The Future of NMR Metabolomics in Cancer Therapy: Towards Personalizing Treatment and Developing Targeted Drugs?

Abstract
:1. Introduction
1.1. Nuclear Magnetic Resonance Spectroscopy
1.2. NMR Metabolomics
1.3. Targeted Therapeutics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Type | Drug | Cancer | Reference |
|---|---|---|---|---|
| Estrogen Receptor Inhibitors | Selective estrogen receptor modulator (SERM) | Tamoxifen citrate (Nolvadex®) | Ductal carcinoma in situ (DCIS) post radiation and surgery | [56,57] |
| Signal transduction inhibitors | Small-molecule drug | Imatinib mesylate (Gleevec®) | Multiple tumors and disorders * | [64] |
| Small-molecule drug | Lapatinib (Tykerb®) | HER-2-/hormone-positive advanced breast cancer | [58] | |
| Small-molecule drug | Vandetanib (Caprelsa®) | Unresectable and advanced medullary thyroid cancer | [55] | |
| Apoptosis inducers | Proteasome inhibitor | Bortezomib (Velcade®) | Multiple myeloma and mantle cell lymphoma (post first-line therapy) | [65] |
1.4. Drug Discovery and Development
2. Investigating Existing and Potential Anti-Cancer Agents
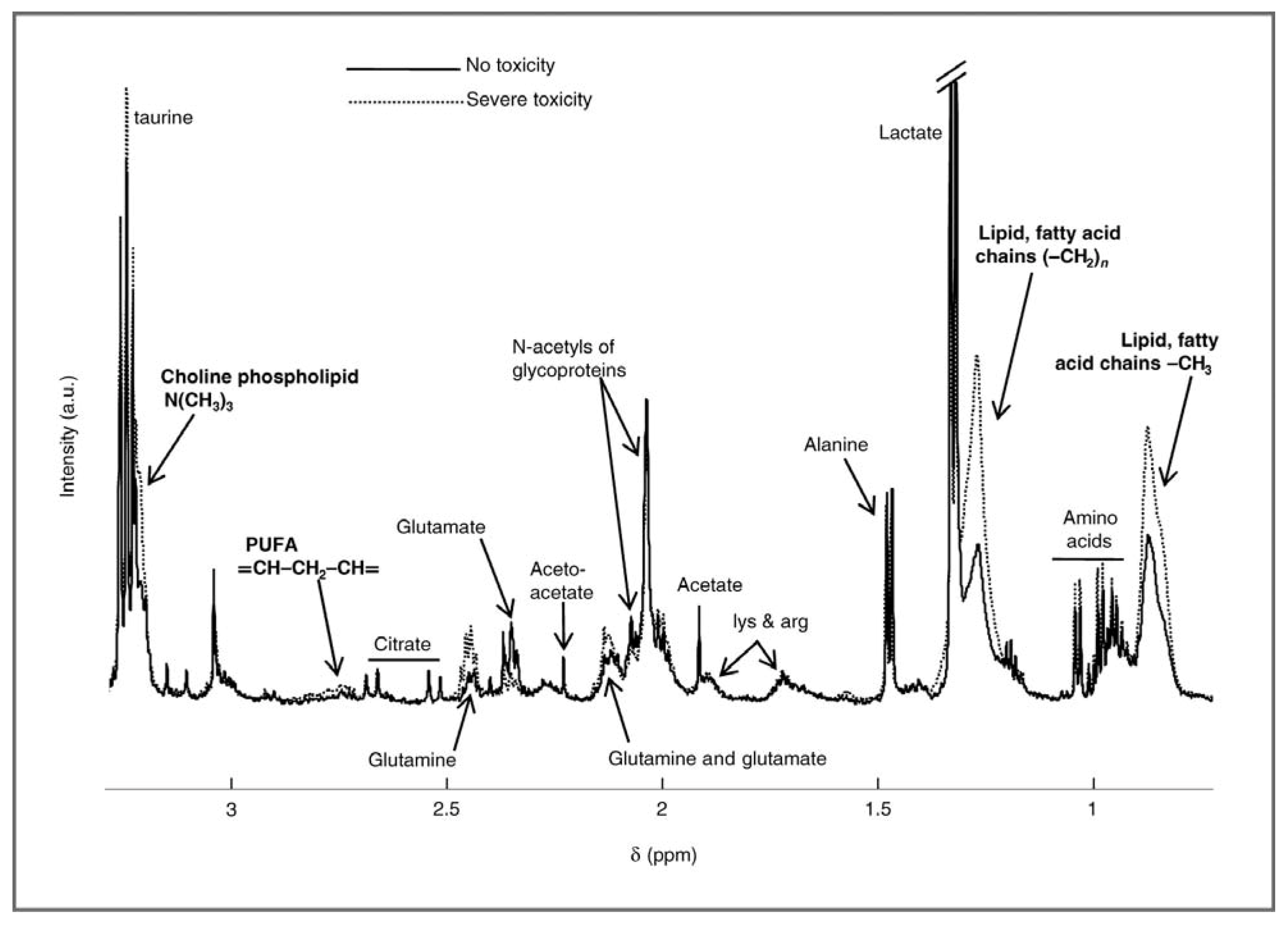
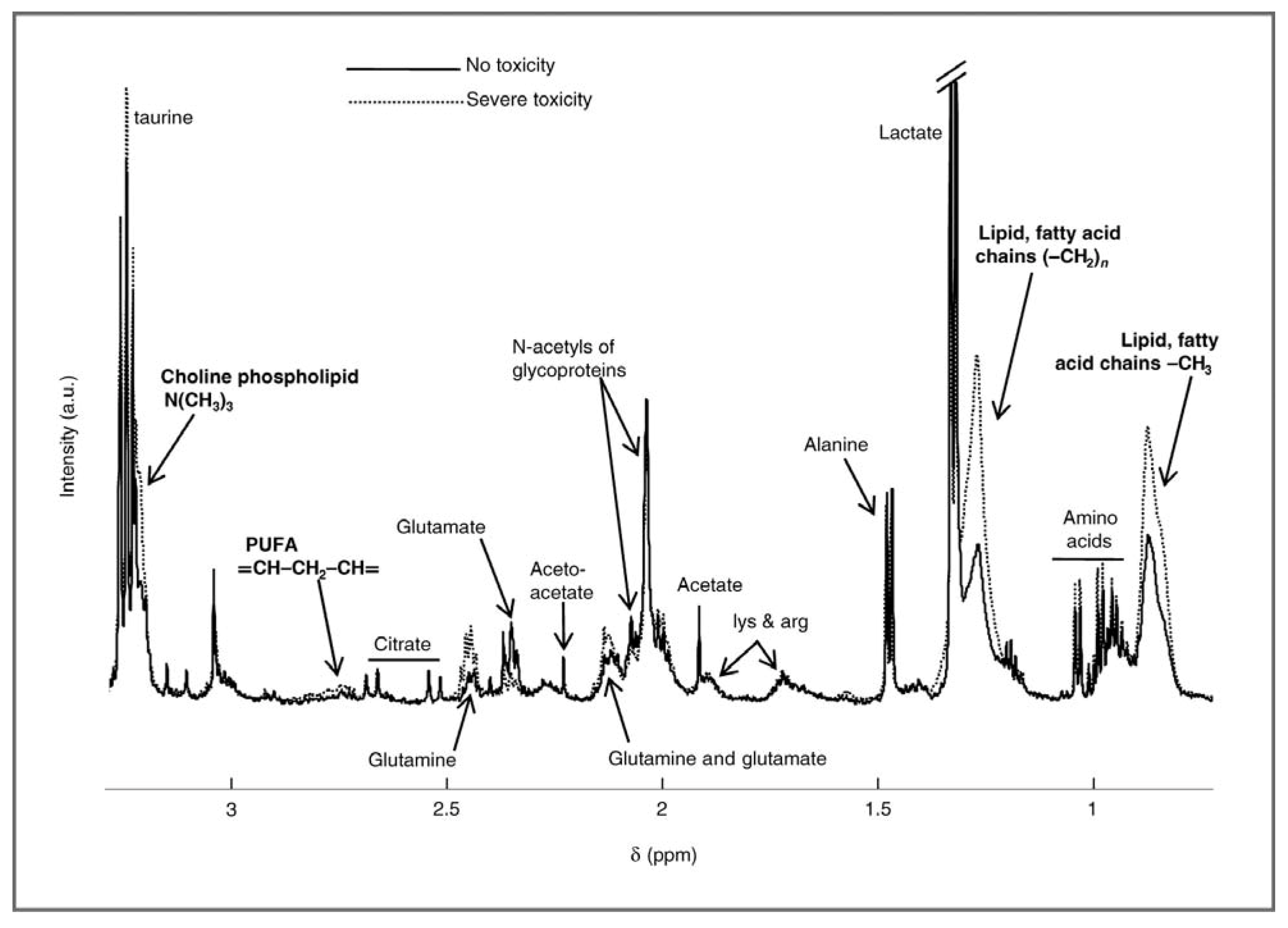
2.1. Evaluating Toxicity
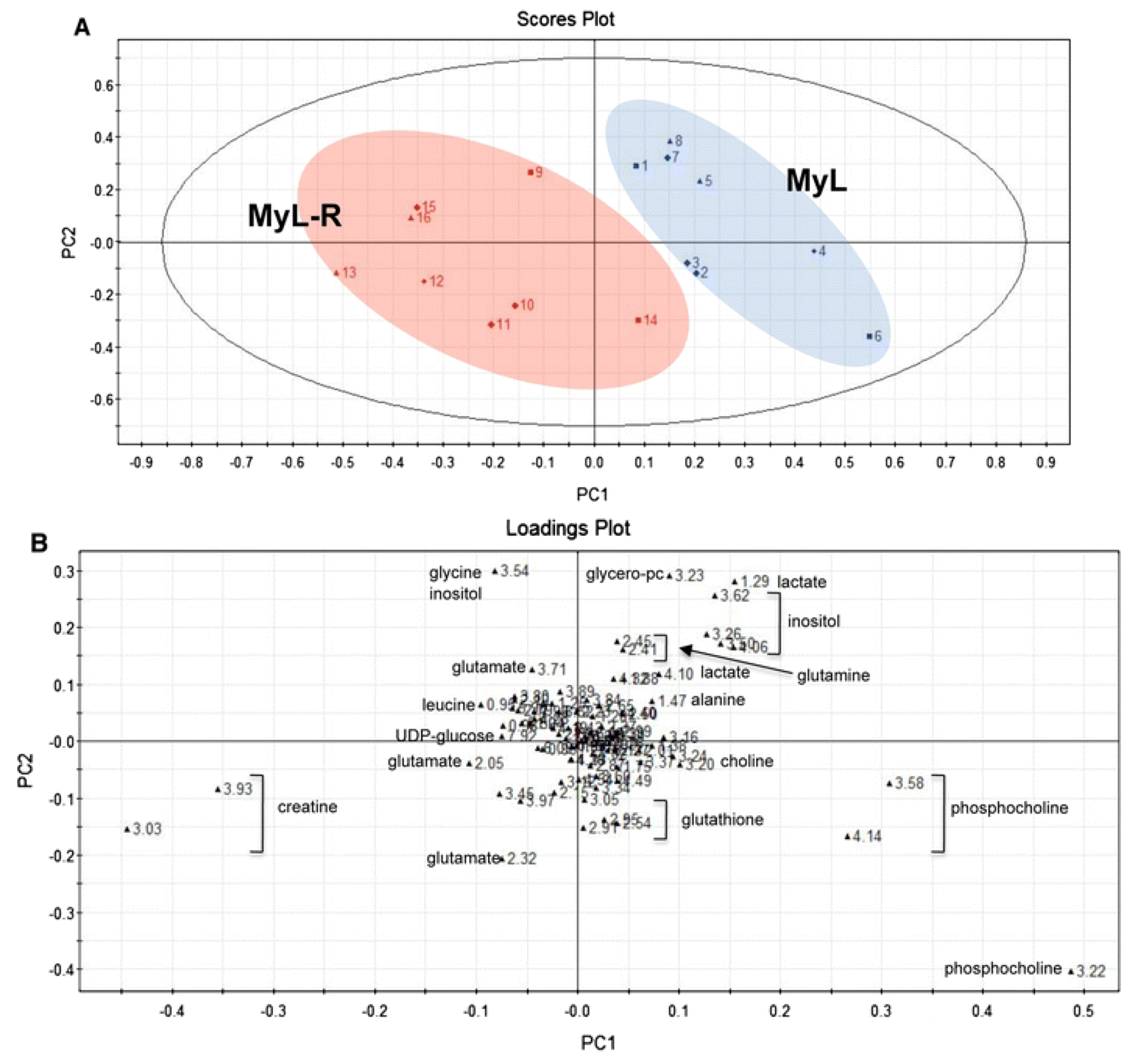
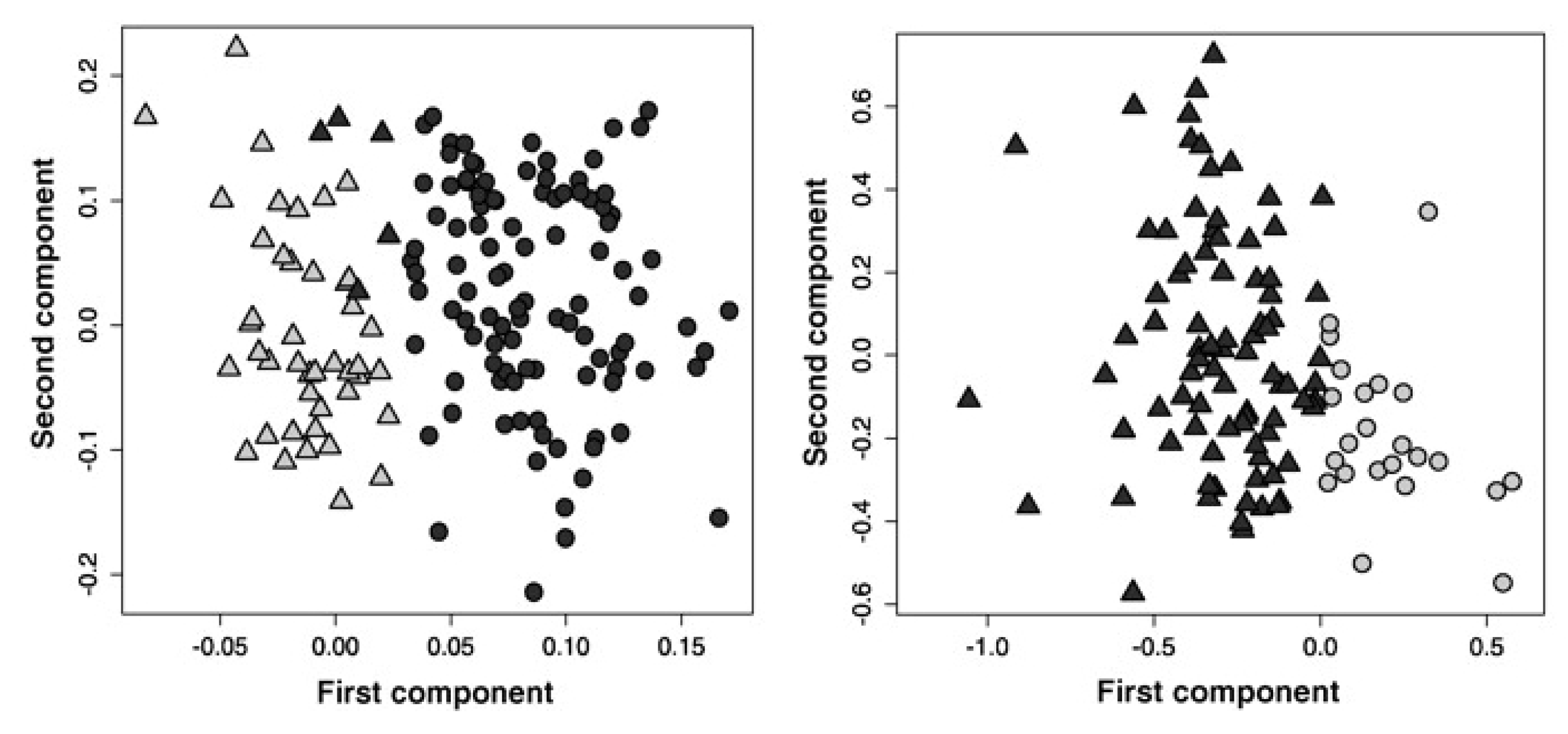
2.2. Evaluating Resistance and Sensitivity
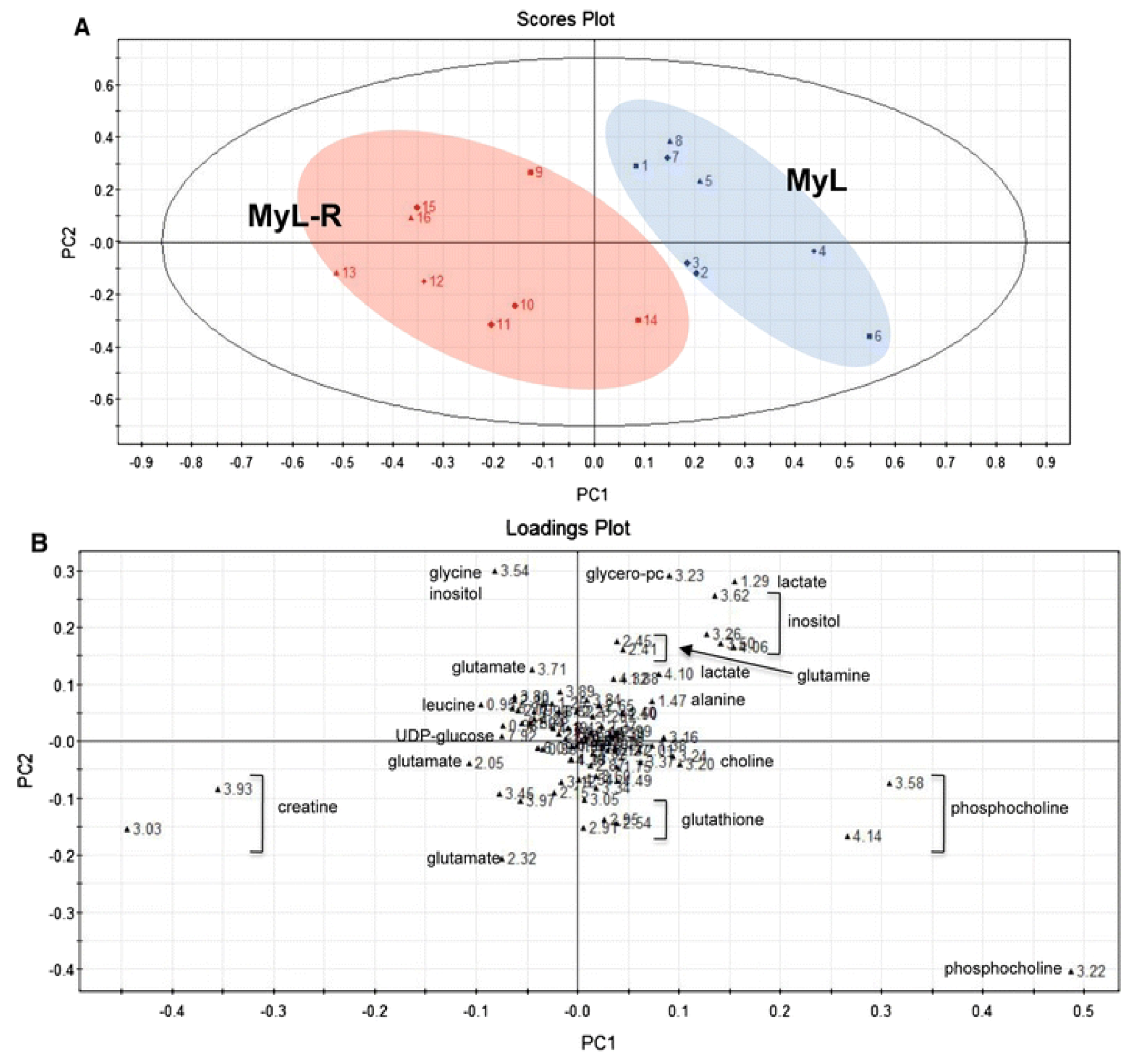
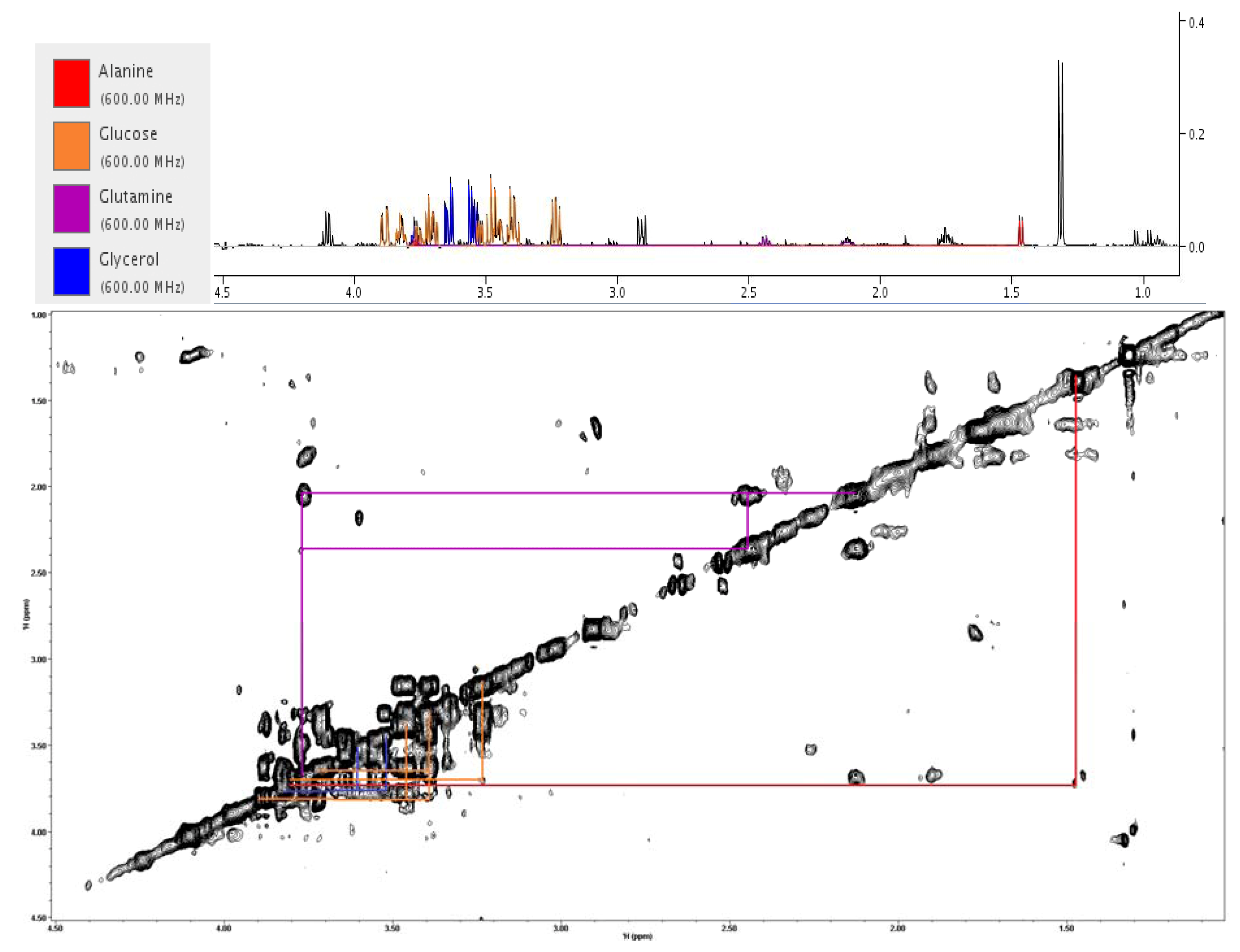
2.3. Treatment Response to Different Drugs
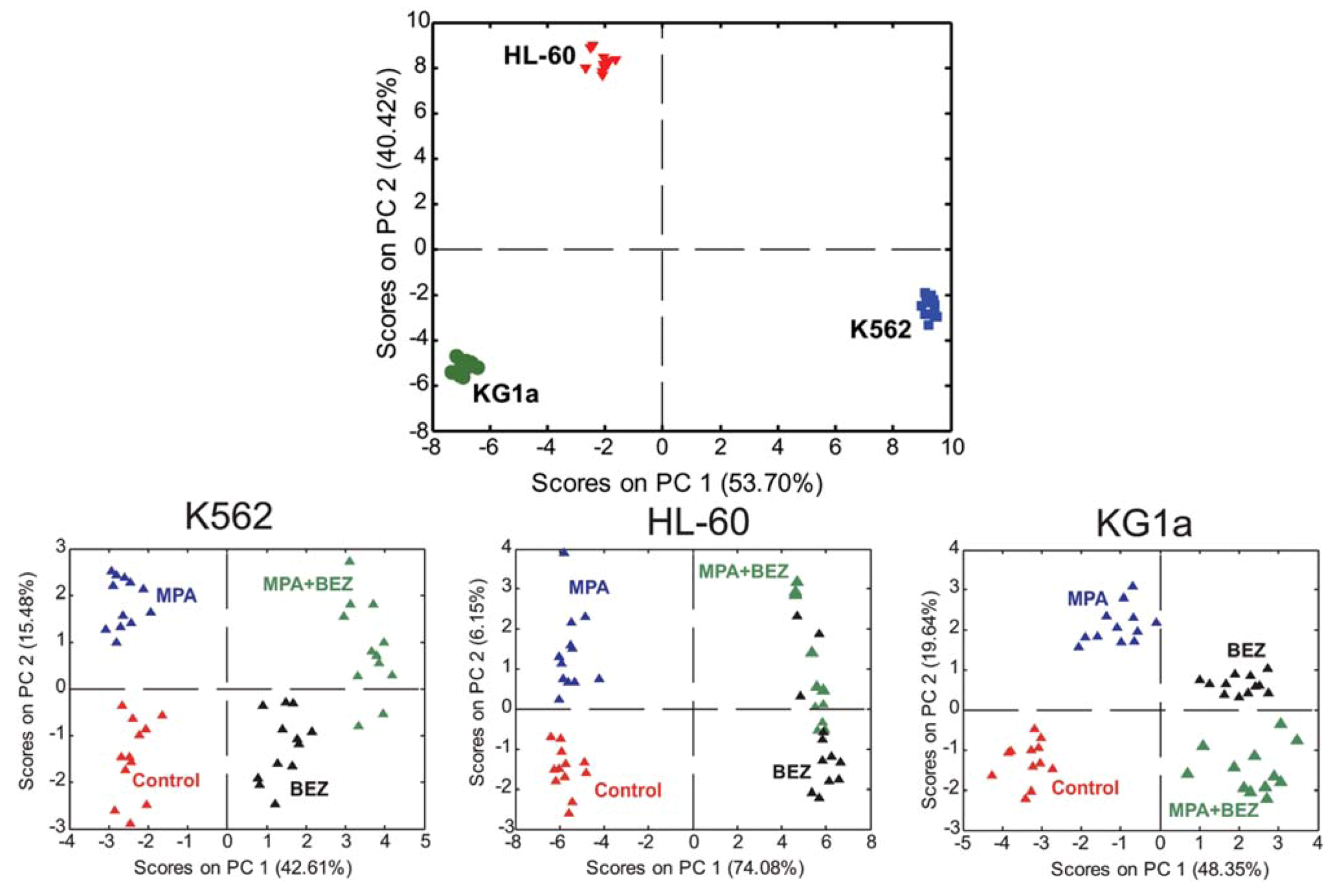
2.4. Treatment Response in Different Cell Lines
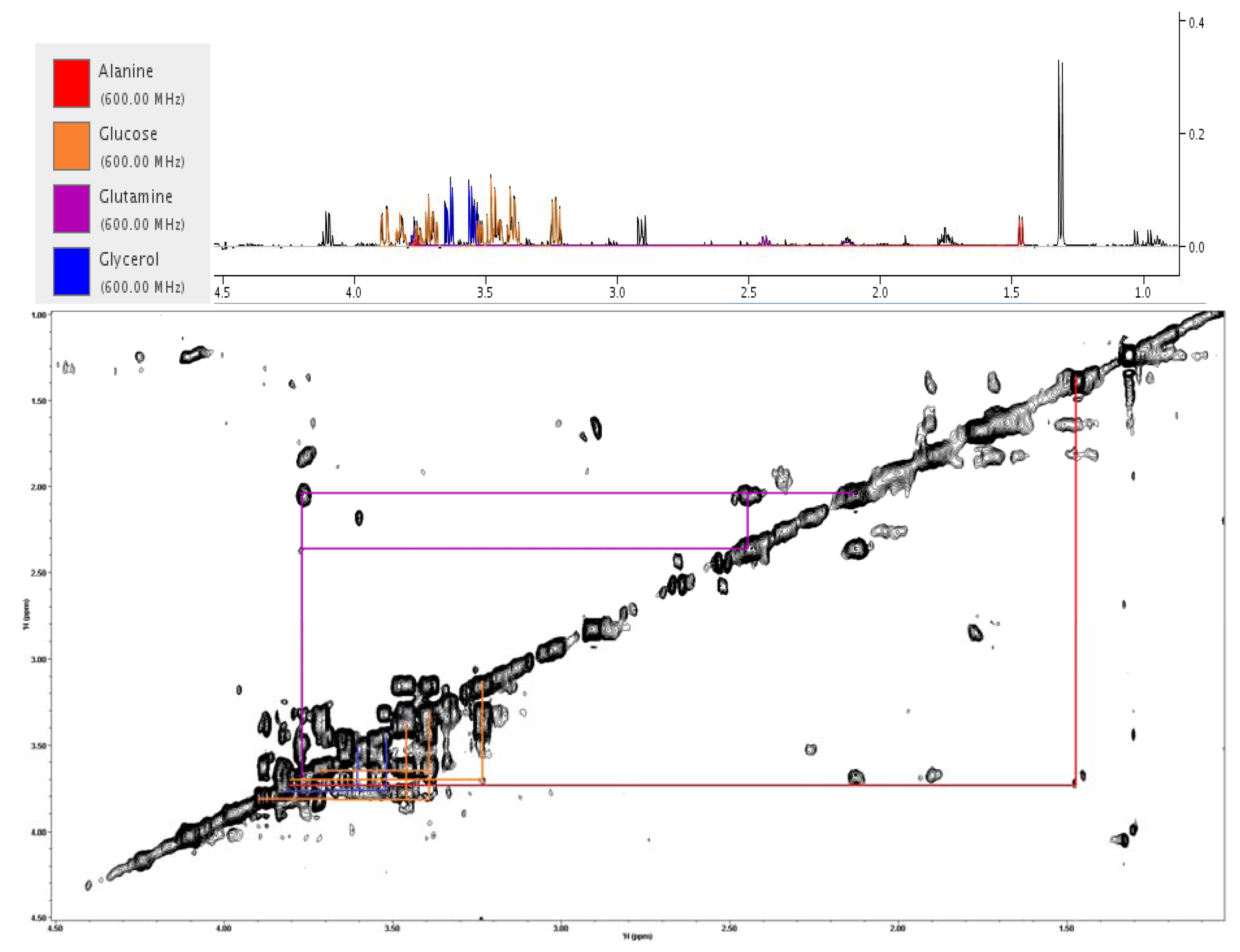
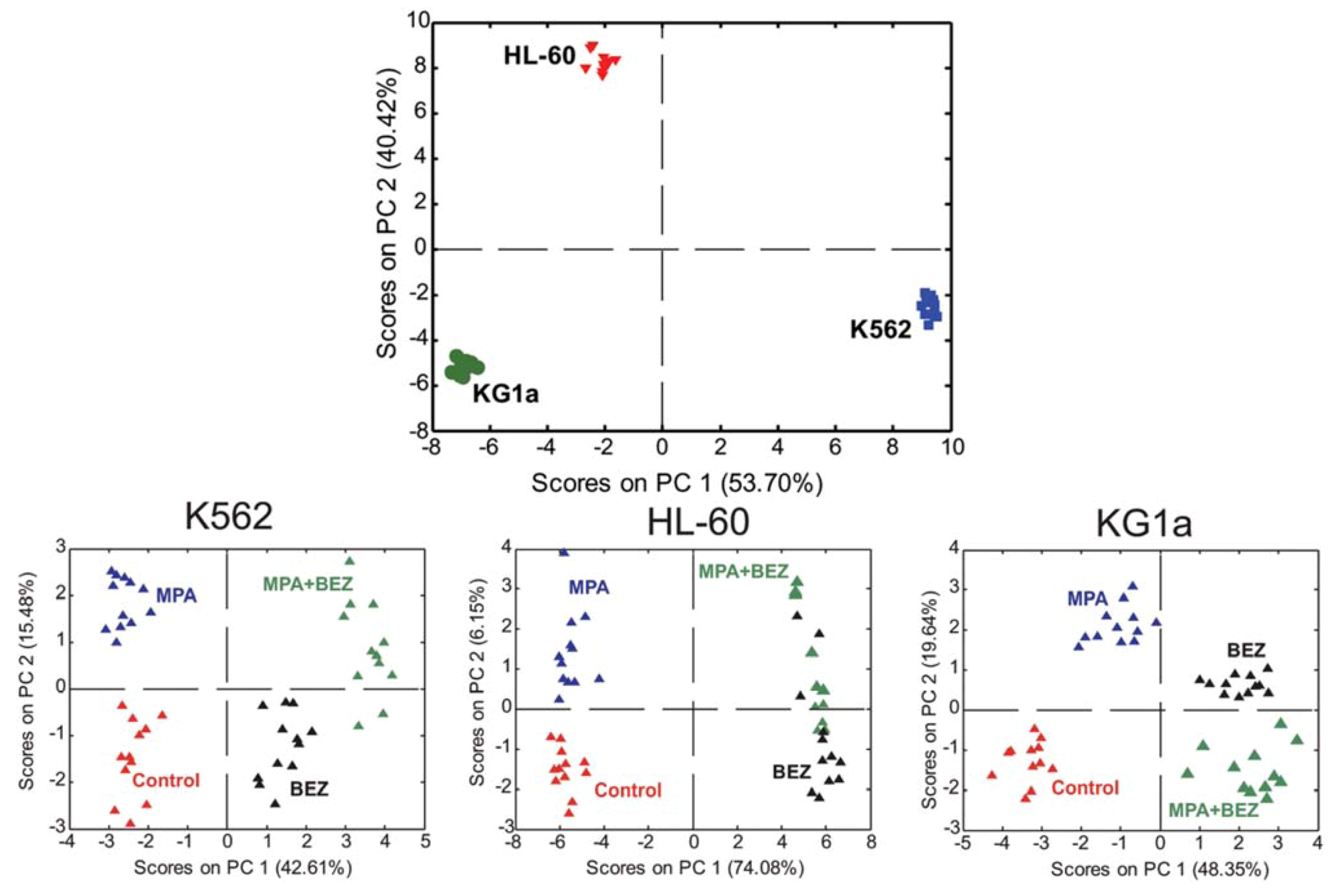
2.5. Evaluating Dose Response
2.6. Evaluation Unconventional Therapies
3. Personalizing and Stratifying Medicine
3.1. Survival and Outcome
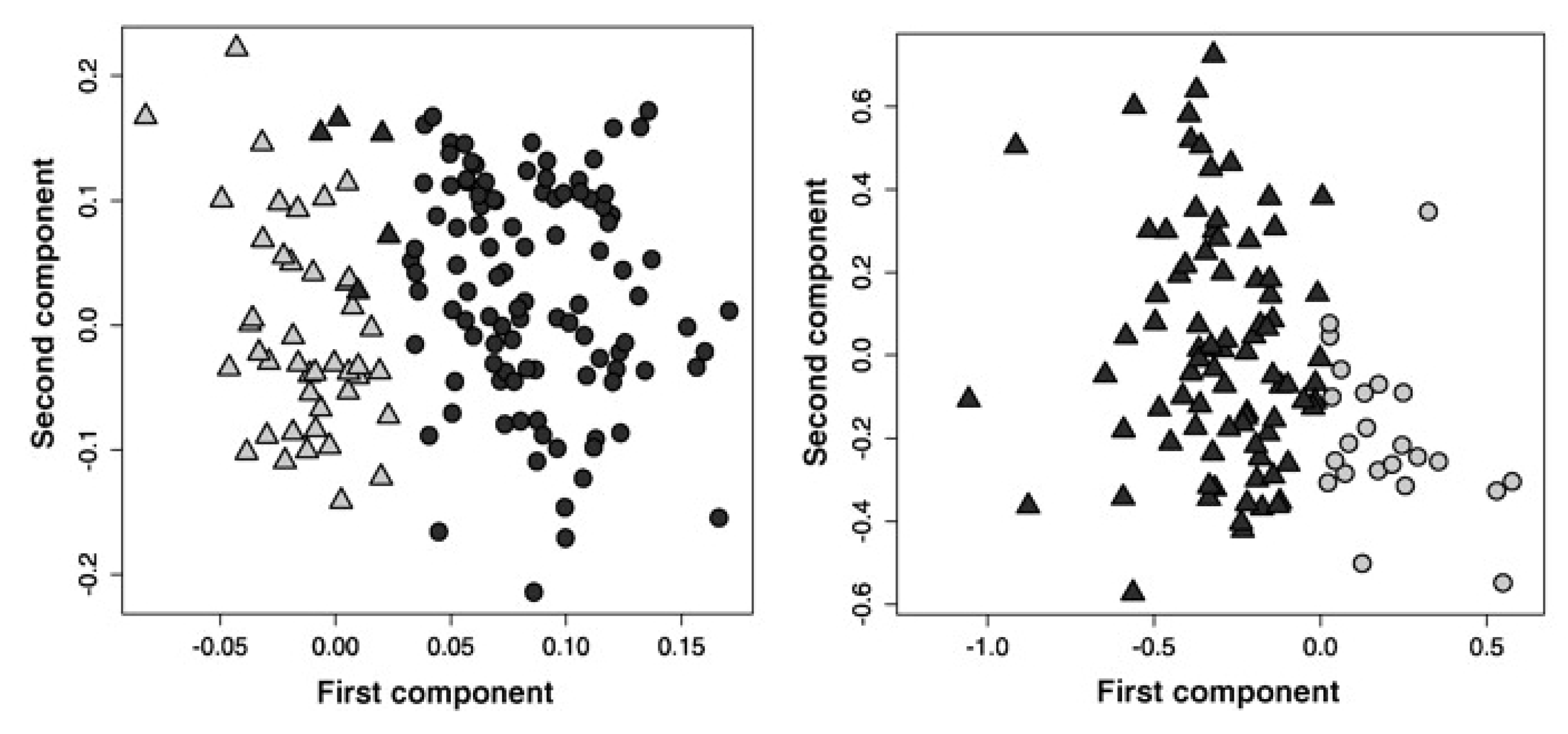
3.2. Identifying Subtypes
3.3. Case Reports
4. Discussion and Conclusions
Acknowledgments
Conflict of Interest
References
- Global Industry Analyst Inc. Available online: http://www.strategyr.com/Metabolomics_Market_Report.asp/ (accessed on 21 January 2013).
- Ben Sellem, D.; Elbayed, K.; Neuville, A.; Moussallieh, F.M.; Lang-Averous, G.; Piotto, M.; Bellocq, J.P.; Namer, I.J. Metabolomic characterization of ovarian epithelial carcinomas by hrmas-NMR spectroscopy. J. Oncol. 2011. [Google Scholar] [CrossRef]
- Weljie, A.M.; Bondareva, A.; Zang, P.; Jirik, F.R. 1H NMR metabolomics identification of markers of hypoxia-induced metabolic shifts in a breast cancer model system. J. Biomol. NMR 2011, 49, 185–193. [Google Scholar] [CrossRef]
- Bathe, O.F.; Shaykhutdinov, R.; Kopciuk, K.; Weljie, A.M.; McKay, A.; Sutherland, F.R.; Dixon, E.; Dunse, N.; Sotiropoulos, D.; Vogel, H.J. Feasibility of identifying pancreatic cancer based on serum metabolomics. Cancer Epidemiol. Biomar. Prev. 2011, 20, 140–147. [Google Scholar] [CrossRef]
- Tiziani, S.; Lopes, L.; Günther, U.L. Early stage diagnosis of oral cancer using 1H NMR–based metabolomics. Neoplasia 2009, 11, 269–276. [Google Scholar]
- Hasim, A.; Ma, H.; Mamtimin, B.; Abudula, A.; Niyaz, M.; Zhang, L.W.; Anwer, J.; Sheyhidin, I. Revealing the metabonomic variation of EC using 1H-NMR spectroscopy and its association with the clinicopathological characteristics. Mol. Biol. Rep. 2012, 39, 8955–8964. [Google Scholar] [CrossRef]
- Carrola, J.; Rocha, C.M.; Barros, A.S.; Gil, A.M.; Goodfellow, B.K.; Carreira, I.M.; Bernardo, J.; Gomes, A.; Sousa, S.; Carvalho, L.; et al. Metabolic signatures of lung cancer in biofluids: NMR-based metabonomics of urine. J. Proteome Res. 2011, 10, 221–230. [Google Scholar] [CrossRef]
- Teahan, O.; Bevan, C.L.; Waxman, J.; Keun, H.C. Metabolic signatures of malignant progression in prostate epithelial cells. Int. J. Biochem. Cell. Biol. 2011, 43, 1002–1009. [Google Scholar] [CrossRef]
- Cao, M.; Zhao, L.; Chen, H.; Xue, W.; Lin, D. NMR-based metabolomic analysis of human bladder cancer. Anal. Sci. 2012, 28, 451–456. [Google Scholar] [CrossRef]
- Chun, E.; Chan, Y.; Koon Koh, P.; Mal, M.; Yean Cheah, P.; Weng Eu, K.; Backshall, A.; Cavill, R.; Nicholson, J.K.; Keun, H.C. Metabolic profiling of human colorectal cancer using high-resolution Magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy and gas chromatography mass spectrometry (GC/MS). J. Proteome Res. 2009, 8, 352–361. [Google Scholar] [CrossRef]
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.; Buie, W.D.; Maclean, A.; Dixon, E.; Sutherland, F.R.; Molckovsky, A.; Vogel, H.J.; Bathe, O.F. Serum metabolomic profile as a means to distinguish stage of colorectal cancer. Genome Med. 2012, 4, 42. [Google Scholar] [CrossRef]
- Slupsky, C.M.; Steed, H.; Wells, T.H.; Dabbs, K.; Schepansky, A.; Capstick, V.; Faught, W.; Sawyer, M.B. Urine metabolite analysis offers potential early diagnosis of ovarian and breast cancers. Clin. Cancer Res. 2010, 16, 5835–5841. [Google Scholar] [CrossRef]
- Fong, M.Y.; McDunn, J.; Kakar, S.S. Identification of metabolites in the normal ovary and their transformation in primary and metastatic ovarian cancer. PloS One 2011, 6, e19963. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [Green Version]
- Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [Google Scholar] [CrossRef]
- Critical Path Opportunities Report. U.S Department of Health and Human Services Food and Drug Administration. 2006. Available online: http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/UCM077254.pdf (accessed on 21 January 2013).
- Orphanos, G.; Kountourakis, P. Targeting the HER2 receptor in metastatic breast cancer. Hematol. Oncol. Stem Cell Ther. 2012, 5, 127–137. [Google Scholar]
- Aiello, M.; Vella, N.; Cannavo, C.; Scalisi, A.; Spandidos, D.A.; Toffoli, G.; Buonadonna, A.; Libra, M.; Stivala, F. Role of genetic polymorphisms and mutations in colorectal cancer therapy (Review). Mol. Med. Rep. 2011, 4, 203–208. [Google Scholar]
- National Cancer Institute. Available online: http://m.cancer.gov/topics/factsheets/targeted/ (accessed on 21 January 2013).
- Lyng, H.; Sitter, B.; Bathen, T.F.; Jensen, L.R.; Sundfor, K.; Kristensen, G.B.; Gribbestad, I.S. Metabolic mapping by use of high-resolution magic angle spinning 1H NMR spectroscopy for assessment of apoptosis in cervical carcinomas. BMC Cancer 2007, 7. [Google Scholar] [CrossRef]
- Blankenberg, F.G.; Katsikis, P.D.; Storrs, R.W.; Beaulieu, C.; Spielman, D.; Chen, J.Y.; Naumovski, L.; Tait, J.F. Quantitative analysis of apoptotic cell death using proton nuclear magnetic resonance spectroscopy. Blood 1997, 89, 3778–3786. [Google Scholar]
- Lindon, J.C.; Keun, H.C.; Ebbels, T.M.D.; Pearce, J.M.T.; Holmes, E.; Nicholson, J.K. The consortium for metabonomic toxicology (COMET): Aims, activities and achievements. Pharmacogenomics 2005, 6, 691–699. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wu, X.; Wang, X. Urine metabolomics. Clin. Chim. Acta 2012, 414, 65–69. [Google Scholar] [CrossRef]
- Weljie, A.M.; Dowlatabadi, R.; Miller, B.J.; Vogel, H.J.; Jirik, F.R. An inflammatory arthritis-associatedmetabolite biomarker pattern revealed by 1H NMR spectroscopy. J. Proteome Res. 2007, 6, 3456–3464. [Google Scholar] [CrossRef]
- Daykin, C.A.; Foxall, P.J.; Connor, S.C.; Lindon, J.C.; Nicholson, J.K. The comparison of plasma deproteinization methods for the detection of low-molecular-weight metabolites by 1H nuclear magnetic resonance spectroscopy. Anal. Biochem. 2002, 304, 220–230. [Google Scholar] [CrossRef]
- Tiziani, S.; Emwas, A.H.; Lodi, A.; Ludwig, C.; Bunce, C.M.; Viant, M.R.; Gunther, U.L. Optimized metabolite extraction from blood serum for 1H nuclear magnetic resonance spectroscopy. Anal. Biochem. 2008, 377, 16–23. [Google Scholar] [CrossRef]
- Soininen, P.; Kangas, A.J.; Würtz, P.; Tukiainen, T.; Tynkkynen, T.; Laatikainen, R.; Järvelin, M-J.; Kähönen, M.; Lehtimäki, T.; Viikari, J.; et al. High-throughput serum NMR metabonomics for cost-effective holistic studies on systemic metabolism. Analyst 2009, 134, 1781–1785. [Google Scholar] [CrossRef]
- Schicho, R.; Shaykhutdinov, R.; Ngo, J.; Nazyrova, A.; Schneider, C.; Panaccione, R.; Kaplan, G.G.; Vogel, H.J.; Storr, M. Quantitative metabolomic profiling of serum, plasma, and urine by 1H NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J. Proteome Res. 2012. [Google Scholar]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted Profiling: quantitative analysis of 1H NMR metabolomics data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Foxhall, P.J.D. 750 MHz 1H and 1H-13C NMR spectroscopy of human blood plasma. Anal. Chem. 1995, 67, 793–811. [Google Scholar] [CrossRef]
- Van, Q.N.; Chmurny, G.N.; Veenstra, T.D. The depletion of protein signals in metabonomics analysis with the WET–CPMG pulse sequence. Biochem. Biophys. Res. Commun. 2003, 301, 952–959. [Google Scholar] [CrossRef]
- Ludwig, C.; Viant, M.R. Two-dimensional J-resolved NMR spectroscopy: review of a key methodology in the metabolomics toolbox. Phytochem. Anal.: PCA 2010, 21, 22–32. [Google Scholar] [CrossRef]
- Fonville, J.M.; Maher, A.D.; Coen, M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Evaluation of full-resolution J-resolved 1H NMR projections of biofluids for metabonomics information retrieval and biomarker identification. Anal. Chem. 2010, 82, 1811–1821. [Google Scholar] [CrossRef]
- Viant, M.R. Improved methods for the acquisition and interpretation of NMR metabolomic data. Biochem. Biophys. Res. Commun. 2003, 310, 943–948. [Google Scholar] [CrossRef]
- Wang, Y.; Bollard, M.E.; Keun, H.; Antti, H.; Beckonert, O.; Ebbels, T.M.; Lindon, J.C.; Holmes, E.; Tang, H.; Nicholson, J.K. Spectral editing and pattern recognition methods applied to high-resolution magic-angle spinning 1H nuclear magnetic resonance spectroscopy of liver tissues. Anal. Biochem. 2003, 323, 26–32. [Google Scholar] [CrossRef]
- Bertram, H.C.; Eggers, N.; Eller, N. Potential of human saliva for nuclear magnetic resonance-based metabolomics and for health-related biomarker identification. Anal. Chem. 2009, 81, 9188–9193. [Google Scholar] [CrossRef]
- Monleon, D.; Morales, J.M.; Barrasa, A.; Lopez, J.A.; Vazquez, C.; Celda, B. Metabolite profiling of fecal water extracts from human colorectal cancer. NMR Biomed. 2009, 22, 342–348. [Google Scholar] [CrossRef]
- Wishart, D.S.; Lewis, M.J.; Morrissey, J.A.; Flegel, M.D.; Jeroncic, K.; Xiong, Y.; Cheng, D.; Eisner, R.; Gautam, B.; Tzur, D.; et al. The human cerebrospinal fluid metabolome. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 871, 164–173. [Google Scholar] [CrossRef]
- Hügle, T.; Kovats, H.; Heijnen, I.A.; Daikeler, T.; Baisch, U.; Hicks, J.M.; Valderrabano, V. Synovial fluid metabolomics in different forms of arthritis assessed by nuclear magnetic resonance spectroscopy. Clin. Exp. Rheumatol. 2012, 30, 240–245. [Google Scholar]
- Moestue, S.; Sitter, B.; Frost Bathen, T.; Tessem, M-B.; Gribbestad, I.S. HR MAS MR spectroscopy in metabolic characterization of cancer. Curr. Top. Med. Chem. 2011, 11, 2–26. [Google Scholar]
- Ferentz, A.E.; Wagner, G. NMR spectroscopy : A multifaceted approach to macromolecular structure. Q Rev. Biophys. 2000, 3, 29–65. [Google Scholar] [CrossRef]
- Bax, A.; Grishaev, A. Weak alignment NMR: A hawk-eyed view of biomolecular structure. Curr. Opin. Struct. Biol. 2005, 15, 563–570. [Google Scholar] [CrossRef]
- Tugarinov, V.; Hwang, P.M.; Kay, L.E. Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu. Rev. Biochem. 2004, 73, 107–146. [Google Scholar] [CrossRef]
- Pellecchia, M.; Sem, D.S.; Wuthrich, K. NMR in drug discovery. Nat. Rev. Drug Discov. 2002, 1, 211–219. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. NMR-based screening in drug discovery. Q Rev. Biophys. 1999, 32, 211–240. [Google Scholar] [CrossRef]
- Jourdan, C.; Petersen, A.K.; Gieger, C.; Doring, A.; Illig, T.; Wang-Sattler, R.; Meisinger, C.; Peters, A.; Adamski, J.; Prehn, C.; et al. Body fat free mass is associated with the serum metabolite profile in a population-based study. PloS One 2012, 7, e40009. [Google Scholar]
- Putri, S.P.; Nakayama, Y.; Matsuda, F.; Uchikata, T.; Kobayashi, S.; Matsubara, A.; Fukusaki, E. Current metabolomics: Practical applications. J. Biosci. Bioeng. 2013, 579–589. [Google Scholar]
- Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics 2011, 7, 307–328. [Google Scholar] [CrossRef]
- Griffin, J.L.; Shockcor, J.P. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004, 4, 551–561. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Conelly, J.; Lindon, J.C.; Holmes, E. Metabonomics: A platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 2002, 1, 153–161. [Google Scholar] [CrossRef]
- Gowda, G.A.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Exp. Rev. Mol. Diagn 2008, 8, 617–633. [Google Scholar] [CrossRef]
- Bathen, T.F.; Sitter, B.; Sjobakk, T.E.; Tessem, M.B.; Gribbestad, I.S. Magnetic resonance metabolomics of intact tissue: A biotechnological tool in cancer diagnostics and treatment evaluation. Cancer Res. 2010, 70, 6692–6696. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.I.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar]
- Martin, P.; Oliver, S.; Kennedy, S.J.; Partridge, E.; Hutchison, M.; Clarke, D.; Giles, P. Pharmacokinetics of vandetanib: Three phase I studies in healthy subjects. Clin. Ther. 2012, 34, 221–237. [Google Scholar] [CrossRef]
- Cohen, M.H.; Hirschfeld, S.; Flamm Honig, S.; Ibrahim, A.; Johnson, J.R.; O’Leary, J.J.; White, R.M.; Williams, G.A.; Pazdur, R. Drug approval summaries: arsenic trioxide, tamoxifen citrate, anastrazole, paclitaxel, bexarotene. Oncologist 2001, 6, 4–11. [Google Scholar]
- Schnackenberg, L.; Beger, R.D.; Dragan, Y. NMR-based metabonomic evaluation of livers from rats chronically treated with tamoxifen, mestranol, and phenobarbital. Metabolomics 2005, 1, 87–94. [Google Scholar] [CrossRef]
- Tenori, L.; Oakman, C.; Claudino, W.M.; Bernini, P.; Cappadona, S.; Nepi, S.; Biganzoli, L.; Arbushites, M.C.; Luchinat, C.; Bertini, I.; et al. Exploration of serum metabolomic profiles and outcomes in women with metastatic breast cancer: a pilot study. Mol. Oncol. 2012, 6, 437–444. [Google Scholar] [CrossRef]
- National Cancer Institute. Available online: http://www.cancer.gov/newscenter/qa/2001/gleevecqa/ (accessed on: 21 January 2013).
- Druker, B.J.; Guillot., F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.N.; Silver, R.T.; Goldman, J.M.; Stone, R.M. A five-year follow-UP of patients receiving imatinib for chronic myeloid leukemia. N Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef]
- Hochhaus, A.; O'Brien, S.G.; Guilhot, F.; Druker, B.J.; Branford, S.; Foroni, L.; Goldman, J.M.; Muller, M.C.; Radich, J.P.; Rudoltz, M.; et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009, 23, 1054–1061. [Google Scholar] [CrossRef]
- Saito, S.; Nakata, K.; Kajiura, S.; Ando, T.; Hosokawa, A.; Sugiyama, T. Long-term follow-up outcome of imatinib mesylate treatment for recurrent and unresectable gastrointestinal stromal tumors. Digestion 2013, 87, 47–52. [Google Scholar] [CrossRef]
- Dewar, B.J.; Keshari, K.; Jeffries, R.; Dzeja, P.; Graves, L.M.; Macdonald, J.M. Metabolic assessment of a novel chronic myelogenous leukemic cell line and an imatinib resistant subline by 1H NMR spectroscopy. Metabolomics 2010, 6, 439–450. [Google Scholar]
- Dengler, M.A.; Staiger, A.M.; Gutekunst, M.; Hofmann, U.; Doszczak, M.; Scheurich, P.; Schwab, M.; Aulitzky, W.E.; van der Kuip, H. Oncogenic stress induced by acute hyper-activation of Bcr-Abl leads to cell death upon induction of excessive aerobic glycolysis. PloS One 2011, 6, e25139. [Google Scholar]
- Spratlin, J.L.; Pitts, T.M.; Kulikowski, G.N.; Morelli, M.P.; Tentler, J.J.; Serkova, N.J.; Eckhardt, S.G. Synergistic activity of histone deacetylase and proteasome inhibition against pancreatic and hepatocellular cancer cell lines. Anticancer Res. 2011, 31, 1093–1104. [Google Scholar]
- National Center for Advancing Translational Sciences. Available online: http://www.ncats.nih.gov/research/reengineering/process.html/ (acessed on: 21 January 2013).
- Cavill, R.; Keun, H.C.; Holmes, E.; Lindon, J.C.; Nicholson, J.K.; Ebbels, T.M. Genetic algorithmsfor simultaneous variable and sample selection in metabonomics. Bioinformatics 2009, 25, 112–118. [Google Scholar]
- Coen, M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. NMR-based metabolic profiling and metabonomic approaches to problems in molecular toxicology. Chem. Res. Toxicol. 2008, 21, 9–27. [Google Scholar] [CrossRef]
- Verstappen, C.C.P.; Heimans, J.J.; Hoekman, K.; Postma, T.J. Neurotoxic complications of chemotherapy in patients with cancer. Drugs 2003, 63, 1549–1563. [Google Scholar] [CrossRef]
- Lyman, G.H. Impact of chemotherapy dose intensity on cancer patient outcomes. J. Natl. Compr. Cancer Network 2009, 7, 99–108. [Google Scholar]
- Sorg, B.L.; Hull, W.E.; Kliem, H.C.; Mier, W.; Wiessler, M. Synthesis and NMR characterization of hydroxyurea and mesylglycol glycoconjugates as drug candidates for targeted cancer chemotherapy. Carbohydr Res. 2005, 340, 181–189. [Google Scholar] [CrossRef]
- Naser-Hijazi, B.; Berger, M.R.; Schmähl, D.; Schlag, P.; Hull, W.E. Locoregional administration of 5-fluoro-2'-deoxyuridine (FdUrd) in Novikoff hepatoma in the rat: effects of dose and infusion time on tumor growth and on FdUrd metabolite levels in tumor tissue as determined by 19F-NMR spectroscopy. J. Cancer Res. Clin. 1991, 117, 295–304. [Google Scholar]
- Backshall, A.; Sharma, R.; Clarke, S.J.; Keun, H.C. Pharmacometabonomic profiling as a predictor of toxicity in patients with inoperable colorectal cancer treated with capecitabine. Clin. Cancer Res. 2011, 17, 3019–3028. [Google Scholar] [CrossRef]
- Adamski, J.; Suhre, K. Metabolomics platforms for genome wide association studies-linking the genome to the metabolome. Curr. Opin. Biotechnol. 2013, 24, 39–47. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, Y.; Wu, C.; Zhao, J.; Yu, S.; Yuan, B.; Yan, X.; Liao, M. Study of a novel indolin-2-ketone compound Z24 induced hepatotoxicity by NMR-spectroscopy-based metabonomics of rat urine, blood plasma, and liver extracts. Toxicol. Appl. Pharmacol. 2006, 215, 71–82. [Google Scholar]
- Wei, S.; Liu, L.; Zhang, J.; Bowers, J.; Gowda, G.A.; Seeger, H.; Fehm, T.; Neubauer, H.J.; Vogel, U.; Clare, S.E.; Raftery, D. Metabolomics approach for predicting response to neoadjuvant chemotherapy for breast cancer. Mol. Oncol. 2012. [Google Scholar] [CrossRef]
- El-Deredy, W.; Ashmore., S.M.; Branston, N.M.; Darling, J.L.; Williams, S.R.; Thomas, D.G.T. Pretreatment prediction of the chemotherapeutic response of human glioma cell cultures using nuclear magnetic resonance spectroscopy and artifical neural networks. Cancer Res. 1997, 57, 4196–4199. [Google Scholar]
- Triba, M.N.; Starzec, A.; Bouchemal, N.; Guenin, E.; Perret, G.Y.; Le Moyec, L. Metabolomic profiling with NMR discriminates between biphosphonate and doxorubicin effects on B16 melanoma cells. NMR Biomed. 2010, 23, 1009–1016. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Loiseau, D.; Rio, P.; Demidem, A.; Barthomeuf, C.; Stepien, G.; Morvan, D. Quantitative two-dimensional HRMAS 1H-NMR spectroscopy-based metabolite profiling of human cancer cell lines and response to chemotherapy. Magn. Reson. Med. 2010, 63, 1172–1183. [Google Scholar] [CrossRef]
- Tiziani, S.; Lodi, A.; Khanim, F.L.; Viant, M.R.; Bunce, C.M.; Gunther, U.L. Metabolomic profiling of drug responses in acute myeloid leukaemia cell lines. PloS One 2009, 4, e4251. [Google Scholar]
- Mailloux, J.M.; Bériault, R.; Lemire, J.; Singh, R.; Chénier, D.R.; Hamel, R.H.; Appanna, V.D. The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PloS One 2007, 2, e690. [Google Scholar]
- Sonneveld, P.; Schmidt-Wolf, I.G.; van der Holt, B.; El Jarari, L.; Bertsch, U.; Salwender, H.; Zweegman, S.; Vellenga, E.; Broyl, A.; Blau, I.W.; et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/ GMMG-HD4 trial. J. Clin. Oncol. 2012, 30, 2946–2955. [Google Scholar] [CrossRef]
- Giaccone, G.; Rajan, A.; Berman, A.; Kelly, R.J.; Szabo, E.; Lopez-Chavez, A.; Trepel, J.; Lee, M.J.; Cao, L.; Espinoza-Delgado, I.; et al. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J. Clin. Oncol. 2011, 29, 2052–2059. [Google Scholar] [CrossRef]
- US National Institutes of Health. Available online: http://clinicaltrials.gov/ct2/show/NCT00431340?term=belinostat&rank=16/ (acessed on 21 January 2013).
- Pan, X.; Wilson, M.; Mirbahai, L.; McConville, C.; Arvanitis, T.N.; Griffin, J.L.; Kauppinen, R.A.; Peet, A.C. In vitro metabonomic study detects increases in UDP-GlcNAc and UDP-GalNAc, as early phase markers of cisplatin treatment response in brain tumor cells. J. Proteome Res. 2011, 10, 3493–3500. [Google Scholar] [CrossRef]
- Gu, Y.; Mi, W.; Ge, Y.; Liu, H.; Fan, Q.; Han, C.; Yang, J.; Han, F.; Lu, X.; Yu, W. GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res. 2010, 70, 6344–6351. [Google Scholar] [CrossRef]
- Brooks, S.A.; Carter, T.M.; Bennett, E.P.; Clausen, H.; Mandel, U. Immunolocalisation of members of the polypeptide N-acetylgalactosaminyl transferase (ppGalNAc-T) family is consistent with biologically relevant altered cell surface glycosylation in breast cancer. Acta Histochem. 2007, 109, 273–284. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Morvan, D.; Chollet, P.; Barthomeuf, C. Pharmacometabolomics of docetaxel-treated human MCF7 breast cancer cells provides evidence of varying cellular responses at high and low doses. Breast Cancer Res. Treat. 2010, 120, 613–626. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Lim, S.; Barthomeuf, C.; Morvan, D. Biochemical disorders induced by cytotoxic marine natural products in breast cancer cells as revealed by proton NMR spectroscopy-based metabolomics. Biochem. Pharmacol. 2010, 80, 1170–1179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Sun, H.; Wang, Z.; Sun, W.; Wang, P.; Wang, X. Metabolomics: Towards understanding traditional Chinese medicine. Planta Med. 2010, 76, 2026–2035. [Google Scholar] [CrossRef]
- Halouska, S.; Fenton, R.J.; Barletta, R.G.; Powers, R. Predicting the in vivo mechanism of action for drug leads using NMR metabolomics. ACS Chem. Biol. 2012, 7, 166–171. [Google Scholar] [CrossRef]
- Bertini, I.; Cacciatore, S.; Jensen, B.V.; Schou, J.V.; Johansen, J.S.; Kruhoffer, M.; Luchinat, C.; Nielsen, D.L.; Turano, P. Metabolomic NMR fingerprinting to identify and predict survival of patients with metastatic colorectal cancer. Cancer Res. 2012, 72, 356–364. [Google Scholar]
- Lodi, A.; Tiziani, S.; Khanim, F.L.; Gunther, U.L.; Viant, M.R.; Morgan, G.J.; Bunce, C.M.; Drayson, M.T. Proton NMR-based metabolite analyses of archived serial paired serum and urine samples from myeloma patients at different stages of disease activity identifies acetylcarnitine as a novel marker of active disease. PloS One 2013, 8, e56422. [Google Scholar]
- US Food and Drug Administration. Available online: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm193900.html/ (accessed on 21 January 2013).
- Borgan, E.; Lindholm, E.M.; Moestue, S.; Maelandsmo, G.M.; Lingjaerde, O.C.; Gribbestad, I.S.; Borresen-Dale, A.L.; Engebraaten, O.; Sorlie, T. Subtype-specific response to bevacizumab is reflected in the metabolome and transcriptome of breast cancer xenografts. Mol. Oncol. 2012. [Google Scholar]
- Abaffy, T.; Moller, M.; Riemer, D.D.; Milikowski, C.; Defazio, R.A. A case report—Volatile metabolomic signature of malignant melanoma using matching skin as a control. J. Cancer Sci. Ther. 2011, 3, 140–144. [Google Scholar]
- Vriens, D.; de Geus-Oei, L.F.; Heerschap, A.; van Laarhoven, H.W.; Oyen, W.J. Vascular and metabolic response to bevacizumab-containing regimens in two patients with colorectal liver metastases measured by dynamic contrast-enhanced MRI and dynamic 18F-FDG-PET. Clin. Colorectal. Cancer 2011, 10, E1–E5. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Palmnas, M.S.A.; Vogel, H.J. The Future of NMR Metabolomics in Cancer Therapy: Towards Personalizing Treatment and Developing Targeted Drugs? Metabolites 2013, 3, 373-396. https://doi.org/10.3390/metabo3020373
Palmnas MSA, Vogel HJ. The Future of NMR Metabolomics in Cancer Therapy: Towards Personalizing Treatment and Developing Targeted Drugs? Metabolites. 2013; 3(2):373-396. https://doi.org/10.3390/metabo3020373
Chicago/Turabian StylePalmnas, Marie S.A., and Hans J Vogel. 2013. "The Future of NMR Metabolomics in Cancer Therapy: Towards Personalizing Treatment and Developing Targeted Drugs?" Metabolites 3, no. 2: 373-396. https://doi.org/10.3390/metabo3020373
APA StylePalmnas, M. S. A., & Vogel, H. J. (2013). The Future of NMR Metabolomics in Cancer Therapy: Towards Personalizing Treatment and Developing Targeted Drugs? Metabolites, 3(2), 373-396. https://doi.org/10.3390/metabo3020373