghi, E.; Ottaviano, E.; Burlina, A.; Biasucci, G. Nutrition, Microbiota and Role of Gut-Brain Axis in Subjects with Phenylketonuria (PKU): A Review. Nutrients 2020, 12, 3319. [Google Scholar] [CrossRef]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef] [PubMed]
- Poeggeler, B.; Sambamurti, K.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Pappolla, M.A. A novel endogenous indole protects rodent mitochondria and extends rotifer lifespan. PLoS ONE 2010, 5, e10206. [Google Scholar] [CrossRef] [PubMed]
- Bercik, P.; Verdu, E.F.; Foster, J.A.; Macri, J.; Potter, M.; Huang, X.; Malinowski, P.; Jackson, W.; Blennerhassett, P.; Neufeld, K.A.; et al. Chronic gastrointestinal inflammation induces anxiety-like behavior and alters central nervous system biochemistry in mice. Gastroenterology 2010, 139, 2102–2112.e1. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, F.; Severance, E.; Yolken, R. The microbiome, immunity, and schizophrenia and bipolar disorder. Brain Behav. Immun. 2017, 62, 46–52. [Google Scholar] [CrossRef]
- Xu, M.; Xu, X.; Li, J.; Li, F. Association between Gut Microbiota and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2019, 10, 473. [Google Scholar] [CrossRef]
- Agostoni, C.; Marangoni, F.; Riva, E.; Giovannini, M.; Galli, C. Plasma arachidonic acid and serum thromboxane B2 concentrations in phenylketonuric children negatively correlate with dietary compliance. Prostaglandins Leukot. Essent. Fat. Acids 1997, 56, 219–222. [Google Scholar] [CrossRef]
- Galli, C.; Agostoni, C.; Mosconi, C.; Riva, E.; Salari, P.C.; Giovannini, M. Reduced plasma C-20 and C-22 polyunsaturated fatty acids in children with phenylketonuria during dietary intervention. J. Pediatr. 1991, 119, 562–567. [Google Scholar] [CrossRef]
- Giovannini, M.; Biasucci, G.; Agostoni, C.; Luotti, D.; Riva, E. Lipid status and fatty acid metabolism in phenylketonuria. J. Inherit. Metab. Dis. 1995, 18, 265–272. [Google Scholar] [CrossRef]
- Moseley, K.; Koch, R.; Moser, A.B. Lipid status and long-chain polyunsaturated fatty acid concentrations in adults and adolescents with phenylketonuria on phenylalanine-restricted diet. J. Inherit. Metab. Dis. 2002, 25, 56–64. [Google Scholar] [CrossRef] [PubMed]
- van Gool, C.J.; van Houwelingen, A.C.; Hornstra, G. The essential fatty acid status in phenylketonuria patients under treatment. J. Nutr. Biochem. 2000, 11, 543–547. [Google Scholar] [CrossRef]
- Pöge, A.P.; Bäumann, K.; Müller, E.; Leichsenring, M.; Schmidt, H.; Bremer, H.J. Long-chain polyunsaturated fatty acids in plasma and erythrocyte membrane lipids of children with phenylketonuria after controlled linoleic acid intake. J. Inherit. Metab. Dis. 1998, 21, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo, P.; Perteagudo, L.; Rodríguez Soriano, J.; Vilaseca, A.; Campistol, J. Polyunsaturated fatty acid status in patients with phenylketonuria. J. Inherit. Metab. Dis. 1994, 17, 704–709. [Google Scholar] [CrossRef]
- Koletzko, B.; Sauerwald, T.; Demmelmair, H.; Herzog, M.; von Schenck, U.; Böhles, H.; Wendel, U.; Seidel, J. Dietary long-chain polyunsaturated fatty acid supplementation in infants with phenylketonuria: A randomized controlled trial. J. Inherit. Metab. Dis. 2007, 30, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Campoy, C.; Escolano-Margarit, M.V.; Anjos, T.; Szajewska, H.; Uauy, R. Omega 3 fatty acids on child growth, visual acuity and neurodevelopment. Br. J. Nutr. 2012, 107 (Suppl. S2), S85–S106. [Google Scholar] [CrossRef]
- Decsi, T.; Koletzko, B. Role of Long-Chain Polyunsaturated Fatty Acids in Early Human Neurodevelopment. Nutr. Neurosci. 2000, 3, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Koletzko, B.; Lien, E.; Agostoni, C.; Böhles, H.; Campoy, C.; Cetin, I.; Decsi, T.; Dudenhausen, J.W.; Dupont, C.; Forsyth, S.; et al. The roles of long-chain polyunsaturated fatty acids in pregnancy, lactation and infancy: Review of current knowledge and consensus recommendations. J. Perinat. Med. 2008, 36, 5–14. [Google Scholar] [CrossRef]
- Bazan, N.G. Omega-3 fatty acids, pro-inflammatory signaling and neuroprotection. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 136–141. [Google Scholar] [CrossRef]
- Agostoni, C.; Massetto, N.; Biasucci, G.; Rottoli, A.; Bonvissuto, M.; Bruzzese, M.G.; Giovannini, M.; Riva, E. Effects of long-chain polyunsaturated fatty acid supplementation on fatty acid status and visual function in treated children with hyperphenylalaninemia. J. Pediatr. 2000, 137, 504–509. [Google Scholar] [CrossRef]
- Agostoni, C.; Riva, E.; Biasucci, G.; Luotti, D.; Bruzzese, M.G.; Marangoni, F.; Giovannini, M. The effects of n-3 and n-6 polyunsaturated fatty acids on plasma lipids and fatty acids of treated phenylketonuric children. Prostaglandins Leukot. Essent. Fat. Acids 1995, 53, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Beblo, S.; Reinhardt, H.; Demmelmair, H.; Muntau, A.C.; Koletzko, B. Effect of fish oil supplementation on fatty acid status, coordination, and fine motor skills in children with phenylketonuria. J. Pediatr. 2007, 150, 479–484. [Google Scholar] [CrossRef]
- Beblo, S.; Reinhardt, H.; Muntau, A.C.; Mueller-Felber, W.; Roscher, A.A.; Koletzko, B. Fish oil supplementation improves visual evoked potentials in children with phenylketonuria. Neurology 2001, 57, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Koletzko, B.; Beblo, S.; Demmelmair, H.; Müller-Felber, W.; Hanebutt, F.L. Does dietary DHA improve neural function in children? Observations in phenylketonuria. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 159–164. [Google Scholar] [CrossRef]
- Agostoni, C.; Scaglioni, S.; Bonvissuto, M.; Bruzzese, M.G.; Giovannini, M.; Riva, E. Biochemical effects of supplemented long-chain polyunsaturated fatty acids in hyperphenylalaninemia. Prostaglandins Leukot. Essent. Fat. Acids 2001, 64, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Agostoni, C.; Verduci, E.; Massetto, N.; Fiori, L.; Radaelli, G.; Riva, E.; Giovannini, M. Long term effects of long chain polyunsaturated fats in hyperphenylalaninemic children. Arch. Dis. Child. 2003, 88, 582–583. [Google Scholar] [CrossRef]
- Drzymała-Czyż, S.; Kałużny, Ł.; Krzyżanowska-Jankowska, P.; Walkowiak, D.; Mozrzymas, R.; Walkowiak, J. Deficiency of long-chain polyunsaturated fatty acids in phenylketonuria: A cross-sectional study. Acta Biochim. Pol. 2018, 65, 303–308. [Google Scholar] [CrossRef]
- Guerra, I.M.S.; Diogo, L.; Pinho, M.; Melo, T.; Domingues, P.; Domingues, M.R.; Moreira, A.S.P. Plasma Phospholipidomic Profile Differs between Children with Phenylketonuria and Healthy Children. J. Proteome Res. 2021, 20, 2651–2661. [Google Scholar] [CrossRef]
- Colomé, C.; Artuch, R.; Lambruschini, N.; Cambra, F.J.; Campistol, J.; Vilaseca, M. Is there a relationship between plasma phenylalanine and cholesterol in phenylketonuric patients under dietary treatment? Clin. Biochem. 2001, 34, 373–376. [Google Scholar] [CrossRef]
- Couce, M.L.; Vitoria, I.; Aldámiz-Echevarría, L.; Fernández-Marmiesse, A.; Roca, I.; Llarena, M.; Sánchez-Pintos, P.; Leis, R.; Hermida, A. Lipid profile status and other related factors in patients with Hyperphenylalaninaemia. Orphanet J. Rare Dis. 2016, 11, 123. [Google Scholar] [CrossRef]
- Schulpi, K.H.; Scarpalezou, A. Triglycerides, cholesterol, HDL, LDL, and VLDL cholesterol in serum of phenylketonuric children under dietary control. Clin. Pediatr. 1989, 28, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Schulpis, K.H.; Karakonstantakis, T.; Bartzeliotou, A.; Karikas, G.A.; Papassotiriou, I. The association of serum lipids, lipoproteins and apolipoproteins with selected trace elements and minerals in phenylketonuric patients on diet. Clin. Nutr. 2004, 23, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Azabdaftari, A.; van der Giet, M.; Schuchardt, M.; Hennermann, J.B.; Plöckinger, U.; Querfeld, U. The cardiovascular phenotype of adult patients with phenylketonuria. Orphanet J. Rare Dis. 2019, 14, 213. [Google Scholar] [CrossRef]
- Lavoie, S.M.; Harding, C.O.; Gillingham, M.B. Normal fatty acid concentrations in young children with phenylketonuria (PKU). Top. Clin. Nutr. 2009, 24, 333–340. [Google Scholar] [CrossRef]
- Fernandez-Crespo, S.; Vazquez-Agra, N.; Marques-Afonso, A.T.; Cruces-Sande, A.; Martinez-Olmos, M.A.; Araujo-Vilar, D.; Hermida-Ameijeiras, A. The value of waist circumference as a preditor of cardiovascular risk in adult patients with classic phenylketonuria. Med. Clin. 2023, 161, 470–475. [Google Scholar] [CrossRef]
- Rocha, J.C.; van Spronsen, F.J.; Almeida, M.F.; Soares, G.; Quelhas, D.; Ramos, E.; Guimarães, J.T.; Borges, N. Dietary treatment in phenylketonuria does not lead to increased risk of obesity or metabolic syndrome. Mol. Genet. Metab. 2012, 107, 659–663. [Google Scholar] [CrossRef]
- Després, J.P. Body fat distribution and risk of cardiovascular disease: An update. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Piché, M.E.; Tchernof, A.; Després, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Hennermann, J.B.; Roloff, S.; Gellermann, J.; Vollmer, I.; Windt, E.; Vetter, B.; Plöckinger, U.; Mönch, E.; Querfeld, U. Chronic kidney disease in adolescent and adult patients with phenylketonuria. J. Inherit. Metab. Dis. 2013, 36, 747–756. [Google Scholar] [CrossRef]
- Rocha, J.C.; Martins, M.J. Oxidative stress in phenylketonuria: Future directions. J. Inherit. Metab. Dis. 2012, 35, 381–398. [Google Scholar] [CrossRef]
- Gündüz, M.; Çakar, S.; Kuyum, P.; Makay, B.; Arslan, N. Comparison of atherogenic risk factors among poorly controlled and well-controlled adolescent phenylketonuria patients. Cardiol. Young 2016, 26, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.V.; McStravick, N.; Ripley, S.; Weetch, E.; Donald, S.; Adam, S.; Micciche, A.; Boocock, S.; MacDonald, A. Body mass index in adult patients with diet-treated phenylketonuria. J. Hum. Nutr. Diet. 2013, 26 (Suppl. S1), 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bickert, A.; Ginkel, C.; Kol, M.; vom Dorp, K.; Jastrow, H.; Degen, J.; Jacobs, R.L.; Vance, D.E.; Winterhager, E.; Jiang, X.C.; et al. Functional characterization of enzymes catalyzing ceramide phosphoethanolamine biosynthesis in mice. J. Lipid Res. 2015, 56, 821–835. [Google Scholar] [CrossRef]
- Fall, T.; Salihovic, S.; Brandmaier, S.; Nowak, C.; Ganna, A.; Gustafsson, S.; Broeckling, C.D.; Prenni, J.E.; Kastenmüller, G.; Peters, A.; et al. Non-targeted metabolomics combined with genetic analyses identifies bile acid synthesis and phospholipid metabolism as being associated with incident type 2 diabetes. Diabetologia 2016, 59, 2114–2124. [Google Scholar] [CrossRef]
- Bao, L.; Zhang, Y.; Yan, S.; Yan, D.; Jiang, D. Lysophosphatidylcholine (17:0) Improves HFD-Induced Hyperglycemia & Insulin Resistance: A Mechanistic Mice Model Study. Diabetes Metab. Syndr. Obes. 2022, 15, 3511–3517. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Yang, H.; Yang, Z.; Jia, Q.; Liu, L.; Kong, L.; Cui, H.; Ding, S.; Qin, Q.; Zhang, N.; et al. Integrated biomarker profiling of the metabolome associated with impaired fasting glucose and type 2 diabetes mellitus in large-scale Chinese patients. Clin. Transl. Med. 2021, 11, e432. [Google Scholar] [CrossRef]
- Haid, M.; Muschet, C.; Wahl, S.; Römisch-Margl, W.; Prehn, C.; Möller, G.; Adamski, J. Long-Term Stability of Human Plasma Metabolites during Storage at −80 °C. J. Proteome Res. 2017, 17, 203–211. [Google Scholar] [CrossRef]
- Wagner-Golbs, A.; Neuber, S.; Kamlage, B.; Christiansen, N.; Bethan, B.; Rennefahrt, U.; Schatz, P.; Lind, L. Effects of Long-Term Storage at −80 °C on the Human Plasma Metabolome. Metabolites 2019, 9, 99. [Google Scholar] [CrossRef]
- Buchanan, J.L.; Vaquerano, J.T.; Taylor, E.B. Isolated Effects of Plasma Freezing versus Thawing on Metabolite Stability. Metabolites 2022, 12, 1098. [Google Scholar] [CrossRef]
- An, Z.; Shi, C.; Li, P.; Liu, L. Stability of Amino Acids and Related Amines in Human Serum under Different Preprocessing and Pre-Storage Conditions Based on iTRAQ®-LC-MS/MS. Biol. Open 2021, 10, bio055020. [Google Scholar] [CrossRef]
- Heiskanen, L.A.; Suoniemi, M.; Ta, H.X.; Tarasov, K.; Ekroos, K. Long-Term Performance and Stability of Molecular Shotgun Lipidomic Analysis of Human Plasma Samples. Anal. Chem. 2013, 85, 8757–8763. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolomics | Lipidomics | |||
|---|---|---|---|---|
| PKU | Control | PKU | Control | |
| n | 35 | 20 | 22 | 22 |
| Age (years) | 29 ± 7 | 34 ± 5 | 33 ± 6 | 34 ± 5 |
| Sex | ||||
| male | 18 (51%) | 12 (60%) | 8 (36%) | 8 (36%) |
| female | 17 (49%) | 8 (40%) | 14 (64%) | 14 (64%) |
| Lifetime Phe (µmol/L) | 439 [223–1001], n = 33 | - | 446 [255–734], n = 20 | - |
| Concurrent Phe (µmol/L) | 616 [176–1250], n = 35 | 645 [176–1250], n = 22 | ||
| BH4 treatment | 8/35 | - | 2/22 | - |
| Plasma total cholesterol (mmol/L) | 4.39 ± 0.81 | 4.65 ± 0.66 | ||
| Plasma total triglyceride (mmol/L) | 1.52 ± 0.55 | 1.32 ± 0.74 | ||
| Matrix for the current study | Serum | Serum | EDTA-plasma | EDTA-plasma |
| Lipidomics | Metabolomics | |||||
|---|---|---|---|---|---|---|
| ESI(+) | ESI(−) | ESI(+) | ESI(−) | |||
| Batch A | Batch B | Batch A | Batch B | |||
| Number of features detected | 15,220 | 5223 | 3627 | 2258 | ||
| Primary exclusion criteria | ||||||
| An isotopic abundance of 100 | 3570 | 527 | 1291 | 571 | ||
| Predominant adduct with z = 2 or 3 | 3285 | 0 | 157 | 0 | ||
| Features with 80% missing values | 1320 | 12 | 314 | 430 | 422 | 314 |
| CV > 30% | 2607 | 568 | 923 | 1145 | 507 | 562 |
| Number of features passing primary criteria | 4438 | 4416 | 725 | 503 | ||
| OPLS-DA | ||||||
| VIP > 0.5 | 3226 | 3243 | 628 | 281 | ||
| Secondary exclusion criteria after OPLS-DA | ||||||
| The final number of unique features | 210 | 64 | 51 | 67 | ||
| Total a | Expected | Hits b | Raw p c | Holm p d | FDR e | |
|---|---|---|---|---|---|---|
| Phenylalanine metabolism | 8 | 0.047 | 3 | 7.64 × 10−6 | 0.000612 | 0.000612 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 4 | 0.023 | 2 | 0.000181 | 0.0143 | 0.00726 |
| Nicotinate and nicotinamide metabolism | 15 | 0.088 | 2 | 0.00307 | 0.239 | 0.0819 |
| Arginine and proline metabolism | 36 | 0.211 | 2 | 0.0173 | 1 | 0.346 |
| Caffeine metabolism | 10 | 0.059 | 1 | 0.0571 | 1 | 0.914 |
| Arginine biosynthesis | 14 | 0.082 | 1 | 0.0792 | 1 | 1 |
| Pantothenate and CoA biosynthesis | 20 | 0.117 | 1 | 0.111 | 1 | 1 |
| Glutathione metabolism | 28 | 0.164 | 1 | 0.153 | 1 | 1 |
| Glycine, serine and threonine metabolism | 33 | 0.193 | 1 | 0.178 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weerd, J.C.v.d.; Wegberg, A.M.J.v.; Boer, T.S.; Engelke, U.F.H.; Coene, K.L.M.; Wevers, R.A.; Bakker, S.J.L.; Blaauw, P.d.; Groen, J.; Spronsen, F.J.v.; et al. Impact of Phenylketonuria on the Serum Metabolome and Plasma Lipidome: A Study in Early-Treated Patients. Metabolites 2024, 14, 479. https://doi.org/10.3390/metabo14090479
Weerd JCvd, Wegberg AMJv, Boer TS, Engelke UFH, Coene KLM, Wevers RA, Bakker SJL, Blaauw Pd, Groen J, Spronsen FJv, et al. Impact of Phenylketonuria on the Serum Metabolome and Plasma Lipidome: A Study in Early-Treated Patients. Metabolites. 2024; 14(9):479. https://doi.org/10.3390/metabo14090479
Chicago/Turabian StyleWeerd, Jorine C. van der, Annemiek M. J. van Wegberg, Theo S. Boer, Udo F. H. Engelke, Karlien L. M. Coene, Ron A. Wevers, Stephan J. L. Bakker, Pim de Blaauw, Joost Groen, Francjan J. van Spronsen, and et al. 2024. "Impact of Phenylketonuria on the Serum Metabolome and Plasma Lipidome: A Study in Early-Treated Patients" Metabolites 14, no. 9: 479. https://doi.org/10.3390/metabo14090479