Abstract
Escherichia coli is a widely used microorganism in biotechnological processes. An obvious goal for current scientific and technical research in this field is the search for new tools to optimize productivity. Usually glucose is the preferred carbon source in biotechnological applications. In E. coli, glucose is taken up by the phosphoenolpyruvate-dependent glucose phosphotransferase system (PTS). The regulation of the ptsG gene for the glucose transporter is very complex and involves several regulatory proteins. Recently, a novel posttranscriptional regulation system has been identified which consists of a small regulatory RNA SgrS and a small regulatory polypeptide called SgrT. During the accumulation of glucose-6-phosphate or fructose-6-phosphate, SgrS is involved in downregulation of ptsG mRNA stability, whereas SgrT inhibits glucose transport activity by a yet unknown mechanism. The function of SgrS has been studied intensively. In contrast, the knowledge about the function of SgrT is still limited. Therefore, in this paper, we focused our interest on the regulation of glucose transport activity by SgrT. We identified the SgrT target sequence within the glucose transporter and characterized the interaction in great detail. Finally, we suggest a novel experimental approach to regulate artificially carbohydrate uptake in E. coli to minimize metabolic overflow in biotechnological applications.
1. Introduction
With its fast growth and simple cultivation Escherichia coli is a widely used microorganism in biotechnological processes and in industrial microbiology. One of the most important applications of recombinant DNA technology is the genetic manipulation of E. coli K-12 for the production of human insulin [1]. Modified E. coli strains are also currently used for the synthesis of different enzymes, amino acids and other peptide hormones. To maximize productivity, i.e., the yield in relation to duration and costs, it is essential to permanently optimize the biotechnological process. One major problem during high density growth of E. coli K-12 is the production and excretion of acetate, which affects growth and recombinant protein expression [2,3]. To circumvent this, different growth strategies [3] have been applied as well as targeted changes in central carbon metabolism [2,4] or control of the glucose transport process has been modified [5,6]. Especially the latter approach seems to be very helpful since acetate excretion mainly occurs when the transport rate exceeds the metabolism which causes a temporal metabolic imbalance.
In E. coli, glucose is taken up by the phosphoenolpyruvate (PEP)-dependent glucose-phosphotransferase-system (Glc-PTS) [7]. Phosphotransferase systems usually consist of two cytoplasmic energy-coupling proteins, Enzyme I (EI, gene ptsI) and Histidine-containing protein (HPr, gene ptsH), and in particular for E. coli K-12 of a range of more than 20 different carbohydrate specific Enzymes II (EIIs), which catalyze concomitant carbohydrate transport and phosphorylation [8]. The first step in the PTS-typical phosphorylation-chain is catalyzed by EI, a PEP-dependent protein-kinase. The use of PEP, an intermediate of glycolysis, as a phosphoryl group donor couples tightly carbohydrate transport and metabolism. In the case of the Glc-PTS, the phosphate group is subsequently transferred from EI~P to HPr, from HPr~P to the soluble EIIAGlc (sometimes also called EIIACrr, gene crr), and finally from EIIAGlc~P to the glucose-specific membrane protein EIICBGlc (gene ptsG), which is responsible for glucose uptake and phosphorylation. EIICBGlc (50.7 kDa) consists of two functional domains, the membrane bound EIICGlc domain (41.1 kDa) and the cytosolic EIIBGlc domain (9.6 kDa). The EIICGlc domain forms a stable homodimer in the membrane and is responsible for glucose uptake, whereas the EIIBGlc is located in the cytoplasm and phosphorylates the glucose [9]. Both domains are connected through a flexible linker. The linker is surface exposed, since a proteolytic cleavage within the linker is possible [10]. Phosphorylation of EIICBGlc protects against protease cleavage, suggesting a conformational change of this region during glucose uptake [10]. The linker shows the highly conserved amino acid sequence KTPGRED (aa 382-388) which is present in most of the PTS transport proteins of the glucose/N-acetyl-glucosamine family. The function of this motif was unclear so far [7]. This motif appears to be nonessential for transport, since alanine substitutions show no or only a slight effect with the exception of EIICBGlcG385A which exhibited a highly reduced phosphorylation activity of less than 10% of wild type activity [10,11]. Only a complete deletion of this sequence led to a total loss of transport and phosphorylation activity [12].
Regulation of the ptsG gene for the EIICBGlc is very complex and occurs both at the levels of transcription and posttranscriptional control. The major specific regulator of ptsG expression is the repressor Mlc (mnemonic for makes large colonies, previously DgsA, gene dgsA), which is inactivated by glucose in the medium. In contrast to other repressors, induction of Mlc is not catalyzed by direct binding of glucose, or by any other small molecular inducer. Instead, as part of an unusual regulatory mechanism, the membrane-bound EIICBGlc binds Mlc, but only when it is in its dephosphorylated form. Thus, in the absence of glucose, Mlc binds to its target promoter/operator ptsGop, while in the presence of glucose, the dephosphorylated EIICBGlc sequesters the repressor away from its operator, allowing enhanced ptsG transcription [13,14,15,16]. Besides this main regulation via the glucose repressor Mlc, several other global factors were identified. These are cAMP-CAP [17], ArcA [18], SoxS [19], Fis [20] and two alternating sigma factors σ32 [21] and σS [22]. In addition to these transcriptional regulation mechanisms, a posttranscriptional regulation system, the so-called sgrRST-system [23,24], was identified as regulating ptsG mRNA stability as well as transport activity of EIICBGlc. Accumulation of glucose-6-phosphate (Glc6-P) or fructose-6-phosphate (Fru6-P) in the cell activates the transcriptional activator SgrR which, in turn, is responsible for the activation of the small regulatory sRNA SgrS [24]. SgrS itself has two functions. On the one hand, it is capable of forming Hfq-dependent RNA-RNA hybrids with the ptsG mRNA, a first step in RNaseE dependent specific degradation of this mRNA. On the other hand, the sgrS gene encodes a small protein of 43 amino acids called SgrT which is responsible for downregulation of glucose transport [25].
The recent discoveries of these two posttranscriptional regulatory mechanisms provide new approaches to control glucose uptake under various growth conditions. For example Negrete et al. managed to reduce acetate excretion of glucose fermenting E. coli cells by overexpressing SgrS [26]. Likewise, exclusive overproduction of SgrT led to a drastic reduction of cell growth in minimal medium with glucose as a sole carbon source [25]. This gave the first hint that the Glc-PTS might be a direct target of SgrT and that the functions of SgrS and SgrT are redundant. Subsequently, Gabor et al. [27] repeated this growth experiment using minimal medium with sucrose as a single carbon source. For this experiment, an E. coli derivative was used, which shares all the components of the typical PTS cascade with the Glc-PTS (EI, HPr, EIIAGlc) with the exception of the sucrose specific EIIBCScr [28]. This transporter protein like the EIICBGlc belongs to the glucose/N-acetyl-glucosamine–sucrose/ß-glucosides superfamily of EII proteins [29]. However, the sucrose specific transporter has a different order of the two functional domains and lacks a conserved KTPGRED motif in the linker region between these two sites. Overproduction of SgrT did not interfere with cell growth in minimal medium with sucrose providing a first hint that indeed EIICBGlc and no other component of the Glc-PTS might be the SgrT target [27].
In this study we focused our interest on the regulation of EIICBGlc activity by SgrT. We identified the SgrT target sequence within EIICBGlc and characterized the interaction between the glucose transporter and the small regulatory peptide in great detail. This may eventually lead to novel approaches to minimize metabolic overflow and thus improve the feasibility of the use of E. coli in biotechnological applications.
2. Results and Discussion
2.1. SgrT Binds to Dephosphorylated EIICBGlc in in vivo Crosslinking assays
In order to test the assumption of a direct protein–protein interaction between SgrT and EIICBGlc, we performed an in vivo crosslinking experiment with paraformaldehyde. With this method, even weak in vivo interactions between two proteins are detectable in the case that the proteins are in close proximity to each other (2 Å or less) [30]. To identify the two interaction partners in subsequent Western blots, both proteins were tagged with different flags (EIICBGlc-5His, SgrT-3HA). Both tagged proteins were fully functional in complementation assays, e.g., glucose transport in a ptsG deletion background in the case of the EIICBGlc-5His protein or reduction of growth in minimal medium with glucose as a sole carbon source in the case of the SgrT-3HA peptide [27]. The two proteins were expressed in a double deletion strain (JKA12) and the cells were treated with paraformaldehyde. The cells were disrupted by sonification, membrane proteins were solubilized and EIICBGlc-His and proteins binding to it were purified with Ni-NTA agarose. The resulting Western blot analysis showed a strong copurification of SgrT and thus an interaction of SgrT and EIICBGlc in the presence of glucose in the medium (Figure 1A, lane 2). Interestingly, only a very weak interaction could be detected in cells grown in the absence of glucose (Figure 1A, lane 1). No signals for SgrT-3HA were obtained in a sgrTHA deletion background (Figure 1A, lane 3) or in a sgrTHA+/ ptsGHis (Figure 1A, lane 4) deletion strain. The latter result demonstrates that the detection of SgrT-3HA clearly depends on the presence of EIICBGlc.
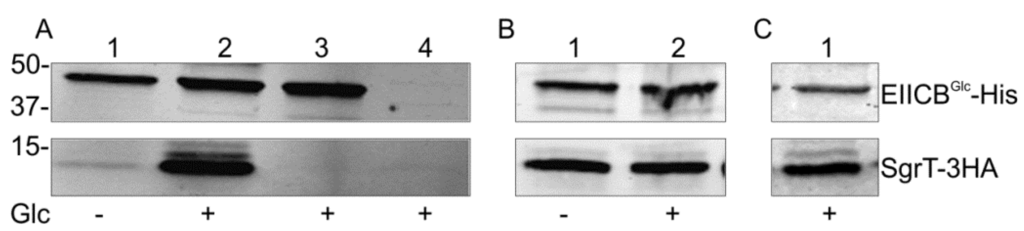
Figure 1.
Crosslinking experiments with EIICBGlc and SgrT in different genetic backgrounds.
Figure 1.
Crosslinking experiments with EIICBGlc and SgrT in different genetic backgrounds.
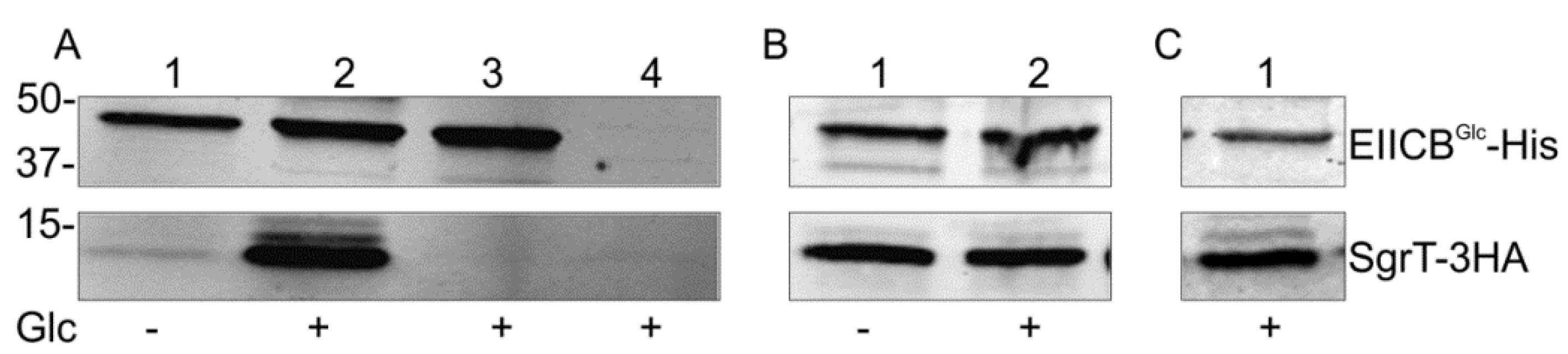
- (a) Lanes 1 and 2 show crosslinking experiments with strain JKA12 (LJ110ΔptsG::cat ΔsgrRST::neo) transformed with two plasmids expressing EIICBGlc-His (pRR48GH) and SgrT-3HA (pACYC184sgrT3HA). Cells were grown in the absence or presence of glucose as indicated; molecular weight markers are given on the left side (in kDa). The results show an interaction of SgrT and EIICBGlc in the presence of glucose. Control experiments are illustrated in lane 3 (JKA12 transformed with pRR48GH and pACYC184) and lane 4 (JKA12 transformed with pRR48 and pACYC184sgrT3HA). In both cases, no signals for SgrT-3HA could be observed.
- (b) Lanes 1 and 2 show crosslinking experiments with the ptsHIcrr deletion strain LJ140 transformed with pRR48GH and pACYC184sgrT3HA. Cells were grown in the absence or presence of glucose as indicated.
- (c) Lane 1 shows a crosslinking experiment with the dgsA deletion strain LJB17 transformed with pRR48GH and pACYC184sgrT3HA. Cells were grown in the presence of glucose. This result indicates an Mlc-independent interaction between EIICBGlc and SgrT.
Glucose uptake leads to a net dephosphorylation of EIICBGlc and to conformational changes of the transporter during the uptake process. To test whether dephosphorylation and no glucose induced conformational change of the transporter is sufficient for SgrT binding, this experiment was repeated in a ptsHIcrr deletion strain (LJ140), where no phosphorylation of EIICBGlc can occur. The results shown in Figure 1B indicate an interaction between SgrT and EIICBGlc both in the presence and in the absence of glucose, indicating that SgrT binds to dephosphorylated EIICBGlc with a much higher preference and that conformational changes of the EIICBGlc induced by glucose transport are not involved in SgrT binding. Dephosphorylated EIICBGlc also binds and sequesters the glucose repressor Mlc in the process of ptsG induction. To see whether SgrT binding to dephosphorylated EIICBGlc depends on the presence of Mlc, we repeated the crosslinking experiment in an mlc (dgsA) deletion background. As shown in Figure 1C, an Mlc-independent interaction between EIICBGlc and SgrT was detected. This finding supports the idea of a direct SgrT-EIICBGlc interaction.
2.2. SgrT Binds to Full Length EIICBGlc and to Its Truncated EIIC-Linker Derivative in Bimolecular Fluorescence Complementation Assays
The previous results showed that SgrT interacts with the unphosphorylated full-length EIICBGlc in crosslinking assays. In order to narrow the region of the EIICBGlc interaction side, we performed bimolecular fluorescence complementation assays [31] with different subdomains of the glucose transporter. In these assays, both proteins of interest are linked to one half of a green fluorescent protein (Gfp) protein. In case of interaction, both halves regenerate a fluorescent full-length protein.
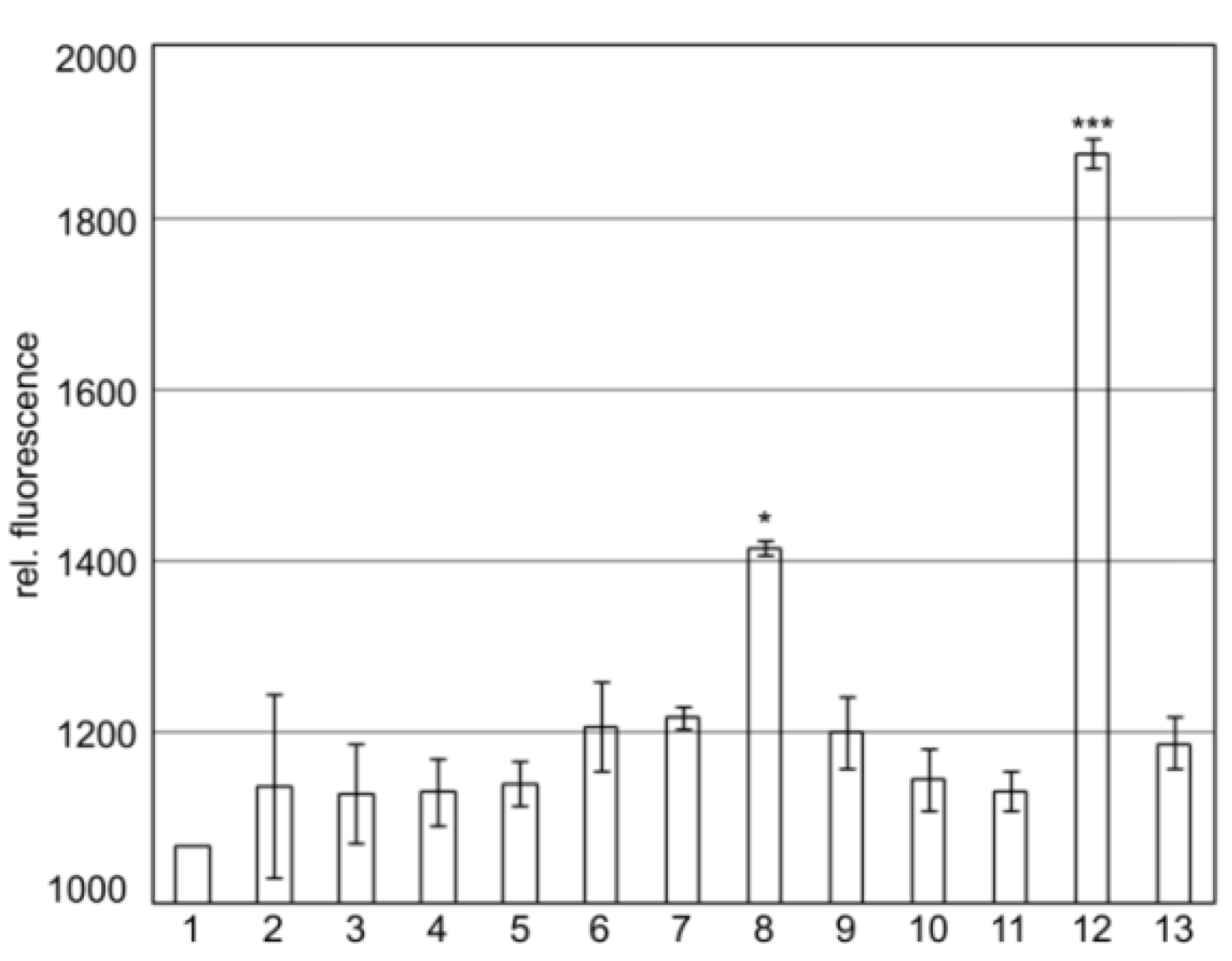
Results shown in Figure 2 indicate that SgrT interacts with the full-length EIICBGlc protein (Figure 2, lane 8) as well as with the EIICGlc-linker domain without EIIBGlc (Figure 2, lane 12). The interaction between SgrT and EIICGlc-linker is even higher compared to the full length protein. This might indicate that a deletion of the EIIBGlc-domain exposes the linker, which thus becomes a better target for SgrT. In contrast, no interaction between SgrT and the EIICGlc domain without the linker could be observed (Figure 2, lane 11). Interestingly, there was also no interaction between the soluble EIIBGlc with or without the linker domain and SgrT (Figure 2, lanes 9 and 10). This could be a hint that either the C-domain also plays at least some role in interaction or that a membrane environment is required for the interplay.
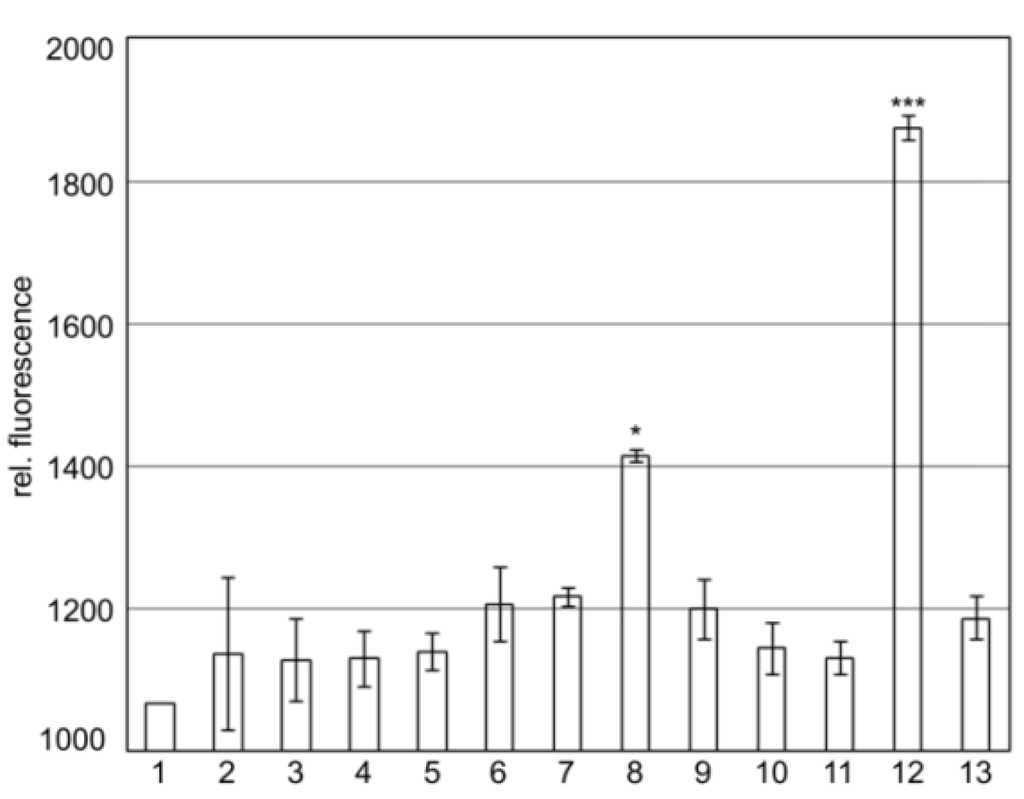
Figure 2.
Bimolecular fluorescence complementation assays with different EIICBGlc derivatives and SgrT.
Figure 2.
Bimolecular fluorescence complementation assays with different EIICBGlc derivatives and SgrT.
The relative fluorescence units were measured for different EIICBGlc derivatives and SgrT both fused to one half of the green fluorescent protein to determine the amounts of bimolecular fluorescence complementations. Strain JKA17 (BL21(λDE3)ΔptsG::cat) was transformed with various plasmids expressing different Gfp-fusion proteins. Equal amounts of cells were used and each culture was inoculated and measured at least three times. For determination of background fluorescence, a leucin-zipper fused to the N- or C-terminal part of GFP was used as follows: Z-NGFP (pET11a-Z-NGFP) and Z-CGFP (pMRBAD-Z-CGFP). For description of plasmid construction and experimental procedure, see experimental section.
Results are given for the following sample combinations:
1. Z-NGfp/EIICBGlc-CGfp; 2. SgrT-NGfp/Z-CGfp; 3. Z-NGfp/EIIBGlc-CGfp; 4. Z-NGfp/Linker-EIIBGlc-CGfp; 5. Z-NGfp/EIICGlc-CGfp; 6. Z-NGfp/EIICGlc-Linker-CGfp; 7. Z-NGfp/EIICGlc-Linker-P384R-CGfp; 8. SgrT-NGfp/EIICBGlcCGfp; 9. SgrT-NGfp/EIIBGlc-CGfp; 10. SgrT-NGfp/Linker-EIIBGlc-CGfp; 11. SgrT-NGfp/EIICGlc-CGfp; 12. SgrT-NGfp/EIICGlc-Linker-CGfp; 13. SgrT-NGfp/EIICGlc-Linker-P384R-CGfp.
The results indicate that there is relative background fluorescence up to 1200 units in control cultures (lanes 1 to 7). The same relative fluorescence was detected for the combinations of SgrT-NGfp with EIIBGlc-CGfp (lane 9), Linker-EIIBGlc-CGfp (lane 10), EIICGlc-CGfp (lane 11) and EIICGlc-linker-P384R-CGfp (lane 13), meaning that there is no interaction between SgrT and these EIICBGlc-domains. A significantly higher relative fluorescence was detected between SgrT-NGfp and EIICBGlc-CGfp (lane 8) and EIICGlc-linker-CGfp (lane 12), respectively. These results indicate an interaction between SgrT and the full-length protein EIICBGlc or the Linker-EIICGlc-domain, respectively. Significance levels: *p = 0.05, ***p = 0.001.
2.3. The KTPGRED Motif in the Linker Region of EIICBGlc is the Main SgrT Target Sequence
In a previously published experiment, we identified the single amino acid substitution P384R in EIICBGlc, which caused a complete release of SgrT inhibition during growth in minimal medium with glucose as a sole carbon source [27]. The amino acid P384 is located within the conserved KTPGRED motif. The function of this region was unknown until now, but it seems to play an important role in SgrT regulation. Accordingly, as indicated in Figure 2, lane 13, no bimolecular fluorescence complementation was detected for SgrT-NGfp and EIICGlc-linker-P384R-CGfp.
To identify other functionally important amino acid residues, we performed SgrT-EIICBGlc crosslinking assays with single amino acid substitutions in the KTPGRED motif of the glucose transporter. All amino acid residues of this motif were replaced by the small and hydrophobic amino acid residue alanine. In addition, EIICBGlc P384R was also reanalyzed in this test. The obtained EIICBGlc derivatives were capable of complementing a ptsG deletion strain on a MacConkey glucose (McCGlc) plate, even EIICBGlc G385A (data not shown). This indicates that under high glucose concentrations (1% in McC plates) the residual activity of all mutants is sufficient to complement transport activity and that all proteins are folded correctly. Similarly important is the fact that all proteins were stable and could be purified easily. Cells overexpressing SgrT and the respective EIICBGlc derivative were grown in rich medium in the presence of glucose and treated with paraformaldehyde. Subsequently, cells were disrupted and EIICBGlc-His was purified with Ni-NTA agarose. Respective SgrT co-purifications were visualized by Western blot analysis. As shown in Figure 3A the strongest effect was exhibited by the P384R substitution, which completely abolished the interaction between the two proteins. Strong effects were also caused by the substitutions T383A, P384A, G385A, R386A and E387A. Compared to the wild type protein almost no effects were obtained for the substitutions K382A and D388A. This might indicate that the crucial residues for the EIICBGlc - SgrT interaction are in the center of this sequence motif.
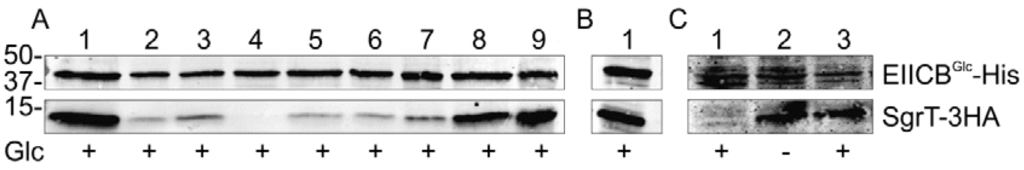
Figure 3.
Crosslinking experiments with different KTPGRED mutants of EIICBGlc and SgrT.
Figure 3.
Crosslinking experiments with different KTPGRED mutants of EIICBGlc and SgrT.
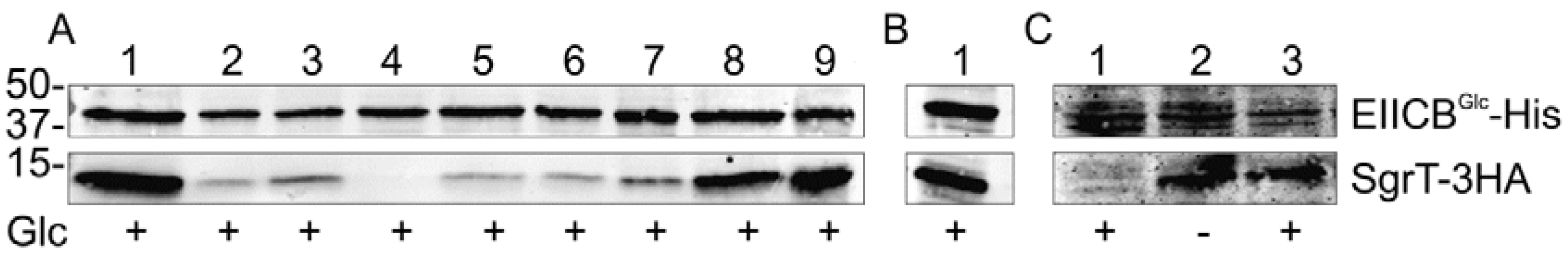
- (a) This part of the figure shows crosslinking experiments with strain JKA12 (LJ110ΔptsG::cat ΔsgrRST::neo) transformed with two plasmids expressing SgrT-3HA (pACYC184sgrT3HA) and wild type EIICBGlc-His (lane 9) or different EIICBGlc-His-derivatives (lanes 1-8). Cells were grown in the presence of glucose as indicated; molecular weight markers are given on the left side (in kDa). The following EIICBGlc derivatives were used in combination with SgrT-3HA:EIICBGlc-K382A-His; 2. EIICBGlc-T383A-His; 3. EIICBGlc-P384A-His; 4. EIICBGlc-P384R-His; 5. EIICBGlc-G385A-His; 6. EIICBGlc-R386A-His; 7. EIICBGlc-E387A-His; 8. EIICBGlc-D388A-His; 9. EIICBGlc-His (wild type). These results indicate that the crucial residues for the interaction between the two proteins are in the center of the KTPGRED motif.
- (b) Lane 1 shows a crosslinking experiment with strain JKA12 expressing SgrT-3HA (pACYC184sgrT3HA) and the so called “relaxed” mutant EIICBGlc-V12F-His (pRR48GH-V12F). Cells were grown in the presence of glucose. These results indicate an interaction between SgrT and the “relaxed” derivative of EIICBGlc.
- (c) This part of the figure shows crosslinking experiments between SgrT-3HA and the “locked in” mutant EIICBGlc-K150E-His (pRR48GH-K150E) in different genetic backgrounds. Lane 1 shows a sample of strain JKA12 expressing SgrT-3HA and EIICBGlc-K150E-His, lanes 2 and 3 exhibit samples of LJ140 expressing the same proteins. Cells were grown in the absence or presence of glucose as indicated. These results indicate no interaction between SgrT and EIICBGlcK150E in a PTS-positive strain, but a strong interaction in a ptsHIcrr deletion background.
The mutation P384R in EIICBGlc has previously been described to cause a so-called “relaxed” conformation [16], which allows a facilitated transport of substrates like mannose, glucosamine or fructose. The exact nature of the conformational difference, however, is still unclear. To test whether this “relaxed” conformation in general interferes with the SgrT interaction, we tested the EIICBGlcV12F derivative in the crosslinking assay. This mutation has also been attributed with properties which cause the same “relaxed” phenotype and thus belongs to the same class of mutants [10]. As shown in Figure 3B, in contrast to EIICBGlcP384R the unphosphorylated V12F derivative exhibited a strong interaction with SgrT, which means that a “relaxed” conformation per se has no influence on the interplay with this small regulatory peptide.
A different class of mutations, such as those caused by the substitution K150E, leads to a so-called “locked-in” conformation of EIICBGlc. In this case, the transporter can be phosphorylated but cannot transfer the phosphate group to the bound glucose molecule [10]. Thus, the transporter remains phosphorylated and cells carrying this mutation are not capable of transporting glucose by the Glc-PTS. Accordingly, no interaction between SgrT and EIICBGlcK150E could be detected in PTS-positive strains in the presence of glucose (Figure 3C, lane 1). In contrast, the introduction of a ptsHIcrr deletion, however, as for the wild type protein, caused a constitutive interaction between SgrT and EIICBGlcK150E (Figure 3C, lanes 2 and 3) which again emphasizes the strong correlation between the phosphorylation level and the ability to interact with SgrT.
2.4. Recruitment of SgrT to the Membrane by EIICBGlc Can Be Visualized By in vivo Fluorescence Microscopy
For further analysis of the interaction between SgrT and EIICBGlc we performed fluorescence microscopy to find out more about the distribution pattern of the two proteins in living cells. As shown in Figure 4, plasmid encoded EIICBGlc tagged with Gfp was homogeneously distributed in the cytoplasmic membrane (4B), whereas SgrT tagged with Gfp could be detected in an EIICBGlc-negative strain only in the cytosol (4D). In contrast, in E. coli ptsG+ cells that were grown in the presence of glucose, the localization of SgrT-Gfp clearly shifted to the membrane, which indicates a sequestration of SgrT by unphosphorylated EIICBGlc (4F). In accordance with the previously obtained results of the crosslinking experiments, EIICBGlcP384R, unlike the wild type protein, was not capable of sequestering SgrT-Gfp (4H), which, yet again, indicates the missing interaction between the two proteins.
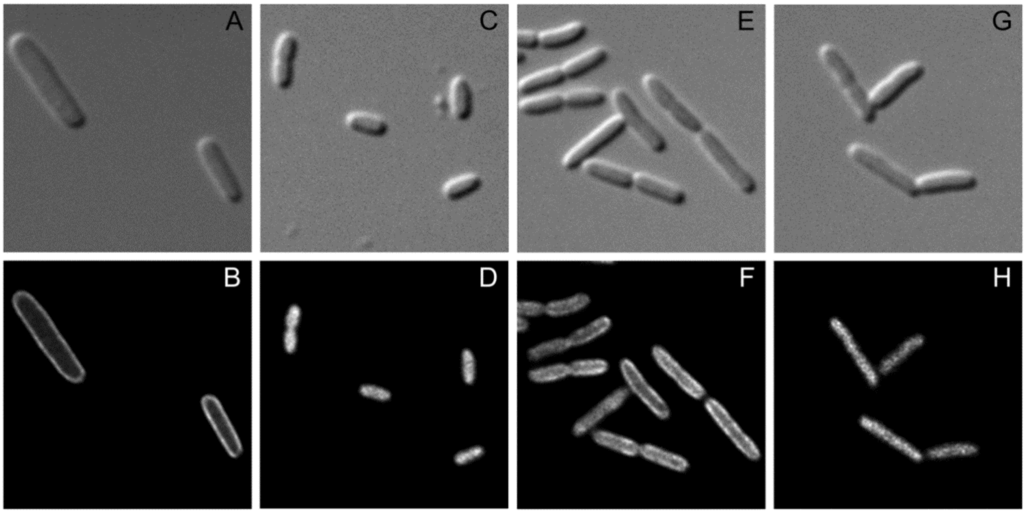
Figure 4.
Fluorescence microscopy for the determination of EIICBGlc and SgrT localization.
Figure 4.
Fluorescence microscopy for the determination of EIICBGlc and SgrT localization.
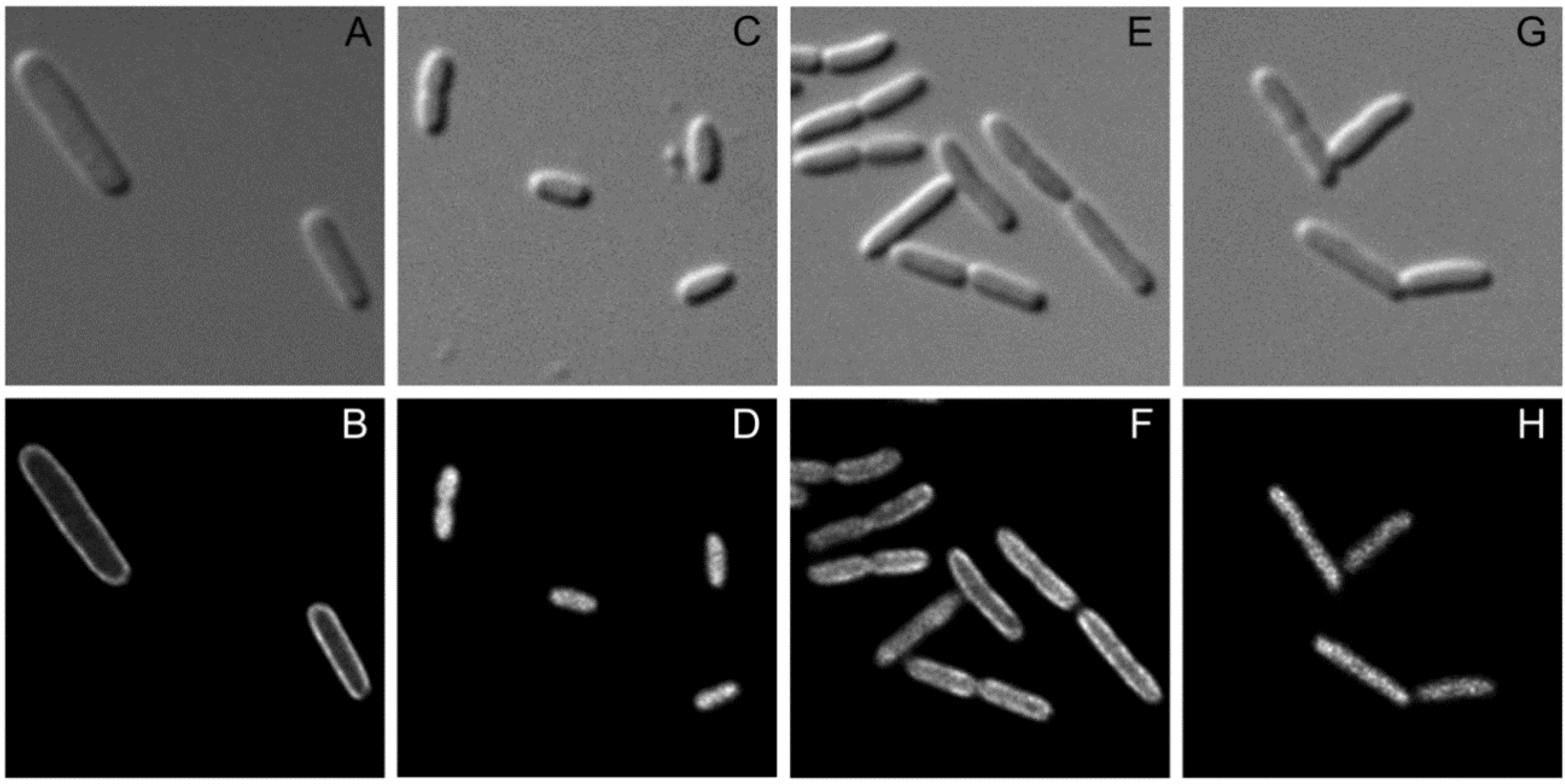
Bright field (upper lane) and fluorescence microscopy (lower lane) were performed with three different strains expressing EIICBGlc or SgrT derivatives tagged with Gfp.
A and B: JKA12 (ΔptsG::cat ΔsgrRST::neo) expressing EIICBGlc-Gfp; C and D: JKA12 expressing SgrT-Gfp; E and F: JKA1 (ptsG+ΔsgrRST::neo) expressing SgrT-Gfp. G and H: JKA18 (ptsGP384RΔsgrRST::neo) expressing EIICBGlcP384R and SgrT-Gfp. All cells were grown in minimal medium with 0.2% glucose. These results indicate a sequestration of SgrT by unphosphorylated EIICBGlc (wild type), but not by EIICBGlcP384R in living cells.
2.5. Discussion
Overflow metabolism, which is accompanied in E. coli by acetate production, is a metabolic phenomenon which takes place when the rates of carbohydrate transport and glycolysis exceed a critical value due to high growth rates under aerobic growth conditions [32]. Acetate is produced from acetyl-CoA via acetyl-phosphate. Thus, under conditions of high glycolytic flux overflow metabolism directs a portion of the excess acetyl-CoA to acetate production. In addition, other byproducts such as succinate, lactate, pyruvate, or methylglyoxalate can also be produced under these conditions. During overflow metabolism, not all of the substrate is converted into biomass which constitutes an enormous disadvantage for biotechnological processes. Accordingly, the phenomenon of overflow metabolism has been investigated in greater depth during the past years in an effort to make industrial biotechnology more cost-efficient and economically advantageous [33].
The preferred carbon source in biotechnological applications is glucose, which in E. coli is usually taken up by the glucose-phosphotransferase system. The regulation of the ptsG gene encoding the glucose transporter EIICBGlc is extremely complex. At least seven different proteins are involved in the transcriptional control (reviewed in [7]). Moreover, several research groups have identified and characterized the function of the sgrRST-system, which creates a sophisticated posttranscriptional feedback regulation mechanism of glucose transport during intracellular Glc6-P or Fru6-P accumulation [23,24,26,27,34,35]. Whereas the small regulatory RNA SgrS destabilizes ptsG m-RNA, the function of the small regulatory peptide SgrT, which is simultaneously encoded by the sgrS gene, has not been very well characterized thus far.
In this paper we could demonstrate for the first time by several experimental approaches, that the highly conserved KTPGRED motif in the linker region between the EIICGlc and the EIIBGlc domains of the glucose transporter constitutes the SgrT target site. Furthermore, using site-directed mutagenesis, we were able to identify the most important residues for this protein–protein interaction. These findings finally provide a good explanation for the existence of this highly conserved motif within an otherwise non-conserved region of the protein. Moreover, we could demonstrate that according to the physiological needs, almost exclusively dephosphorylated EIICBGlc interacts with SgrT. During glucose uptake EIICBGlc conducts several conformational changes which result in glucose translocation and phosphorylation [36]. Glucose is bound with high affinity on the periplasmic site of the inner membrane. Subsequently, the protein conformation changes into an occluded state (the glucose is completely surrounded by the protein). Phosphorylation of the substrate again causes a conformational change and thus leads to a decreased affinity and to the release of Glc6-P into the cytoplasm [36]. Erni et al. could demonstrate that the flexible linker which connects the phosphorylated EIIBGlc-domain with the glucose binding EIICGlc-domain conducts severe conformational changes during this transport process [10]. This provides an explanation for the clear differences observed in SgrT binding between phosphorylated and unphosphorylated EIICBGlc.
Furthermore, transport activity of EIICBGlc is influenced by different amino acid substitutions which can be dissected into three groups: The first group consists of mutations which cause “relaxed” substrate specificity. These single amino acid substitutions are scattered over the entire EIICGlc domain and allow facilitated diffusion of substrates like mannose and glucosamine [16,37], fructose [38], ribose and xylose [39], mannitol [40], ribitol und arabinitol, respectively (reviewed in [10]). In contrast, so-called “uncoupled” mutants exhibit a separation of translocation and phosphorylation and the uptake of unphosphorylated glucose, for example, in a ptsHICrr deletion background [29,41]. The third group consists of so-called “locked-in” mutations that displayed a radical decrease in transport, but unchanged phosphorylation activity [42]. Our results indicate that none of these specific or intermediate conformations has an influence on the interaction with SgrT and that the biggest conformational differences seem to exist between the phosphorylated and the unphosphorylated EIICBGlc.
In spite of the SgrS/SgrT feedback regulation loop of glucose transport, E. coli tends to produce acetate during high cell density fermentation. However, the sgrRST-system provides new regulatory tools to artificially modify glucose uptake rates according to biotechnological needs. Negrete et al. already demonstrated that overexpression of SgrS is sufficient to reduce acetate excretion of glucose fermenting E. coli cells [26]. In addition, we and others have demonstrated that the exclusive overproduction of SgrT causes a drastic reduction of bacterial growth in minimal medium with glucose, but not with sucrose as sole carbon source [25,27]. In principle, it should be possible to couple the production of SgrT more strictly to the glycolytic flux, for example by isolating SgrR mutants with enhanced affinity to its molecular inducer. In this case, the slightest accumulation of these metabolites should result in a shutdown of glucose transport and should minimize the overflow. After identification of the SgrT target sequence it should be possible to incorporate this target box into other carbohydrate transport proteins to create an artificial control system in order to achieve the desired uptake rates. Especially sucrose may be a sought-after alternative for a cheap carbohydrate source, as sugar-cane molasses is available in great quantities.
3. Experimental Section
Media and growth conditions. Cells were grown routinely either in Luria broth without glucose and calcium ions (LB0), or in 2xTY medium as described [55]. Antibiotics were used at the following concentrations: tetracycline (Tc) 10 mg/L, kanamycin (Kn) 25 mg/L and ampicillin (Ap) 50 mg/L, respectively. Minimal medium supplemented with 0.2% carbon source was used as indicated [56]. IPTG was used at concentrations of 100µM-500µM for induction of protein production in growth inhibition assays and at a concentration of 1 mM for induction of protein production for crosslinking experiments. Cells were incubated at 37 °C with shaking.
Bacteria strains and plasmids. All strains used were E.coli K-12 derivates. Table 1 and Table 2 list the genotypes and sources of the relevant bacterial strains and plasmids. Oligonucleotides are listed in Table 3 in the supplemental materials. Alleles were moved between strains by P1-transduction (performed as described previously [47]) or inserted via λ Red recombination [44].

Table 1.
Strains and Phages used in this study.
| Strains | Relevant Genotype or Phenotype | Source or Reference |
|---|---|---|
| Escherichia coli | ||
| BL21 (λDE3) | fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS λ DE3 = λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5 | [43] |
| BW25113 | lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 | [44] |
| JKA1 | LJ110 ΔsgrRST::cat+ (CamR) | this study |
| JKA12 | LJ120 ΔsgrRST:: neo+ (KanR) | this study |
| JKA17 | BL21 (λDE3) ΔptsG::cat+ (CamR) | this study |
| JKA18 | LJB5 ΔsgrRST:: neo+ (KanR) | this study |
| JM109 | thi-1Δ(lac-proA,B)U169 gyrA1,96 recA1 endA1 relA1 hsdR17 supE44/F´traD36 proA+B+lacIqlacZΔM15 | [45] |
| K-12 | Wildtyp | K. Jahreis, lab stock |
| LJ110 | W3110 Fnr+ | [16] |
| LJ120 | LJ110 ΔptsG::cat+ (CamR) | [16] |
| LJ140 | LJ110 ΔptsHIcrr::neo+ (KanR) | [16] |
| LJ231 | LJ110 ΔmanXYZ::cat csc+ | K. Jahreis, lab stock |
| LJB5 | LJ231 ptsGP384R Tn10tet+(TetR) | [46] |
| LJB17 | LJ110 dgsA::cat | [47] |
| MG1655 | F-, λ-, rph-1 | Yale E.coli Stock Center |
| W3110 | F-λ- IN(rrnD-rrnE)1 rph-1 | Yale E.coli Stock Center |
| Bacteriophage | ||
| P1kc | lysogen | [48] |
Table 2.
Plasmids used in this study
| Name | Resistance | Properties | Source or Reference |
|---|---|---|---|
| Escherichia coli vectors | |||
| pACYC184 | TcR | [49] | |
| pACYC184sgrT3HA | TcR | SgrT-3HA (tacPO) | this study |
| pBAD24 | ApR | [50] | |
| pBLP2 | ApR | EIICBGlc-GFP | [51] |
| pET11a-link-NGFP | ApR | [52] | |
| pET11a-Z-NGFP | ApR | NGFP-Leucinzipper | [52] |
| pETS | ApR | NGFP-SgrT | this study |
| pKD3 | ApR, CmR | [44] | |
| pKD46 | ApR | [44] | |
| pMRB | KnR | EIIBGlc-CGFP | this study |
| pMRBAD-link-CGFP | KnR | [52] | |
| pMRBAD-Z-CGFP | KnR | Leucinzipper-CGFP | [52] |
| pMRC | KnR | EIICGlc-CGFP | this study |
| pMRCL | KnR | EIICGlc-linker-CGFP | this study |
| pMRCL-P384R | KnR | EIICGlc-linker-CGFP-P384R | this study |
| pMRG | KnR | EIICBGlc-CGFP | this study |
| pMRLB | KnR | Linker-EIIBGlc-CGFP | this study |
| pMRLB-P384R | KnR | Linker-EIIBGlc-CGFP-P384R | this study |
| pRR48 | ApR | Parkinson, LKS | |
| pRR48G | ApR | EIICBGlc | [53] |
| pRR48GH | ApR | EIICBGlc-5His | [53] |
| pRR48GH-D388A | ApR | EIICBGlc-D388A-5His | this study |
| pRR48GH-E387A | ApR | EIICBGlc-E387A-5His | this study |
| pRR48GH-G385A | ApR | EIICBGlc-G385A-5His | this study |
| pRR48GH-I296N | ApR | EIICBGlc-I296N-5His | this study |
| pRR48GH-K150E | ApR | EIICBGlc-K150E-5His | this study |
| pRR48GH-K382A | ApR | EIICBGlc-K382A-5His | this study |
| pRR48GH-P384A | ApR | EIICBGlc-384A-5His | this study |
| pRR48GH-P384R | ApR | EIICBGlc-384R-5His | this study |
| pRR48GH-R386A | ApR | EIICBGlc-R386A-5His | this study |
| pRR48GH-T383A | ApR | EIICBGlc-T383A-5His | this study |
| pRR48GH-V12F | ApR | EIICBGlc-V12F-5His | this study |
| pTM30 | ApR | [54] | |
| pTM30sgrT | ApR | SgrT | this study |
| pTM30sgrT3HA | ApR | SgrT-3HA | this study |
| pTM30sgrTgfp | ApR | SgrT-GFP | this study |
Isolation of chromosomal and plasmid DNA, restriction analysis, PCR and DNA sequencing. All manipulations of chromosomal or recombinant DNA were carried out using standard procedures as described previously [55]. Plasmid DNA was prepared using QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany). Restriction enzymes were purchased from New England Biolabs (Schwalbach, Germany) or Fermentas (St. Leon-Rot, Germany) and used according to supplier recommendations. Oligonucleotides for PCR were purchased from Thermo Fisher Scientific (Ulm, Germany). DNA sequencing was commissioned to Scientific Research and Development (Bad Homburg, Germany). Polymerase chain reactions (PCR) were performed as described by [57] using TaKaRa DNA polymerase from Lonza (Köln, Germany).
Construction of plasmids. The open reading frame of sgrT (sgrT ORF) was amplified using chromosomal DNA of LJ110, which is closely related to wild type E. coli K-12, with forward primer sgrT+, containing a PstI restriction site and reverse primer sgrT-, which had a HindIII restriction site. This PCR product was purified with Wizard DNA purification system (Promega) and cloned into the vector pTM30, resulting in plasmid pTM30sgrT. The expression plasmid pTM30 provides a tac-promoter, an artificial start codon and and an artificial ribosomal binding site as described before [54]. pTM30sgrT3HA carries additional sequences that encode a 3xHA tag (received from pFA6a3HA, Oligonucleotides HA+/-) fused to the C-terminus of SgrT. For pACYC184sgrT3HA the tacPO and sgrT3HA sequence from pTM30sgrT3HA was amplified with oligonucleotides TacPO+ and HA- and cloned into the vector pACYC184. For the bimolecular fluorescence complementation assay the sgrT ORF was amplified by PCR (Oligonucleotides pETS+/-) and cloned into the vector pET11a-link-NGFP [52] using the restriction enzymes XhoI/BamHI. The genes encoding EIIBGlc (aa 389-477), Linker-EIIBGlc (aa 380-477), Linker-EIIBGlc-P384R (aa 380-477), EIICGlc (aa 1-381), EIICGlc-Linker (aa 1-396), EIICGlc-Linker-P384R (aa 1-396) and EIICBGlc (aa 1-477) were also amplified by PCR and purified. The oligonucleotides were used as follows: EIIBGlc (pMRB+/pMRG-), Linker-EIIBGlc (pMRLB+/pMRG-), Linker-EIIBGlc-P384R (pMRLB-P384R+/pMRG-), EIICGlc(pMRG+/pMRC-), EIICGlc-Linker (pMRG+/pMRCL-), EIICGlc-Linker-P384R (pMRG+/pMRCL-P384R-) and EIICBGlc (pMRG+/-). PCR products were cloned into the vector pMRBAD-link-CGFP [52] using the restriction enzymes NcoI/AatII or SphI/AatII, respectively. For fluorescence microscopy, an SgrT-GFP fusion protein was created. The sgrT ORF was amplified with oligonucleotides SgrT+/SgrT2- and a gfp gene was amplified using pBLP2 as template and oligonucleotides Gfp2+/-. Both PCR products were purified and cloned together into pTM30, resulting in pTM30sgrT-gfp. All oligonucleotide sequences used are listed in Table3 in the supplemental materials.
Site-directed mutagenesis. We generated defined mutations in the ptsGHis gene using the pRR48GH plasmid as a template and the Phusion Site-directed Mutagenesis Kit according to standard protocol (Finnzymes, Vantaa, Finland). The oligonucleotides used are listed in Table 3 in the supplemental materials (K150E+/-, V12F+/-, K382A+/ktpg-, T383A+/ktpg-, P384A+/ktpg-, P384R+/ktpg-, G385A+/ktpg-, R386A+/red-, E387A+/red-,D388A+/red-).
Construction of strains. JKA1: In order to characterize the consequence of a sgrRST-deletion we disrupted the sgrRST-wildtype genes via λ Red recombination as described in the protocol of Datsenko and Wanner [44]. Briefly, a chloramphenicol resistance selection marker with flanking regions that are homologous to chromosomal sequences at the 5’ and 3’ end of the sgrRST gene region was amplified from template pKD3 [44] by using the primer pair SgrR+ and SgrS-. The PCR product was purified with the Wizard DNA purification system (Promega), treated with DpnI and further enriched by ethanol precipitation. Subsequently, the DNA-fragment was integrated into the chromosome of BW25113/pKD46 [44] via λ Red recombination, resulting in BW25113ΔsgrRST::cat. JKA1 was created by the transfer of the deletion cassette of BW25113ΔsgrRST::cat into LJ110 via P1-transduction. PCR of flanking regions was used to confirm the correct integration of the desired gene disruption.
JKA12: For crosslinking analysis a strain with a deletion of ptsG and sgrRST gene regions was created. The sgrRST gene region in BW25113/pKD46 was disrupted with a kanamycin resistance cassette via λ Red recombination, using the protocol of Datsenko and Wanner [44]. The deletion cassette of BW25113ΔsgrRST::kan was then inserted via P1-transduction into LJ120, resulting in JKA12. PCR was used to confirm correct integration of the desired gene disruption.
JKA17: To characterize the interaction of SgrT and EIICBGlc with bimolecular fluorescence complementation a BL21 (λDE3) strain with a ptsG-deletion was created via P1-transduction (ΔptsG::cat from LJ120). PCR was used to confirm correct integration of the desired gene disruption.
JKA18: For fluorescence microscopy one strain with a deletion of sgrRST gene region and a chromosomal point mutation in ptsG was constructed. The deletion cassette of BW25113ΔsgrRST::kan was inserted via P1-transduction into LJB5, resulting in JKA18. PCR was used to confirm correct integration of the desired gene disruption. The oligonucleotides are listed in Table 3 in the supplemental materials.
Western blot analyses. For Western blot analysis, bacterial cells were treated as described in the section “crosslinking with paraformaldehyde” or grown overnight in LB0 with ampicillin and inoculated in fresh medium to an OD650 = 0.1. The cultures were grown to early-log phase (OD650 = 0.2) and induced with IPTG. Cells were harvested at an optical density at 650 nm of 1 by centrifugation and resuspended in 100 µL sterile water and 100 µL of SDS-PAGE loading buffer (125 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulphate (SDS), 10% glycerol, 5% ß-mercaptoethanol, 0.01% bromophenol blue). If not described otherwise, samples were heated at 95 °C for 10 min. 15 µL of total cellular proteins were separated by electrophoresis on 0.1% SDS-containing 15% polyacrylamide gels and transferred to a Nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). For the detection of EIICBGlc-His protein derivatives, we used a Penta-His antibody (Qiagen, Hilden, Germany). SgrTec3HA was detected with HA-antibody (kindly provided by Anja Lorberg, University of Osnabrück). Detection of antibody binding was performed using infrared-labeled second antibodies (LI-COR Biosciences, Bad Homburg, Germany).Visualization and quantification were done using an Odyssey infrared imager (LI-COR Biosciences, USA) and the software provided by the supplier (Odyssey 2.1).
Crosslinking with paraformaldehyde. For crosslinking of proteins with paraformaldehyde the general procedure from [30] was followed. Cells were grown overnight in LB0 media with ampicillin and tetracycline and inoculated in 200 mL fresh medium to an OD650 = 0.1. The cultures were grown for one hour at 37 °C and induced with 1mM IPTG. After one hour 0.2% glucose was added to cultures when indicated and cultures were incubated for another hour. Then paraformaldehyde solution (4% in PBS (136 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4, 10 mM Na2HPO4) was added in a concentration of 0.3%. Cultures were incubated for 20 min at 37 °C while shaking and cells were harvested via centrifugation. The pellet was washed in a lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM Imidazol, pH 8.0) and finally resuspended in 5 mL of lysis buffer. 1mM AEBSF was added and cells were disrupted by sonification. Cell debris was removed via centrifugation and the supernatant was used for solubilization of membrane proteins. Therefore 2% triton X-100 was added to the supernatant and incubated at room temperature (RT) for 30 min while mixing. Membranes were removed via ultracentrifugation. The supernatant was then used for protein purification with Ni-NTA Agarose (Qiagen, Hilden, Germany). 1.25 mL Ni-NTA agarose was mixed with 5 mL protein suspension and incubated for one hour at RT. Supernatant was removed via centrifugation and unbound protein was removed using wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM Imidazol, pH 8.0) twice. 625 µL (1/8) Elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM Imidazol, pH 8.0) was used to elute purified protein. The same amount SDS sample buffer was added, proteins heated to 95 °C for 10 min to destroy protein complexes, and equal amounts of proteins were analyzed with Western blot analysis.
Bimolecular fluorescence complementation. For bimolecular fluorescence complementation strain JKA17 was used. Protocol and plasmids were used as described in [31,52]. The cells were inoculated in rich medium with 100 µM IPTG, 0.4% arabinose and 0.2% glucose and incubated for three days at 25 °C while shaking. Cells were harvested via centrifugation and resuspended in 1 mL of lysis buffer [52]. OD420 was determined and equal amounts of cells used for measuring fluorescence activity in a fluorimeter (Fluorometer Fluostar Optima, BMG LABTECH GmbH, Ortenberg, Germany).
Fluorescence microscopy. For standard microscope examination, cells were grown to early logarithmic phase in minimal medium complemented with 0.2% glucose. After 2 hours, 5 µM IPTG or 100 mM arabinose was added, incubated for two hours and cells were fixed on a slide with polylysin. The setup used for fluorescence microscopy consisted of a Zeiss Axioplan2e (Carl Zeiss, Jena, Germany) equipped with a 100× alpha-Plan Fluar objective (NA 1.45) and differential interference contrast (DIC). Images were acquired using a Photometrics CoolSNAP HQ Camera (Roper ScientiWc, Tucson, USA). Fluorescence was excited with a helium lamp and appropriate filter sets were used to adjust excitation and emission wavelengths. The setup was controlled by the Metamorphs v6.2 program (Universal Imaging Corporation, Downingtown, USA). Bright field images were acquired as single planes using t DIC. All fluorescence images were taken from single focal planes and scaled using Metamorphs scale image command. All GFP fusions were taken with 1 sec acquisition time. From all cultures, at least 100 cells were controlled. For unspecific cell wall staining, the cells were incubated with 4µM FM4-64 for 10 min at RT.
4. Conclusions
E. coli tends to produce acetate during high cell density fermentation. Acetate production takes place when the rates of carbohydrate transport and glycolysis exceed a critical value. Many attempts have been performed to couple carbohydrate uptake rates to metabolic flux in order to avoid overflow mechanisms. The sgrRST system provides new regulatory tools to artificially modify glucose uptake rates according to biotechnological needs. Clearly, further fundamental research efforts are necessary to adapt and optimize the sgrRST system as an instrument for fine-tuning carbohydrate uptake in biotechnological applications.
Acknowledgments
We gratefully acknowledge Anna-Katharina Göhler, Elisabeth Gabor and Jürgen Heinisch for helpful discussions, Katrin Fänger for excellent technical support and Lucille Schmieding for help with the manuscript. This work was financially supported by the German Ministry of Education and Research through the FORSYS-program (grant FKZ 0315285C to K. Jahreis).
References
- Ladisch, M.R.; Kohlmann, K.L. Recombinant human insulin. Biotechnol. Prog. 1992, 8, 469–478. [Google Scholar] [CrossRef]
- Contiero, J.; Beatty, C.; Kumari, S.; DeSanti, C.L.; Strohl, W.R.; Wolfe, A. Effects of mutations in acetate metabolism on high-cell-density growth of Escherichia coli. J. Ind. Microbiol. Biot. 2000, 24, 421–430. [Google Scholar] [CrossRef]
- Lee, S.Y. High cell-density culture of Escherichia coli. Trends Biotechnol. 1996, 14, 98–105. [Google Scholar] [CrossRef]
- Phue, J.N.; Lee, S.J.; Kaufman, J.B.; Negrete, A.; Shiloach, J. Acetate accumulation through alternative metabolic pathways in ackA - pta - poxB - triple mutant in Escherichia coli B (BL21). Biotechnol. Lett. 2010, 32, 1897–1903. [Google Scholar] [CrossRef]
- De Anda, R.; Lara, A.R.; Hernandez, V.; Hernandez-Montalvo, V.; Gosset, G.; Bolivar, F.; Ramirez, O.T. Replacement of the glucose phosphotransferase transport system by galactose permease reduces acetate accumulation and improves process performance of Escherichia coli for recombinant protein production without impairment of growth rate. Metab. Eng. 2006, 8, 281–290. [Google Scholar] [CrossRef]
- Flores, N.; Leal, L.; Sigala, J.C.; de Anda, R.; Escalante, A.; Martinez, A.; Ramirez, O.T.; Gosset, G.; Bolivar, F. Growth recovery on glucose under aerobic conditions of an Escherichia coli strain carrying a phosphoenolpyruvate:carbohydrate phosphotransferase system deletion by inactivating arcA and overexpressing the genes coding for glucokinase and galactose permease. J. Mol. Microbiol. Biotechnol. 2007, 13, 105–116. [Google Scholar] [CrossRef]
- Jahreis, K.; Pimentel-Schmitt, E.F.; Bruckner, R.; Titgemeyer, F. Ins and outs of glucose transport systems in eubacteria. FEMS Microbiol. Rev. 2008, 32, 891–907. [Google Scholar] [CrossRef]
- Lengeler, J.W.; Jahreis, K. Bacterial PEP-dependent carbohydrate: phosphotransferase systems couple sensing and global control mechanisms. Contrib. Microbiol. 2009, 16, 65–87. [Google Scholar] [CrossRef]
- Lengeler, J.W.; Jahreis, K. Phosphotransferase systems or PTSs as carbohydrate transport and as signal transduction systems. In Handbook of Biological Physics; Konings, W.N., Kaback, H.S., Lolkema, J.S., Eds.; Elsevier Science: Amsterdam, the Netherlands, 1996; Vol. 2, pp. 573–598. [Google Scholar]
- Erni, B. Glucose Transport by the Bacterial Phosphotransferase System (PTS): An Interface between Energy- and Signal Transduction. In Transport Systems; Winkelmann, G., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003. [Google Scholar] [CrossRef]
- Buhr, A.; Flukiger, K.; Erni, B. The glucose transporter of Escherichia coli. Overexpression, purification, and characterization of functional domains. J. Biol. Chem. 1994, 269, 23437–23443. [Google Scholar]
- Siebold, C.; Flukiger, K.; Beutler, R.; Erni, B. Carbohydrate transporters of the bacterial phosphoenolpyruvate: sugar phosphotransferase system (PTS). FEBS Lett. 2001, 504, 104–111. [Google Scholar]
- Lee, S.J.; Boos, W.; Bouche, J.P.; Plumbridge, J. Signal transduction between a membrane-bound transporter, PtsG, and a soluble transcription factor, Mlc, of Escherichia coli. Embo. J. 2000, 19, 5353–5361. [Google Scholar] [CrossRef]
- Nam, T.W.; Cho, S.H.; Shin, D.; Kim, J.H.; Jeong, J.Y.; Lee, J.H.; Roe, J.H.; Peterkofsky, A.; Kang, S.O.; Ryu, S.; et al. The Escherichia coli glucose transporter enzyme IICB(Glc) recruits the global repressor Mlc. Embo. J. 2001, 20, 491–498. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kimata, K.; Aiba, H. A novel regulatory role of glucose transporter of Escherichia coli: membrane sequestration of a global repressor Mlc. Embo. J. 2000, 19, 5344–5352. [Google Scholar] [CrossRef]
- Zeppenfeld, T.; Larisch, C.; Lengeler, J.W.; Jahreis, K. Glucose transporter mutants of Escherichia coli K-12 with changes in substrate recognition of IICB(Glc) and induction behavior of the ptsG gene. J. Bacteriol. 2000, 182, 4443–4452. [Google Scholar] [CrossRef]
- Jahreis, K. cAMP Signaling in Prokaryotes. In Bacterial Signaling; Krämer, R., Jung, K., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 357–375. [Google Scholar]
- Jeong, J.Y.; Kim, Y.J.; Cho, N.; Shin, D.; Nam, T.W.; Ryu, S.; Seok, Y.J. Expression of ptsG encoding the major glucose transporter is regulated by ArcA in Escherichia coli. J. Biol. Chem. 2004, 279, 38513–38518. [Google Scholar]
- Rungrassamee, W.; Liu, X.; Pomposiello, P.J. Activation of glucose transport under oxidative stress in Escherichia coli. Arch. Microbiol. 2008, 190, 41–49. [Google Scholar] [CrossRef]
- Shin, D.; Cho, N.; Heu, S.; Ryu, S. Selective regulation of ptsG expression by Fis. Formation of either activating or repressing nucleoprotein complex in response to glucose. J. Biol. Chem. 2003, 278, 14776–14781. [Google Scholar]
- Shin, D.; Lim, S.; Seok, Y.J.; Ryu, S. Heat shock RNA polymerase (E sigma(32)) is involved in the transcription of mlc and crucial for induction of the Mlc regulon by glucose in Escherichia coli. J. Biol. Chem. 2001, 276, 25871–25875. [Google Scholar]
- Seeto, S.; Notley-McRobb, L.; Ferenci, T. The multifactorial influences of RpoS, Mlc and cAMP on ptsG expression under glucose-limited and anaerobic conditions. Res. Microbiol. 2004, 155, 211–215. [Google Scholar] [CrossRef]
- Morita, T.; El-Kazzaz, W.; Tanaka, Y.; Inada, T.; Aiba, H. Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli. J. Biol. Chem. 2003, 278, 15608–15614. [Google Scholar]
- Vanderpool, C.K.; Gottesman, S. Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol. Microbiol. 2004, 54, 1076–1089. [Google Scholar] [CrossRef]
- Wadler, C.S.; Vanderpool, C.K. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc. Natl. Acad. Sci. USA 2007, 104, 20454–20459. [Google Scholar] [CrossRef]
- Negrete, A.; Majdalani, N.; Phue, J.N.; Shiloach, J. Reducing acetate excretion from E. coli K-12 by over-expressing the small RNA SgrS. N. Biotechnol. 2011. [Google Scholar] [CrossRef]
- Gabor, E.; Göhler, A.K.; Kosfeld, A.; Staab, A.; Kremling, A.; Jahreis, K. The phosphoenolpyruvate-dependent glucose-phosphotransferase system from Escherichia coli K-12 as the center of a network regulating carbohydrate flux in the cell. Eur. J. Cell Biol. 2011, 90, 711–720. [Google Scholar] [CrossRef]
- Schmid, K.; Ebner, R.; Altenbuchner, J.; Schmitt, R.; Lengeler, J.W. Plasmid-mediated sucrose metabolism in Escherichia coli K12: mapping of the scr genes of pUR400. Mol. Microbiol. 1988, 2, 1–8. [Google Scholar] [CrossRef]
- Lengeler, J.W.; Jahreis, K.; Wehmeier, U.F. Enzymes II of the phosphoenol pyruvate-dependent phosphotransferase systems: their structure and function in carbohydrate transport. Biochim. Biophys. Acta 1994, 1188, 1–28. [Google Scholar] [CrossRef]
- Herzberg, C.; Weidinger, L.A.; Dorrbecker, B.; Hubner, S.; Stulke, J.; Commichau, F.M. SPINE: a method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics 2007, 7, 4032–4035. [Google Scholar] [CrossRef]
- Magliery, T.J.; Wilson, C.G.; Pan, W.; Mishler, D.; Ghosh, I.; Hamilton, A.D.; Regan, L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J. Am. Chem. Soc. 2005, 127, 146–157. [Google Scholar]
- Xu, B.; Jahic, M.; Enfors, S.O. Modeling of overflow metabolism in batch and fed-batch cultures of Escherichia coli. Biotechnol. Prog. 1999, 15, 81–90. [Google Scholar] [CrossRef]
- De Mey, M.; De Maeseneire, S.; Soetaert, W.; Vandamme, E. Minimizing acetate formation in E. coli fermentations. J. Ind. Microbiol. Biotechnol. 2007, 34, 689–700. [Google Scholar] [CrossRef]
- Morita, T.; Aiba, H. Small RNAs making a small protein. Proc. Natl. Acad. Sci. USA 2007, 104, 20149–20150. [Google Scholar] [CrossRef]
- Vanderpool, C.K.; Gottesman, S. The Novel Transcription Factor SgrR Coordinates the Response to Glucose-Phosphate Stress. J. Bacteriol. 2007, 189, 2238–2248. [Google Scholar] [CrossRef]
- Lolkema, J.S.; Dijkstra, D.S.; Robillard, G.T. Mechanics of solute translocation catalyzed by enzyme IImtl of the phosphoenolpyruvate-dependent phosphotransferase system of Escherichia coli. Biochemistry 1992, 31, 5514–5521. [Google Scholar] [CrossRef]
- Notley-McRobb, L.; Ferenci, T. Substrate specificity and signal transduction pathways in the glucose-specific enzyme II (EII(Glc)) component of the Escherichia coli phosphotransferase system. J. Bacteriol. 2000, 182, 4437–4442. [Google Scholar] [CrossRef]
- Kornberg, H.L.; Lambourne, L.T.; Sproul, A.A. Facilitated diffusion of fructose via the phosphoenolpyruvate/glucose phosphotransferase system of Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 1808–1812. [Google Scholar]
- Oh, H.; Park, Y.; Park, C. A mutated PtsG, the glucose transporter, allows uptake of D-ribose. J. Biol. Chem. 1999, 274, 14006–14011. [Google Scholar]
- Begley, G.S.; Warner, K.A.; Arents, J.C.; Postma, P.W.; Jacobson, G.R. Isolation and characterization of a mutation that alters the substrate specificity of the Escherichia coli glucose permease. J. Bacteriol. 1996, 178, 940–942. [Google Scholar]
- Ruijter, G.J.; van Meurs, G.; Verwey, M.A.; Postma, P.W.; van Dam, K. Analysis of mutations that uncouple transport from phosphorylation in enzyme IIGlc of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system. J. Bacteriol. 1992, 174, 2843–2850. [Google Scholar]
- Buhr, A.; Daniels, G.A.; Erni, B. The glucose transporter of Escherichia coli. Mutants with impaired translocation activity that retain phosphorylation activity. J. Biol. Chem. 1992, 267, 3847–3851. [Google Scholar]
- Studier, F.W.; Rosenberg, A.H.; Dunn, J.J.; Dubendorff, J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990, 185, 60–89. [Google Scholar]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Becker, A.K. Das EIICBGlc als Glukose-Sensor in E.coli K-12: Eine molekulargenetische Analyse der Funktion des Enzyms und der Regulation des zugehörigen Gens ptsG. Master Thesis, Universität Osnabrück, Osnabrück, Saxony, Germany, 2000. [Google Scholar]
- Becker, A.K.; Zeppenfeld, T.; Staab, A.; Seitz, S.; Boos, W.; Morita, T.; Aiba, H.; Mahr, K.; Titgemeyer, F.; Jahreis, K. YeeI, a novel protein involved in modulation of the activity of the glucose-phosphotransferase system in Escherichia coli K-12. J. Bacteriol. 2006, 188, 5439–5449. [Google Scholar] [CrossRef]
- Lennox, E.S. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1955, 1, 190–206. [Google Scholar] [CrossRef]
- Chang, A.C.; Cohen, S.N. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 1978, 134, 1141–1156. [Google Scholar]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar]
- Siepelmeyer, J. Entwicklung von Systemen zur quantitativen Messung der intrazellulären cAMP-Konzentration und der in vivo Aktivität der Adenylatzyklase CyaA in einer isogenen Stammreihe von Escherichia coli K-12. Ph.D. Thesis, Universität Osnabrück, Osnabrück, Saxony, Germany, 2000. [Google Scholar]
- Wilson, C.G.; Magliery, T.J.; Regan, L. Detecting protein-protein interactions with GFP-fragment reassembly. Nat. Methods 2004, 1, 255–262. [Google Scholar] [CrossRef]
- Gabor, E. Molekularbiologische Untersuchungen verschiedener Komponenten des Glukose-Phosphotransferasesystems in Escherichia coli K-12 mit Schwerpunkt auf der Strukturanalyse des Transportproteins EIICBGlc. Ph.D. Thesis, Universität Osnabrück, Thesis, Universität Osnabrück, Osnabrück, Saxony, Germany, 2011. [Google Scholar]
- Morrison, T.B.; Parkinson, J.S. Liberation of an interaction domain from the phosphotransfer region of CheA, a signaling kinase of Escherichia coli. Proc. Natl. Acad. Sci. USA 1994, 91, 5485–5489. [Google Scholar] [CrossRef]
- Ausubel, F.M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidmann, J.G.; Smith, J.A.; Struhl, K. Current Protocols in Molecular Biology; Greene Publishing and Wiley-Interscience: New York, NY, USA, 1990. [Google Scholar]
- Tanaka, S.; Lerner, S.A.; Lin, E.C. Replacement of a phosphoenolpyruvate-dependent phosphotransferase by a nicotinamide adenine dinucleotide-linked dehydrogenase for the utilization of mannitol. J. Bacteriol. 1967, 93, 642–648. [Google Scholar]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).