Is N1-Methylnicotinamide a Good Organic Cation Transporter 2 (OCT2) Biomarker?

 , ,
, ,
Abstract
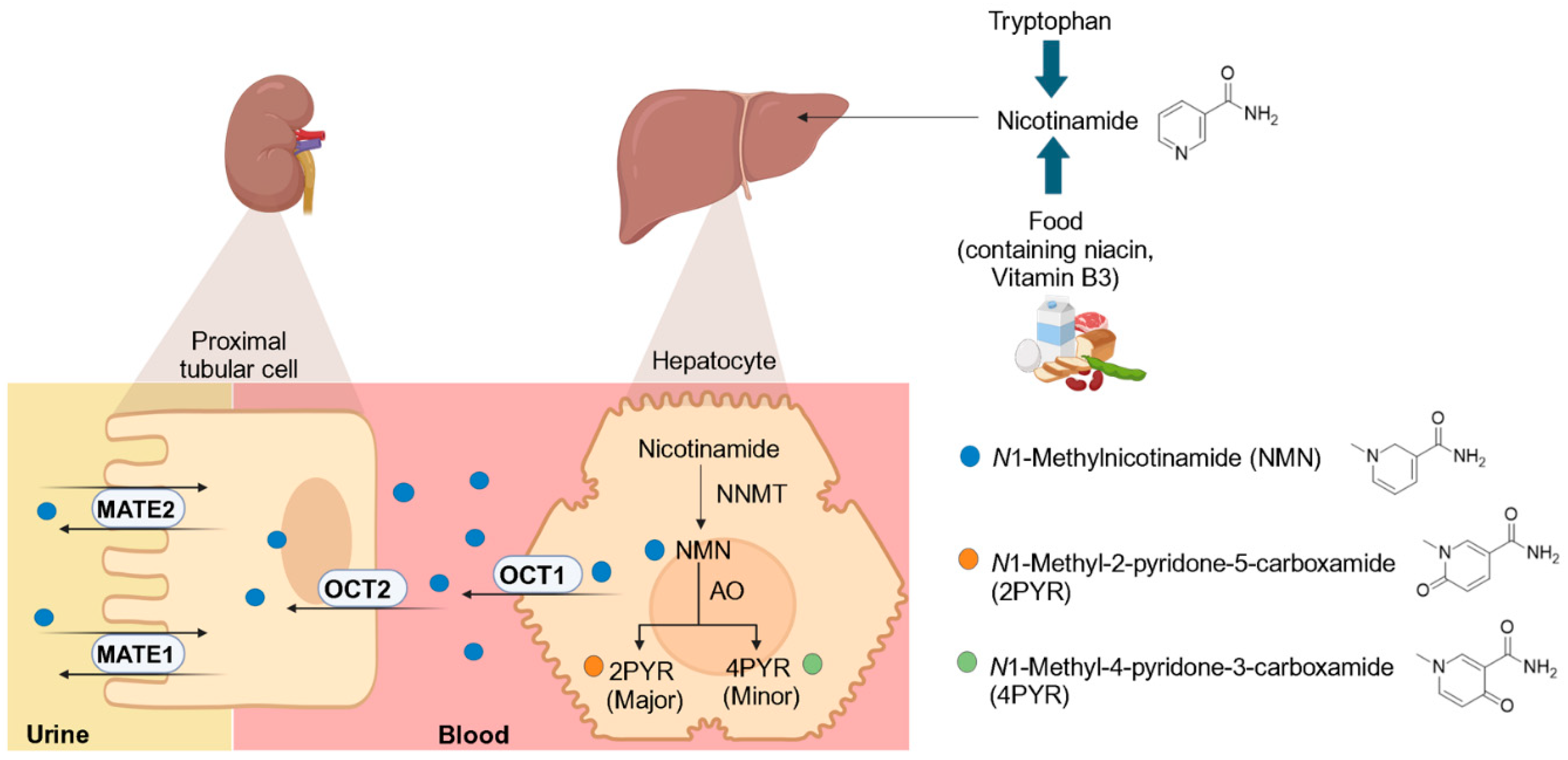
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. A Literature Analysis of the Effect of OCT Inhibition on NMN Plasma Concentration
2.3. N1-Methylnicotinamide Uptake by HEK293-OCT1-Transfected Cells
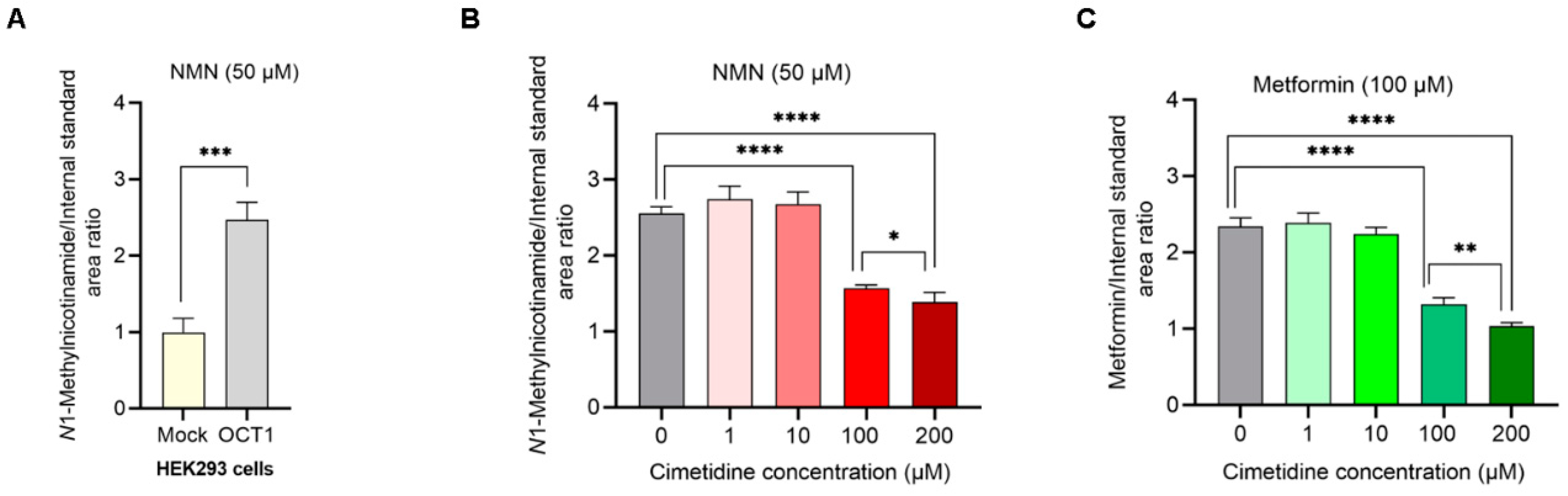
2.4. Quantification of OCT1 Uptake of NMN in the Presence and Absence of Cimetidine Using HEK293 Overexpressing OCT1
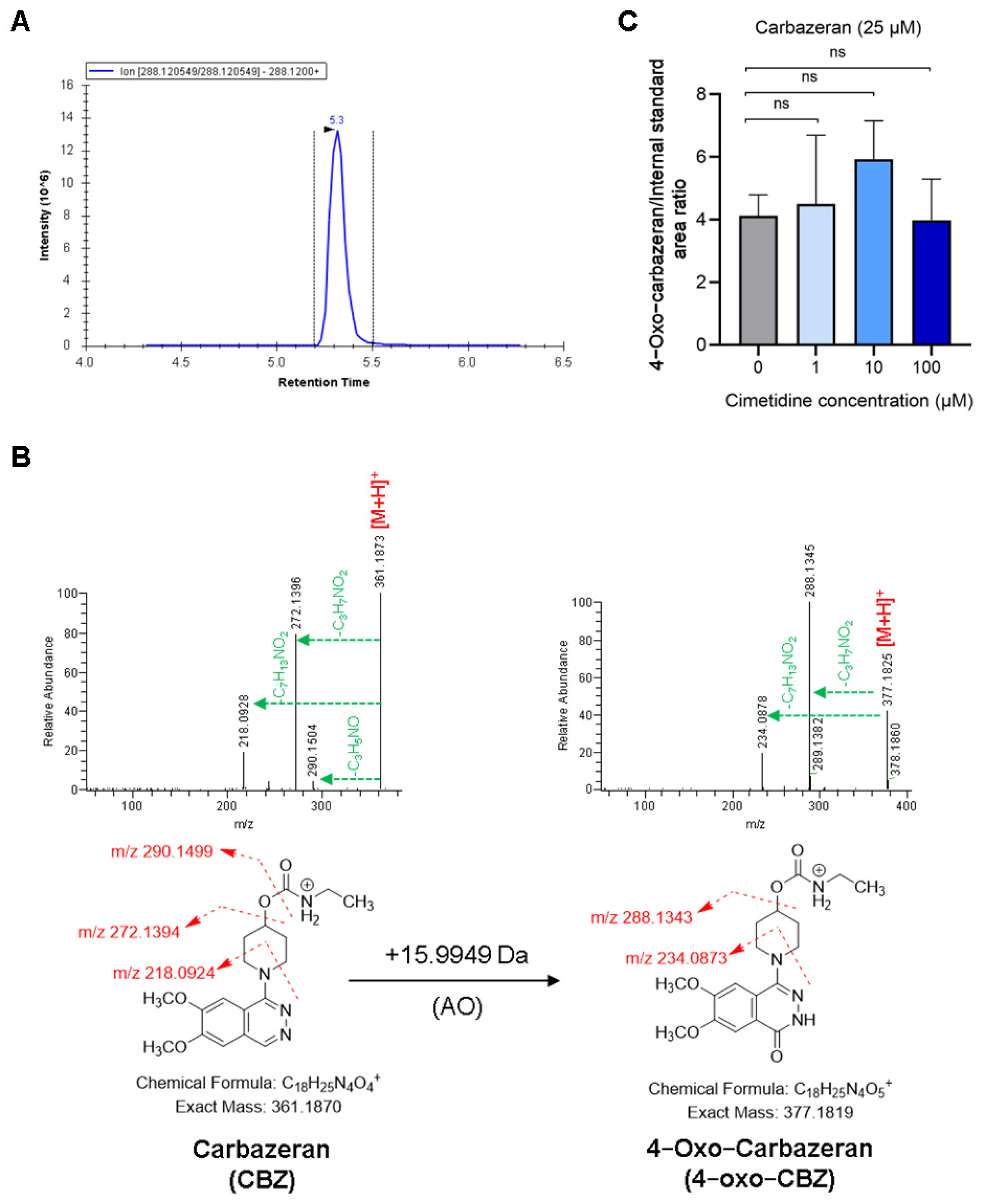
2.5. Quantification of Carbazeran Metabolism by Aldehyde Oxidase in the Presence and Absence of Cimetidine Using Human Liver Cytosol (HLC)
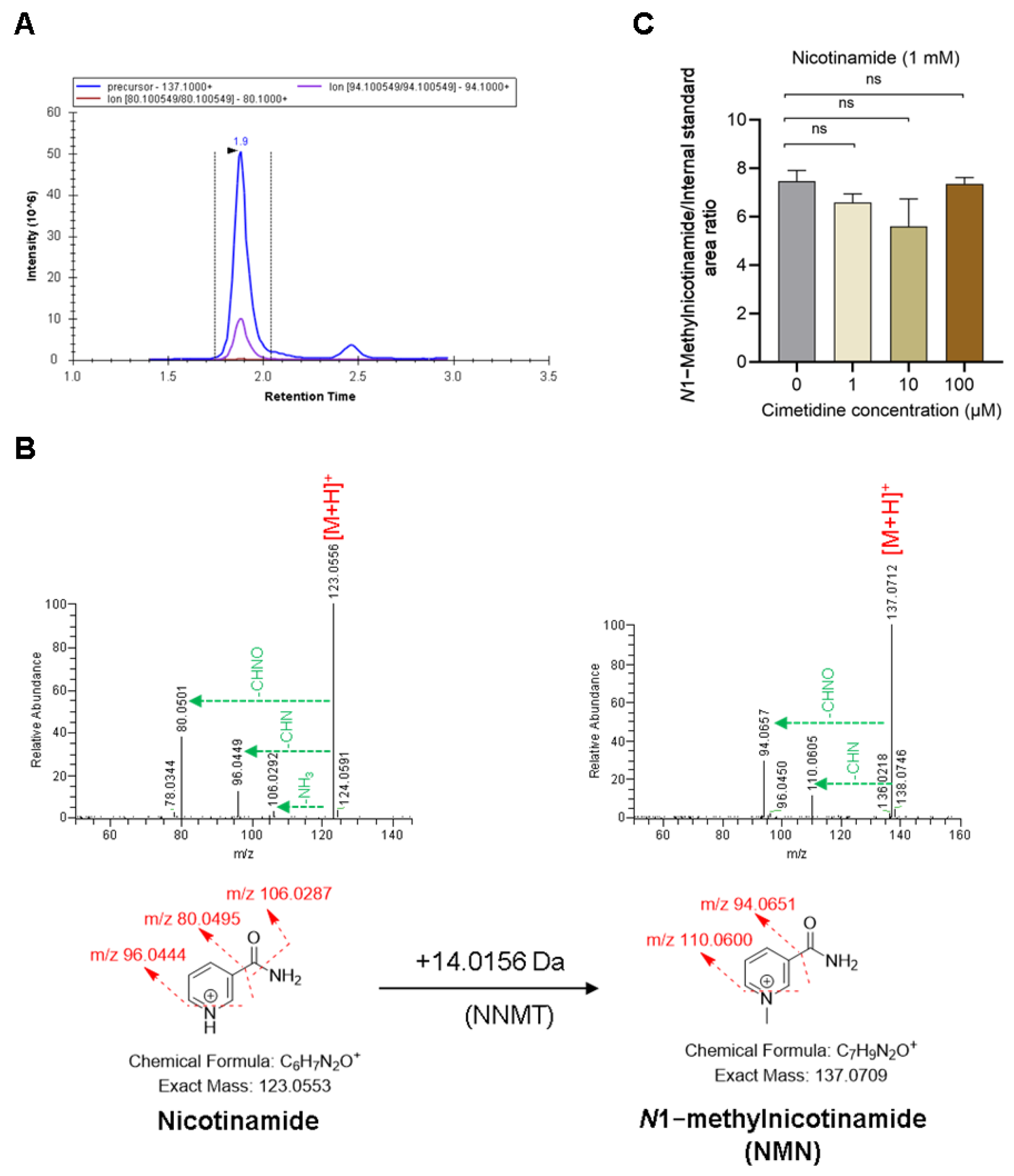
2.6. Quantification of NMN Formation by NNMT in the Presence and Absence of Cimetidine Using Recombinant Human NNMT
2.7. Targeted LC–MS/MS Analysis of Metabolites
2.8. Structural Confirmation of Metabolites Using High Resolution-Mass Spectrometry
2.9. Statistical Analysis
3. Results
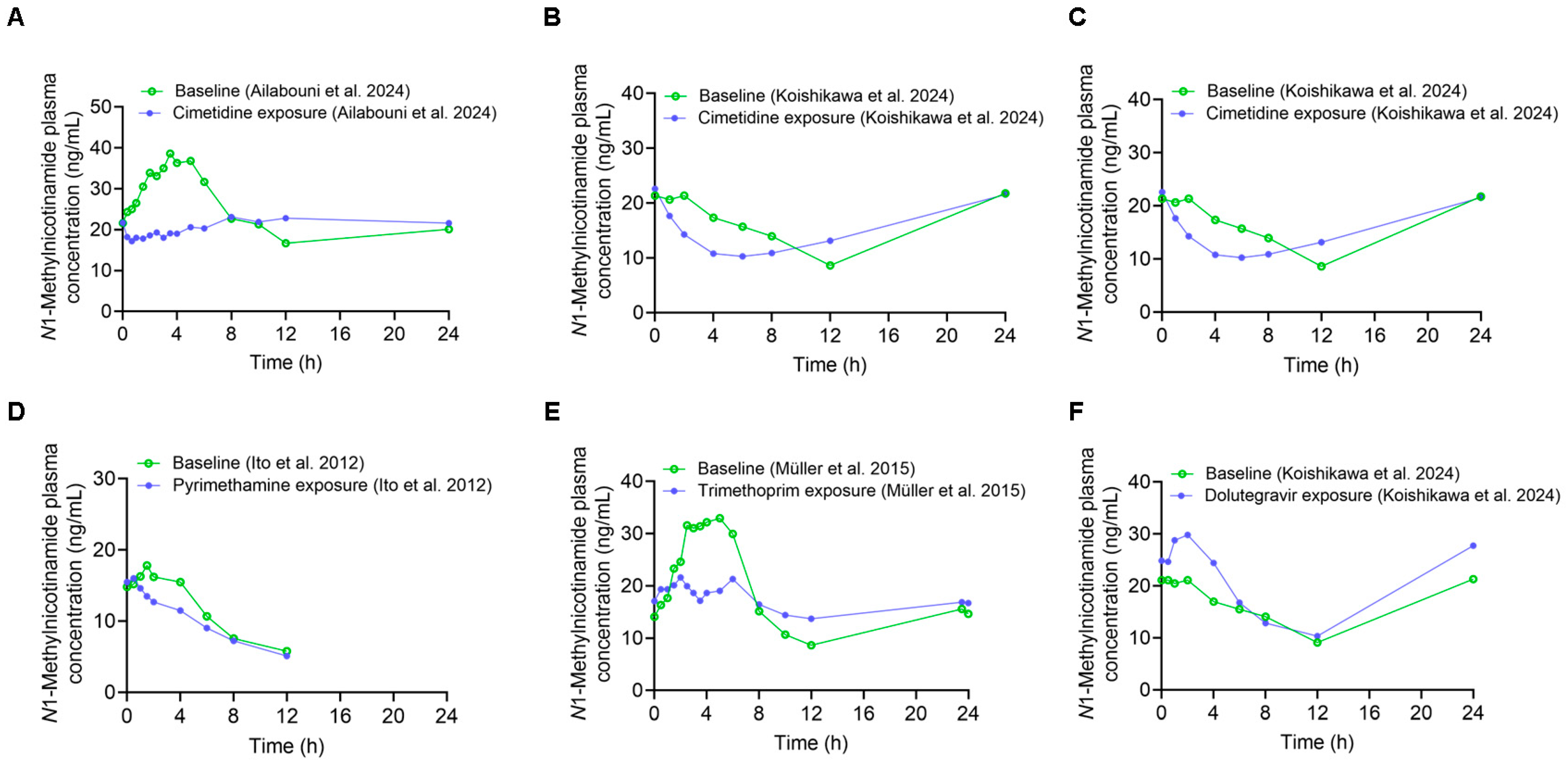
3.1. Effect of OCT Inhibition on NMN Pharmacokinetics
3.2. NMN Is a Substrate of OCT1; Cimetidine Inhibits NMN and Metformin Uptake Mediated by OCT1
3.3. Cimetidine Does Not Affect AO-Mediated Metabolism of CBZ and NMN
3.4. Cimetidine Does Not Affect NNMT Activity-Mediated Formation of NMN
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galetin, A.; Brouwer, K.L.R.; Tweedie, D.; Yoshida, K.; Sjöstedt, N.; Aleksunes, L.; Chu, X.; Evers, R.; Hafey, M.J.; Lai, Y.; et al. Membrane Transporters in Drug Development and as Determinants of Precision Medicine. Nat. Rev. Drug Discov. 2024, 23, 255–280. [Google Scholar] [CrossRef]
- Feng, B.; LaPerle, J.L.; Chang, G.; Varma, M.V. Renal Clearance in Drug Discovery and Development: Molecular Descriptors, Drug Transporters and Disease State. Expert Opin. Drug Metab. Toxicol. 2010, 6, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Sharma, A.; König, J.; Fromm, M.F. Biomarkers for in Vivo Assessment of Transporter Function. Pharmacol. Rev. 2018, 70, 246–277. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Chu, X.; Cook, J.A.; Custodio, J.M.; Galetin, A.; Giacomini, K.M.; Lee, C.A.; Paine, M.F.; Ray, A.S.; Ware, J.A.; et al. ITC Commentary on Metformin Clinical Drug–Drug Interaction Study Design That Enables an Efficacy- and Safety-Based Dose Adjustment Decision. Clin. Pharmacol. Ther. 2018, 104, 781–784. [Google Scholar] [CrossRef]
- Chu, X.; Chan, G.H.; Evers, R. Identification of Endogenous Biomarkers to Predict the Propensity of Drug Candidates to Cause Hepatic or Renal Transporter-Mediated Drug-Drug Interactions. J. Pharm. Sci. 2017, 106, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Madari, S.; Huang, F. Utilising Endogenous Biomarkers in Drug Development to Streamline the Assessment of Drug–Drug Interactions Mediated by Renal Transporters: A Pharmaceutical Industry Perspective. Clin. Pharmacokinet. 2024, 63, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Mathialagan, S.; Feng, B.; Rodrigues, A.D.; Varma, M.V.S. Drug-Drug Interactions Involving Renal OCT2/MATE Transporters: Clinical Risk Assessment May Require Endogenous Biomarker-Informed Approach. Clin. Pharmacol. Ther. 2021, 110, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Nitta, K.; Tayama, Y.; Tanoue, C.; Sugihara, K.; Inoue, T.; Horie, T.; Ohta, S. Aldehyde Oxidase-Catalyzed Metabolism of N1-Methylnicotinamide in Vivo and in Vitro in Chimeric Mice with Humanized Liver. Drug Metab. Dispos. 2008, 36, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kusuhara, H.; Kumagai, Y.; Moriyama, Y.; Inoue, K.; Kondo, T.; Nakayama, H.; Horita, S.; Tanabe, K.; Yuasa, H.; et al. N-Methylnicotinamide Is an Endogenous Probe for Evaluation of Drug-Drug Interactions Involving Multidrug and Toxin Extrusions (MATE1 and MATE2-K). Clin. Pharmacol. Ther. 2012, 92, 635–641. [Google Scholar] [CrossRef]
- Müller, F.; Pontones, C.A.; Renner, B.; Mieth, M.; Hoier, E.; Auge, D.; Maas, R.; Zolk, O.; Fromm, M.F. N1-Methylnicotinamide as an Endogenous Probe for Drug Interactions by Renal Cation Transporters: Studies on the Metformin-Trimethoprim Interaction. Eur. J. Clin. Pharmacol. 2015, 71, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Hohl, K.; Keller, S.; Schmidt-Gerets, S.; Deutsch, B.; Schuler-Metz, A.; Fromm, M.F.; Stopfer, P.; Gessner, A. N1-Methylnicotinamide as Biomarker for MATE-Mediated Renal Drug–Dug Interactions: Impact of Cimetidine, Rifampin, Verapamil, and Probenecid. Clin. Pharmacol. Ther. 2023, 113, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Kimoto, E.; Luo, L.; Mathialagan, S.; Horlbogen, L.M.; Ramanathan, R.; Wood, L.S.; Johnson, J.G.; Le, V.H.; Vourvahis, M.; et al. Identification of Appropriate Endogenous Biomarker for Risk Assessment of Multidrug and Toxin Extrusion Protein-Mediated Drug-Drug Interactions in Healthy Volunteers. Clin. Pharmacol. Ther. 2021, 109, 507–516. [Google Scholar] [CrossRef]
- Ailabouni, A.S.; Singh, D.K.; Thakur, A.; Paine, M.F.; Boone, E.C.; Gaedigk, A.; Prasad, B. Quantitative Contributions of Hepatic and Renal Organic Cation Transporters to the Clinical Pharmacokinetic Cimetidine-Metformin Interaction. bioRxiv, 2024; preprint. [Google Scholar] [CrossRef]
- Koishikawa, T.; Fujiwara, K.; Taskar, K.; Zamek-Gliszczynski, M.J.; Yoshida, K.; Chu, X.; Hirabayashi, H.; Mao, J.; Rockich, K.; Takashima, T.; et al. Effects of Cimetidine and Dolutegravir on the Endogenous Drug–Drug Interaction Biomarkers for Organic Cation Transporter 2 and Multidrug and Toxin Extrusion Protein 1 in Healthy Volunteers. Clin. Pharmacol. Ther. 2024, 117, 523–533. [Google Scholar] [CrossRef]
- Subash, S.; Singh, D.K.; Ahire, D.S.; Khojasteh, S.C.; Murray, B.P.; Zientek, M.A.; Jones, R.S.; Kulkarni, P.; Smith, B.J.; Heyward, S.; et al. Dissecting Parameters Contributing to the Underprediction of Aldehyde Oxidase-Mediated Metabolic Clearance of Drugs. Drug Metab. Dispos. 2023, 51, 1362–1371. [Google Scholar] [CrossRef]
- Behera, D.; Pattem, R.; Gudi, G. Effect of Commonly Used Organic Solvents on Aldehyde Oxidase-Mediated Vanillin, Phthalazine and Methotrexate Oxidation in Human, Rat and Mouse Liver Subcellular Fractions. Xenobiotica 2014, 44, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Van Haren, M.J.; Sastre Toraño, J.; Sartini, D.; Emanuelli, M.; Parsons, R.B.; Martin, N.I. A Rapid and Efficient Assay for the Characterization of Substrates and Inhibitors of Nicotinamide N-Methyltransferase. Biochemistry 2016, 55, 5307–5315. [Google Scholar] [CrossRef]
- International Council for Harmonisation. M12 Drug Interaction Studies Guidance for Industry ICH-Multidisciplinary. 2024. Available online: https://database.ich.org/sites/default/files/ICH_M12_Step4_Guideline_2024_0521.pdf (accessed on 25 May 2024).
- Feng, B.; Varma, M.V. Evaluation and Quantitative Prediction of Renal Transporter-Mediated Drug-Drug Interactions. J. Clin. Pharmacol. 2016, 56, S110–S121. [Google Scholar] [CrossRef]
- Chu, X.; Bleasby, K.; Chan, G.H.; Nunes, I.; Evers, R. The Complexities of Interpreting Reversible Elevated Serum Creatinine Levels in Drug Development: Does a Correlation with Inhibition of Renal Transporters Exist? Drug Metab. Dispos. 2016, 44, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical Studies on Drug–Drug Interactions Involving Metabolism and Transport: Methodology, Pitfalls, and Interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Mathialagan, S.; Bi, Y.-A.; Costales, C.; Kalgutkar, A.S.; Rodrigues, A.D.; Varma, M.V.S. Nicotinic Acid Transport into Human Liver Involves Organic Anion Transporter 2 (SLC22A7). Biochem. Pharmacol. 2020, 174, 113829. [Google Scholar] [CrossRef] [PubMed]
- Musfeld, C.; Me Biollaz, J.; Bélaz, N.; Kesselring, U.W.; Decosterd, L.A. Validation of an HPLC Method for the Determination of Urinary and Plasma Levels of N 1-Methylnicotinamide, an Endogenous Marker of Renal Cationic Transport and Plasma Flow. J. Pharm. Biomed. Anal. 2001, 24, 391–404. [Google Scholar] [CrossRef]
- Weber, W.; Toussaint, S.; Looby, M.; Nitz, M.; Kewitz, H. System Analysis in Multiple Dose Kinetics: Evidence for Saturable Tubular Reabsorption of the Organic Cation N1-Methylnicotinamide in Humans. J. Pharmacokinet. Biopharm. 1991, 19, 553–574. [Google Scholar] [CrossRef]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human Liver Nicotinamide N-Methyltransferase: CDNA Cloning, Expression, and Biochemical Characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef]
- Droździk, M.; Oswald, S.; Droździk, A. Extrahepatic Drug Transporters in Liver Failure: Focus on Kidney and Gastrointestinal Tract. Int. J. Mol. Sci. 2020, 21, 5737. [Google Scholar] [CrossRef]
- Elsby, R.; Chidlaw, S.; Outteridge, S.; Pickering, S.; Radcliffe, A.; Sullivan, R.; Jones, H.; Butler, P. Mechanistic in Vitro Studies Confirm That Inhibition of the Renal Apical Efflux Transporter Multidrug and Toxin Extrusion (MATE) 1, and Not Altered Absorption, Underlies the Increased Metformin Exposure Observed in Clinical Interactions with Cimetidine, Trimethoprim or Pyrimethamine. Pharmacol. Res. Perspect. 2017, 5, e00357. [Google Scholar] [PubMed]
- Wright, S.H.; Secomb, T.W. Novel Method for Kinetic Analysis Applied to Transport by the Uniporter OCT2. Am. J. Physiol. Renal Physiol. 2022, 323, F370–F387. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.J.; Savina, P.M.; Generaux, G.T.; Tracey, H.; Humphreys, J.E.; Kanaoka, E.; Webster, L.O.; Harmon, K.A.; Clarke, J.D.; Polli, J.W. In Vitro Investigations into the Roles of Drug Transporters and Metabolizing Enzymes in the Disposition and Drug Interactions of Dolutegravir, a Hiv Integrase Inhibitor. Drug Metab. Dispos. 2013, 41, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Song, I.H.; Zong, J.; Borland, J.; Jerva, F.; Wynne, B.; Zamek-Gliszczynski, M.J.; Humphreys, J.E.; Bowers, G.D.; Choukour, M. The Effect of Dolutegravir on the Pharmacokinetics of Metformin in Healthy Subjects. J. Acquir. Immune. Defic. Syndr. 2016, 72, 400–407. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef]
- Türk, D.; Müller, F.; Fromm, M.F.; Selzer, D.; Dallmann, R.; Lehr, T. Renal Transporter-Mediated Drug-Biomarker Interactions of the Endogenous Substrates Creatinine and N1-Methylnicotinamide: A PBPK Modeling Approach. Clin. Pharmacol. Ther. 2022, 112, 687–698. [Google Scholar] [CrossRef]
- Subash, S.; Singh, D.K.; Ahire, D.; Khojasteh, S.C.; Murray, B.P.; Zientek, M.A.; Jones, R.S.; Kulkarni, P.; Zubair, F.; Smith, B.J.; et al. Ontogeny of Human Liver Aldehyde Oxidase: Developmental Changes and Implications for Drug Metabolism. Mol. Pharm. 2024, 21, 2740–2750. [Google Scholar] [CrossRef] [PubMed]
- Renwick, A.B.; Ball, S.E.; Tredger, J.M.; Price, R.J.; Walters, D.G.; Kao, J.; Scatina, J.A.; Lake, B.G. Inhibition of Zaleplon Metabolism by Cimetidine in the Human Liver: In Vitro Studies with Subcellular Fractions and Precision-Cut Liver Slices. Xenobiotica 2002, 32, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Huynh, P.; Allen, M.C.; Beedham, C. Human Liver Aldehyde Oxidase: Inhibition by 239 Drugs. J. Clin. Pharmacol. 2004, 44, 7–19. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetic Measures | Precipitant Dosing Regimen | AUC0–24 h (µg·h/L) | AUC0–12 h (µg·h/L) | AUC0–4 h (µg·h/L) | Cmax (µg/L) | CLr (L/h) | Reference |
|---|---|---|---|---|---|---|---|
| Baseline | 400 mg single oral dose | 579.2 | 348.7 | 127.5 | 50.5 | 12.6 | [13] |
| Cimetidine exposure | 530.6 | 253.6 | 74.9 | 31.3 | 14.0 | ||
| Ratio (cimetidine exposure/baseline) | 0.92 | 0.73 | 0.59 | 0.62 | 1.11 | ||
| Baseline | 400 mg single oral dose | 370.9 | 188.6 | 80.7 | N/P | 16.7 | [14] |
| Cimetidine exposure | 359.6 | 151.4 | 61.1 | N/P | 15.5 | ||
| Ratio (cimetidine exposure/baseline) | 0.97 | 0.80 | 0.76 | 0.93 | |||
| Baseline | 400 mg oral dose five times on day 1 | 363.4 | 220.8 | 105.7 | 31.5 | 17.4 | [11] |
| Cimetidine exposure | 313.3 | 155.0 | 68.0 | 28.8 | 12.6 | ||
| Ratio (cimetidine exposure/baseline) | 0.86 | 0.70 | 0.64 | 0.91 | 0.72 | ||
| Baseline | 200 mg oral dose twice daily for 5 days | 396.9 | 253.8 | 99.6 | 40.1 | 15.5 | [10] |
| Trimethoprim exposure | 397.1 | 212.9 | 77.0 | 24.7 | 11.3 | ||
| Ratio (trimethoprim exposure/baseline) | 1.00 | 0.84 | 0.77 | 0.62 | 0.73 | ||
| Baseline | 50 mg single oral dose | N/A | 136 | 64.1 | N/P | 24.18 | [9] |
| Pyrimethamine exposure | N/A | 114 | 53.3 | N/P | 7.14 | ||
| Ratio (pyrimethamine exposure/baseline) | N/A | 0.84 | 0.83 | N/P | 0.30 | ||
| Baseline | 50 mg oral dose twice daily on day 1 | 371.0 | 188.3 | 79.9 | N/P | 16.7 | [14] |
| Dolutegravir exposure | 455.0 | 226.4 | 109.3 | N/P | 14.0 | ||
| Ratio (dolutegravir exposure/baseline) | 1.23 | 1.20 | 1.37 | N/P | 0.84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ailabouni, A.S.; Vijaywargi, G.; Subash, S.; Singh, D.K.; Gaborik, Z.; Prasad, B. Is N1-Methylnicotinamide a Good Organic Cation Transporter 2 (OCT2) Biomarker? Metabolites 2025, 15, 80. https://doi.org/10.3390/metabo15020080
Ailabouni AS, Vijaywargi G, Subash S, Singh DK, Gaborik Z, Prasad B. Is N1-Methylnicotinamide a Good Organic Cation Transporter 2 (OCT2) Biomarker? Metabolites. 2025; 15(2):80. https://doi.org/10.3390/metabo15020080
Chicago/Turabian StyleAilabouni, Anoud Sameer, Gautam Vijaywargi, Sandhya Subash, Dilip Kumar Singh, Zsuzsanna Gaborik, and Bhagwat Prasad. 2025. "Is N1-Methylnicotinamide a Good Organic Cation Transporter 2 (OCT2) Biomarker?" Metabolites 15, no. 2: 80. https://doi.org/10.3390/metabo15020080
APA StyleAilabouni, A. S., Vijaywargi, G., Subash, S., Singh, D. K., Gaborik, Z., & Prasad, B. (2025). Is N1-Methylnicotinamide a Good Organic Cation Transporter 2 (OCT2) Biomarker? Metabolites, 15(2), 80. https://doi.org/10.3390/metabo15020080