Rationales and Approaches for Studying Metabolism in Eukaryotic Microalgae


Abstract
:1. Introduction
2. Microalgae: Potential Candidate Species for Metabolic Engineering

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species (group) | Transfection method a | Transgene integration b | Promoters c | Selection marker d | Reference |
|---|---|---|---|---|---|
| Thalassiosira pseudonana (Diatoms) | PB | NHR | LHCF9 NR | nat | [15] |
| Phaeodactylum tricornutum (Diatoms) | PB | NHR | fcpA | ble nat nptII sat-1 cat | [16,17,18,19] |
| Nannochloropsis gaditana/oculata (Eustigmatophytes) | EP AB | NHR HR Transient | VCP1 VCP2 UEP βTUB HSP70 HSP70-RBCS2 | ble hygR bsr | [11,12,13,14,20] |
| Cyanidioschyzon merolae (Rhodophytes) | EP PEG | HR Transient | URA3 Catalase βTUB | GFP URA3 | [21,22,23] |
| Chlorella (Chlorophytes) | PB PEG EP | HR Transient | CaMV-35S Chlorella virus Chlamydomonas RBCS2 | NR hpt nptII | [24,25,26] |
| Haematococcus pluvialis (Chlorophytes) | PB EP | NHR Transient | SV40 Phytoene desaturase | Modified phytoene desaturase | [27] |
| Dunaliella salina (Chlorophytes) | PB EP GB | NHR Transient | Maize ubiquitin CaMV-35S Chlamydomonas RBCS2 Actin CA NR | bar ble NR | [28,29,30] |
| Chlamydomonas reinhardtii (Chlorophytes) | PB EP GB SCW AB | NH HR (chloroplast) | HSP70A-RBCS2 PSAD β2TUB NR CYC6 | ARG7 NR (nit1) ble aphVIII aph7“ aadA | [31,32,33,34,35,36,37,38,39,40,41] |
| Volvox carteri (Chlorophytes) | PB | NHR | NR | NR | [42] |
3. Systems Biology towards Microalgal Biotechnology
4. Metabolite Profiling Using GC-MS
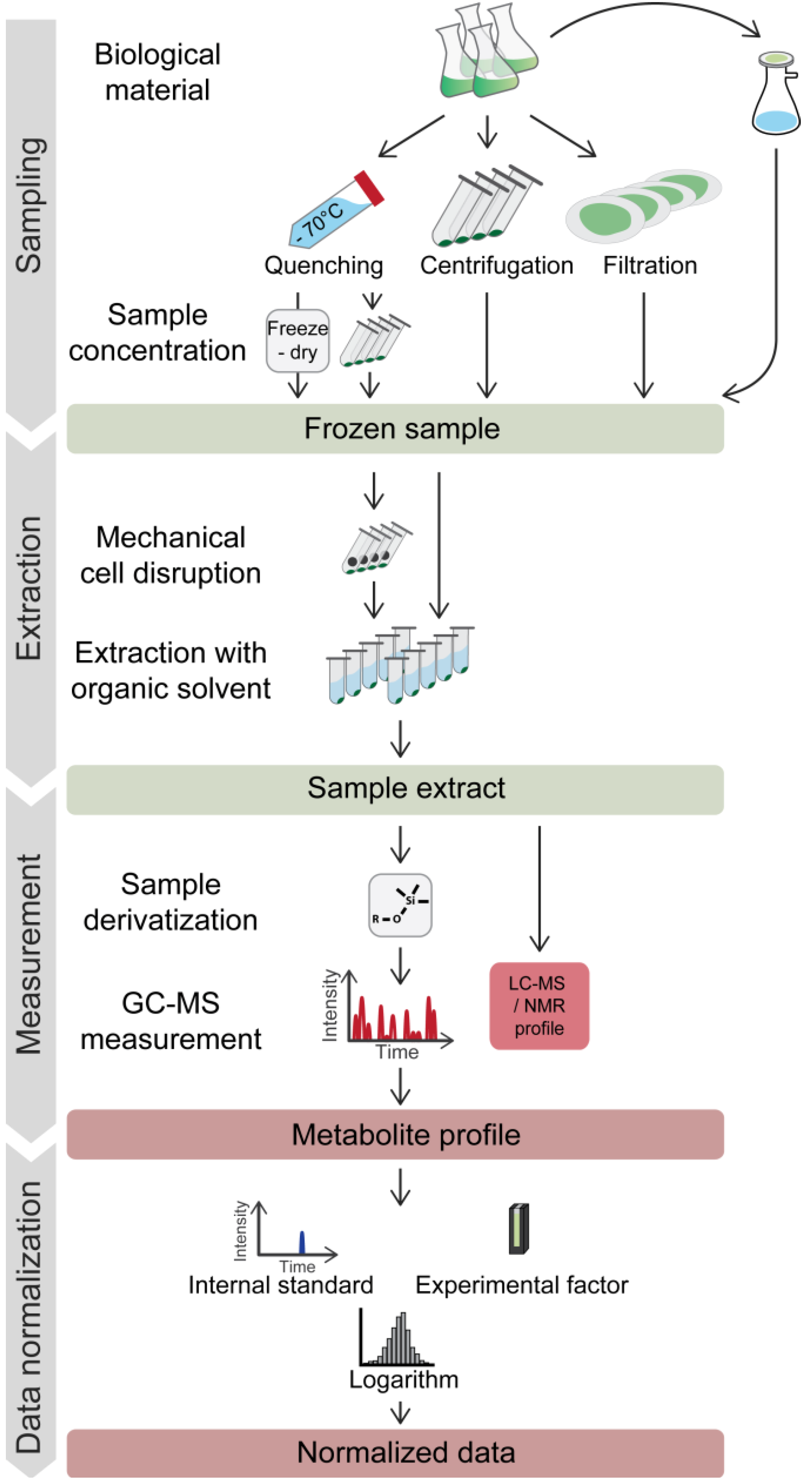
5. From Experiment to Data: Metabolite Profiling Workflow for Chlamydomonas
5.1. Cell Harvest

5.1.1. Quenching before (or without) Separating Cells from Growth Medium
5.1.2. Centrifugation
| Study | Strain used | Harvesting method a | Harvesting conditions b | Harvested cells c | Mechanical cell disruption | Extraction buffer d | Cells/mL extraction buffer | Extract equivalent to cells injected into GC-MS |
|---|---|---|---|---|---|---|---|---|
| [111] | CC 125 | Q | 32.5% MW; −25 °C; 4:1 | × | mortar and pestle | MCW 10:3:1 | 1.20 × 106 | × |
| [112] | CC 125 | Q | 70% MW; −70 °C; 1:1 | 2.50 × 106 | 5-mm steel ball | MCW 5:2:1 | 1.92 × 106 | 1.68 × 104 |
| [113] | CC 503 cw92 mt+ | Q broth | 100% M; −20 °C; 0.43:1 | × | none | MCW 1:1:0 | × | × |
| [115] | Stm6 | C | 3,000 g; 1 min; 4 °C | 3.00 × 107 | none | MCW 1:0:0 | 6.00 × 107 | 1.00 × 107 |
| [116] | CC 406 & Stm6Glc4 | C | 3,000 g; 1 min | 3.60 × 108 | homogenizer, 0.1-mm silica beads | MCW 4:0:1 | 3.60 × 108 | 2.52 × 106 |
| [114] | cw 92 | Q | 32.5 MW; −25 °C; 4:1 | 1.50 × 106 | none | MCW 3:1:1 | × | × |
| [117] | CC 503 cw92 mt+ | Q broth | 100% M; −20 °C; 1:1 | 6.00 × 106 | none | MCW 5:2:1 | 3.00 × 107 | 5.31 × 104 |
| [118] | CC 125 | Q | 70% MW; −80 °C; 3:1 | 2.00 × 106 | sonicator (3 × 30 sec) | MCW 10:3:1 | 2.00 × 106 | 2.00 × 104 |
| [119] | CC 125 | Q | 70% MW; −70 °C; 1:1 | 5.00 × 106 | 5-mm steel ball | MCW 5:2:2 | 6.67 × 106 | 8.97 × 104 |
| [120] | cw 15 | F | × | 3.50 × 107 | none | MCW 5:2:1 | 1.75 × 107 | 1.75 × 105 |
| [121] | CC 503 cw92 mt+ | × | × | 15–25 mg fresh weight | Retsch mill, quartz sand | MCW 5:2:1 (1% acetic acid) | × | × |
| [122] | CC125 | F | 30–45 sec | 2.00 × 107 | mortar and pestle | MCW 0:1:1 | 4.00 × 106 | 1.92 × 105 |
| [123] | CC125 | Q | 70% MW; −70 °C; 1:1 | 7.00 × 106 | 5-mm steel ball | MCW 5:2:2 | 9.33 × 106 | 6.53 × 104 |
| This publication (see Experimental section) | CC 1690 | F | 10–20 sec | 1.00 × 107 | none | MCW 7:3:0 | 1.39 × 107 | 1.50 × 105 |
5.1.3. Fast Filtration
5.1.4. Different Harvesting Methods Produce Distinct Metabolite Profiles of Chlamydomonas Cells

5.2. Metabolite Extraction
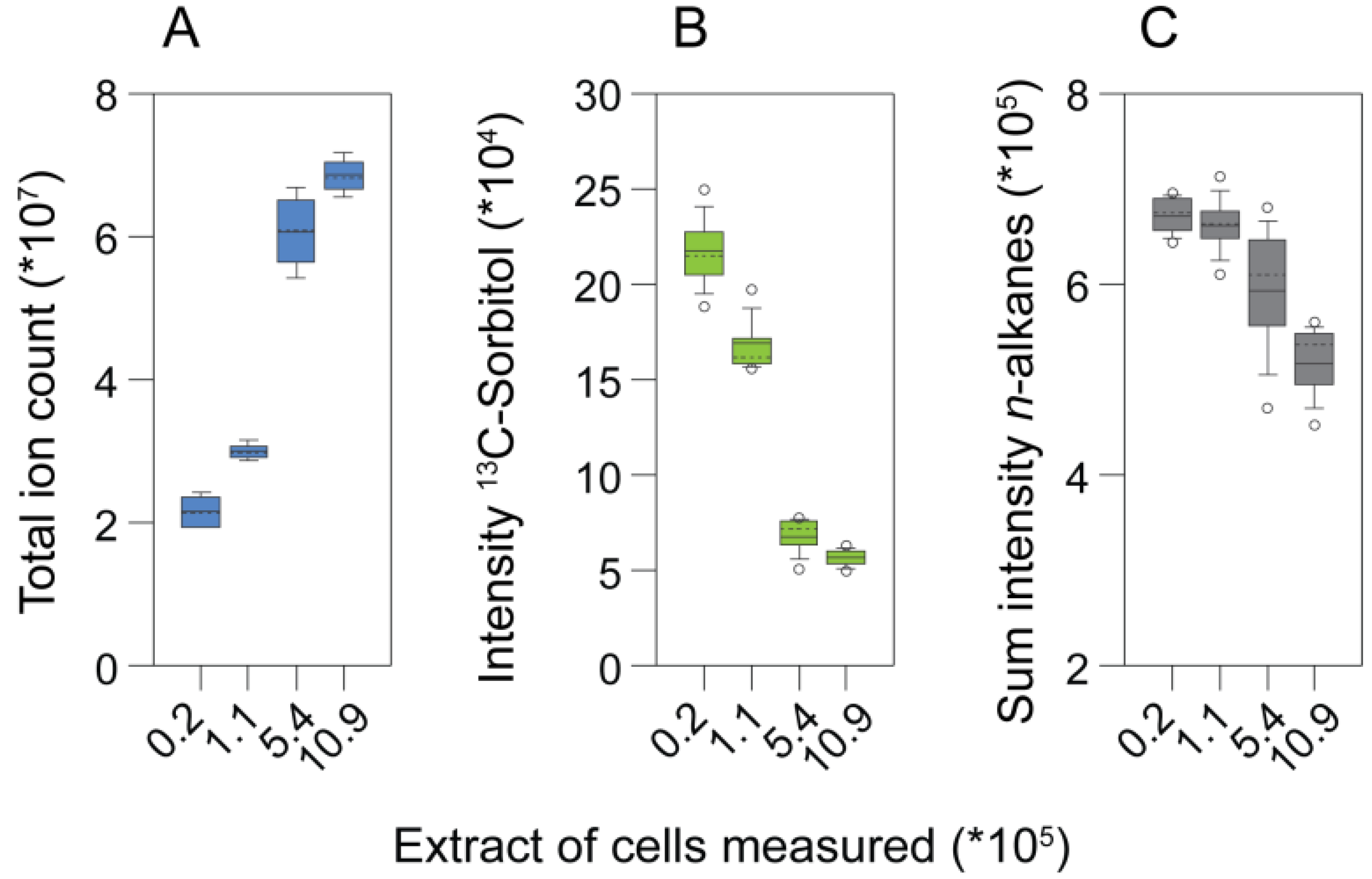
5.3. Sample Amounts, Matrix Effects and Extracellular Metabolites
5.3.1. The Linear Range of Biomass Concentration in Chlamydomonas Metabolite Extracts Is Limited


5.3.2. Extracellular Metabolites and Growth Media Have Strong Impacts on the Sample Matrix in Chlamydomonas Metabolite Extracts

5.4. GC-MS Data Normalization
7. Experimental Section
7.1. Harvesting Methods Comparison Experiment (Figure 2)
7.2. Extract Concentration Experiment (Figure 3 and Figure 4)
7.3. Extracellular Metabolite Experiment (Figure 5)
7.4. Metabolite Derivatization and Measurement
7.5. Data Pre-Processing and Peak Identification
7.6. Data Processing and Visualization
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Georgianna, D.R.; Mayfield, S.P. Exploiting diversity and synthetic biology for the production of algal biofuels. Nature 2012, 488, 329–335. [Google Scholar] [CrossRef]
- Larkum, A.W.; Ross, I.L.; Kruse, O.; Hankamer, B. Selection, breeding and engineering of microalgae for bioenergy and biofuel production. Trends Biotechnol. 2012, 30, 198–205. [Google Scholar] [CrossRef]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal triacylglycerols as feedstocks for biofuel production: Perspectives and advances. Plant J. 2008, 54, 621–639. [Google Scholar] [CrossRef]
- Merchant, S.S.; Kropat, J.; Liu, B.; Shaw, J.; Warakanont, J. TAG, you're it! Chlamydomonas as a reference organism for understanding algal triacylglycerol accumulation. Curr. Opin. Biotechnol. 2012, 23, 352–363. [Google Scholar]
- Barsanti, L.; Coltelli, P.; Evangelista, V.; Frassanito, A.M.; Passarelli, V.; Vesentini, N.; Gualtieri, P. The world of algae. In Algal Toxins: Nature, Occurrence, Effect and Detection; Evangelista, V., Barsanti, L., Frassanito, A.M., Passarelli, V., Gualtieri, P., Eds.; 2008; pp. 1–15. [Google Scholar]
- Leliaert, F.; Smith, D.R.; Moreau, H.; Herron, M.D.; Verbruggen, H.; Delwiche, C.F.; De Clerck, O. Phylogeny and molecular evolution of the green algae. Crit. Rev. Plant Sci 2012, 31, 1–46. [Google Scholar] [CrossRef]
- Pulz, O.; Gross, W. Valuable products from biotechnology of microalgae. Appl. Microbiol. Biotechnol. 2004, 65, 635–648. [Google Scholar] [CrossRef]
- Tirichine, L.; Bowler, C. Decoding algal genomes: tracing back the history of photosynthetic life on Earth. Plant J. 2011, 66, 45–57. [Google Scholar] [CrossRef]
- Leon, R.; Fernandez, E. Nuclear transformation of eukaryotic microalgae: Historical overview, achievements and problems. Adv. Exp. Med. Biol. 2007, 616, 1–11. [Google Scholar] [CrossRef]
- Rodolfi, L.; Chini Zittelli, G.; Bassi, N.; Padovani, G.; Biondi, N.; Bonini, G.; Tredici, M.R. Microalgae for oil: Strain selection, induction of lipid synthesis and outdoor mass cultivation in a low-cost photobioreactor. Biotechnol. Bioeng. 2009, 102, 100–112. [Google Scholar]
- Radakovits, R.; Jinkerson, R.E.; Fuerstenberg, S.I.; Tae, H.; Settlage, R.E.; Boore, J.L.; Posewitz, M.C. Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropis gaditana. Nat. Commun 2012, 3, 686. [Google Scholar] [CrossRef]
- Cha, T.S.; Chen, C.F.; Yee, W.; Aziz, A.; Loh, S.H. Cinnamic acid, coumarin and vanillin: Alternative phenolic compounds for efficient Agrobacterium-mediated transformation of the unicellular green alga, Nannochloropsis sp. J. Microbiol. Methods 2011, 84, 430–434. [Google Scholar] [CrossRef]
- Chen, H.L.; Li, S.S.; Huang, R.; Tsai, H.J. Conditional production of a functional fish growth hormone in the transgenic line of Nannochloropsis oculata (Eustigmatophyceae). J. Phycol 2008, 44, 768–776. [Google Scholar] [CrossRef]
- Kilian, O.; Benemann, C.S.; Niyogi, K.K.; Vick, B. High-efficiency homologous recombination in the oil-producing alga Nannochloropsis sp. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 21265–21269. [Google Scholar] [CrossRef]
- Poulsen, N.; Chesley, P.M.; Kroger, N. Molecular genetic manipulation of the diatom Thalassiosira pseudonana (Bacillariophyceae). J. Phycol 2006, 42, 1059–1065. [Google Scholar] [CrossRef]
- Apt, K.E.; Kroth-Pancic, P.G.; Grossman, A.R. Stable nuclear transformation of the diatom Phaeodactylum tricornutum. Mol. Gen. Genet. 1996, 252, 572–579. [Google Scholar]
- Zaslavskaia, L.A.; Lippmeier, J.C.; Shih, C.; Ehrhardt, D.; Grossman, A.R.; Apt, K.E. Trophic conversion of an obligate photoautotrophic organism through metabolic engineering. Science 2001, 292, 2073–2075. [Google Scholar] [CrossRef]
- Zaslavskaia, L.A.; Lippmeier, J.C.; Kroth, P.G.; Grossman, A.R.; Apt, K.E. Transformation of the diatom Phaeodactylum tricornutum (Bacillariophyceae) with a variety of selectable marker and reporter genes. J. Phycol. 2000, 36, 379–386. [Google Scholar]
- Sakaguchi, T.; Nakajima, K.; Matsuda, Y. Identification of the UMP synthase gene by establishment of uracil auxotrophic mutants and the phenotypic complementation system in the marine diatom, Phaeodactylum tricornutum. Plant Physiol. 2011.
- Li, S.S.; Tsai, H.J. Transgenic microalgae as a non-antibiotic bactericide producer to defend against bacterial pathogen infection in the fish digestive tract. Fish. Shellfish Immunol. 2009, 26, 316–325. [Google Scholar] [CrossRef]
- Minoda, A.; Sakagami, R.; Yagisawa, F.; Kuroiwa, T.; Tanaka, K. Improvement of culture conditions and evidence for nuclear transformation by homologous recombination in a red alga, Cyanidioschyzon merolae 10D. Plant Cell. Physiol. 2004, 45, 667–671. [Google Scholar]
- Ohnuma, M.; Misumi, O.; Fujiwara, T.; Watanabe, S.; Tanaka, K.; Kuroiwa, T. Transient gene suppression in a red alga, Cyanidioschyzon merolae 10D. Protoplasma 2009, 236, 107–112. [Google Scholar] [CrossRef]
- Ohnuma, M.; Yokoyama, T.; Inouye, T.; Sekine, Y.; Tanaka, K. Polyethylene glycol (PEG)-mediated transient gene expression in a red alga, Cyanidioschyzon merolae 10D. Plant Cell. Physiol. 2008, 49, 117–120. [Google Scholar] [CrossRef]
- Dawson, H.N.; Burlingame, R.; Cannons, A.C. Stable transformation of Chlorella: Rescue of nitrate reductase-deficient mutants with the nitrate reductase gene. Curr. Microbiol. 1997, 35, 356–362. [Google Scholar] [CrossRef]
- Hawkins, R.L.; Nakamura, M. Expression of human growth hormone by the eukaryotic alga, Chlorella. Curr. Microbiol. 1999, 38, 335–341. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, Y.T.; Cho, J.J.; Bae, J.H.; Hur, S.B.; Hwang, I.; Choi, T.J. Stable integration and functional expression of flounder growth hormone gene in transformed microalga, Chlorella ellipsoidea. Mar. Biotechnol. (N. Y.) 2002, 4, 63–73. [Google Scholar] [CrossRef]
- Steinbrenner, J.; Sandmann, G. Transformation of the green alga Haematococcus pluvialis with a phytoene desaturase for accelerated astaxanthin biosynthesis. Appl. Environ. Microbiol. 2006, 72, 7477–7484. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Z.; Gao, X.; Li, Q.; Zhang, Q.; Xu, Z. Expression of foreign genes in Dunaliella by electroporation. Mol. Biotechnol. 2005, 30, 185–192. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Xue, L.; Xie, H. A structurally novel salt-regulated promoter of duplicated carbonic anhydrase gene 1 from Dunaliella salina. Mol. Biol. Rep. 2010, 37, 1143–1154. [Google Scholar] [CrossRef]
- Li, J.; Xue, L.; Yan, H.; Wang, L.; Liu, L.; Lu, Y.; Xie, H. The nitrate reductase gene-switch: A system for regulated expression in transformed cells of Dunaliella salina. Gene 2007, 403, 132–142. [Google Scholar] [CrossRef]
- Debuchy, R.; Purton, S.; Rochaix, J.D. The argininosuccinate lyase gene of Chlamydomonas reinhardtii: An important tool for nuclear transformation and for correlating the genetic and molecular maps of the ARG7 locus. EMBO J. 1989, 8, 2803–2809. [Google Scholar]
- Fernandez, E.; Schnell, R.; Ranum, L.P.; Hussey, S.C.; Silflow, C.D.; Lefebvre, P.A. Isolation and characterization of the nitrate reductase structural gene of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA. 1989, 86, 6449–6453. [Google Scholar]
- Kindle, K.L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA. 1990, 87, 1228–1232. [Google Scholar] [CrossRef]
- Stevens, D.R.; Rochaix, J.D.; Purton, S. The bacterial phleomycin resistance gene ble as a dominant selectable marker in Chlamydomonas. Mol. Gen. Genet. 1996, 251, 23–30. [Google Scholar]
- Molnar, A.; Bassett, A.; Thuenemann, E.; Schwach, F.; Karkare, S.; Ossowski, S.; Weigel, D.; Baulcombe, D. Highly specific gene silencing by artificial microRNAs in the unicellular alga Chlamydomonas reinhardtii. Plant J. 2009, 58, 165–174. [Google Scholar] [CrossRef]
- Berthold, P.; Schmitt, R.; Mages, W. An engineered Streptomyces hygroscopicus aph 7" gene mediates dominant resistance against hygromycin B in Chlamydomonas reinhardtii. Protist 2002, 153, 401–412. [Google Scholar] [CrossRef]
- Sizova, I.; Fuhrmann, M.; Hegemann, P. A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii. Gene 2001, 277, 221–229. [Google Scholar] [CrossRef]
- Cerutti, H.; Johnson, A.M.; Gillham, N.W.; Boynton, J.E. Epigenetic silencing of a foreign gene in nuclear transformants of Chlamydomonas. Plant Cell. 1997, 9, 925–945. [Google Scholar] [CrossRef]
- Boynton, J.E.; Gillham, N.W.; Harris, E.H.; Hosler, J.P.; Johnson, A.M.; Jones, A.R.; Randolph-Anderson, B.L.; Robertson, D.; Klein, T.M.; Shark, K.B.; et al. Chloroplast transformation in Chlamydomonas with high velocity microprojectiles. Science 1988, 240, 1534–1538. [Google Scholar]
- Goldschmidt-Clermont, M. Transgenic expression of aminoglycoside adenine transferase in the chloroplast: A selectable marker of site-directed transformation of Chlamydomonas. Nucleic Acids Res. 1991, 19, 4083–4089. [Google Scholar] [CrossRef]
- Schroda, M.; Blocker, D.; Beck, C.F. The HSP70A promoter as a tool for the improved expression of transgenes in Chlamydomonas. Plant J. 2000, 21, 121–131. [Google Scholar]
- Schiedlmeier, B.; Schmitt, R.; Muller, W.; Kirk, M.M.; Gruber, H.; Mages, W.; Kirk, D.L. Nuclear transformation of Volvox carteri. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 5080–5084. [Google Scholar]
- Harris, E.H. Chlamydomonas as a Model Organism. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 363–406. [Google Scholar] [CrossRef]
- Rochaix, J.D. Chlamydomonas, a model system for studying the assembly and dynamics of photosynthetic complexes. FEBS Lett. 2002, 529, 34–38. [Google Scholar] [CrossRef]
- Eberhard, S.; Finazzi, G.; Wollman, F.A. The dynamics of photosynthesis. Annu. Rev. Genet. 2008, 42, 463–515. [Google Scholar] [CrossRef]
- Marshall, W.F.; Rosenbaum, J.L. How centrioles work: Lessons from green yeast. Curr. Opin. Cell. Biol. 2000, 12, 119–125. [Google Scholar] [CrossRef]
- Rosenbaum, J.L.; Witman, G.B. Intraflagellar transport. Nat. Rev. Mol. Cell. Biol. 2002, 3, 813–825. [Google Scholar]
- Pan, J.; Snell, W.J. Signal transduction during fertilization in the unicellular green alga, Chlamydomonas. Curr. Opin. Microbiol. 2000, 3, 596–602. [Google Scholar] [CrossRef]
- Matsuo, T.; Ishiura, M. Chlamydomonas reinhardtii as a new model system for studying the molecular basis of the circadian clock. FEBS Lett. 2011, 585, 1495–1502. [Google Scholar] [CrossRef]
- Mus, F.; Dubini, A.; Seibert, M.; Posewitz, M.C.; Grossman, A.R. Anaerobic acclimation in Chlamydomonas reinhardtii: Anoxic gene expression, hydrogenase induction, and metabolic pathway. J. Biol. Chem. 2007, 282, 25475–25486. [Google Scholar] [CrossRef]
- Melis, A.; Zhang, L.; Forestier, M.; Ghirardi, M.L.; Seibert, M. Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol. 2000, 122, 127–136. [Google Scholar] [CrossRef]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Marechal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef]
- Lumbreras, V.; Stevens, D.R.; Purton, S. Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron. Plant J. 1998, 14, 441–447. [Google Scholar] [CrossRef]
- Fischer, N.; Rochaix, J.D. The flanking regions of PsaD drive efficient gene expression in the nucleus of the green alga Chlamydomonas reinhardtii. Mol. Genet. Genomics 2001, 265, 888–894. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Oertel, W.; Hegemann, P. A synthetic gene coding for the green fluorescent protein (GFP) is a versatile reporter in Chlamydomonas reinhardtii. Plant J. 1999, 19, 353–361. [Google Scholar] [CrossRef]
- Shao, N.; Bock, R. A codon-optimized luciferase from Gaussia princeps facilitates the in vivo monitoring of gene expression in the model alga Chlamydomonas reinhardtii. Curr. Genet. 2008, 53, 381–388. [Google Scholar] [CrossRef]
- Tam, L.W.; Lefebvre, P.A. Cloning of flagellar genes in Chlamydomonas reinhardtii by DNA insertional mutagenesis. Genetics 1993, 135, 375–384. [Google Scholar]
- Sizova, I.; Greiner, A.; Awasthi, M.; Kateriya, S.; Hegemann, P. Nuclear gene targeting in Chlamydomonas using engineered zinc-finger nucleases. Plant J. 2013, 73, 873–882. [Google Scholar] [CrossRef]
- Schroda, M. RNA silencing in Chlamydomonas: Mechanisms and tools. Curr. Genet. 2006, 49, 69–84. [Google Scholar] [CrossRef]
- Schmollinger, S.; Strenkert, D.; Schroda, M. An inducible artificial microRNA system for Chlamydomonas reinhardtii confirms a key role for heat shock factor 1 in regulating thermotolerance. Curr. Genet. 2010, 56, 383–389. [Google Scholar] [CrossRef]
- Zhao, T.; Wang, W.; Bai, X.; Qi, Y. Gene silencing by artificial microRNAs in Chlamydomonas. Plant J. 2009, 58, 157–164. [Google Scholar] [CrossRef]
- Chang, R.L.; Ghamsari, L.; Manichaikul, A.; Hom, E.F.; Balaji, S.; Fu, W.; Shen, Y.; Hao, T.; Palsson, B.O.; Salehi-Ashtiani, K.; et al. Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol. Syst. Biol. 2011, 7, 518. [Google Scholar]
- Fernie, A.R. Grand challenges in plant systems biology: Closing the circle(s). Front. Plant Sci 2012, 3, 35. [Google Scholar] [CrossRef]
- Palsson, B. Metabolic systems biology. FEBS Lett. 2009, 583, 3900–3904. [Google Scholar] [CrossRef]
- Feist, A.M.; Herrgard, M.J.; Thiele, I.; Reed, J.L.; Palsson, B.O. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 2009, 7, 129–143. [Google Scholar]
- Manichaikul, A.; Ghamsari, L.; Hom, E.F.Y.; Lin, C.; Murray, R.R.; Chang, R.L.; Balaji, S.; Hao, T.; Shen, Y.; Chavali, A.K.; et al. Metabolic network analysis integrated with transcript verification for sequenced genomes. Nat. Methods 2009, 6, 589–592. [Google Scholar] [CrossRef]
- Boyle, N.R.; Morgan, J.A. Flux balance analysis of primary metabolism in Chlamydomonas reinhardtii. BMC Syst. Biol. 2009, 3, 4. [Google Scholar] [CrossRef]
- Dal’Molin, C.G.; Quek, L.E.; Palfreyman, R.W.; Nielsen, L.K. AlgaGEM—a genome-scale metabolic reconstruction of algae based on the Chlamydomonas reinhardtii genome. BMC Genomics 2011, 12 (Suppl 4), S5. [Google Scholar]
- Kruse, O.; Rupprecht, J.; Bader, K.P.; Thomas-Hall, S.; Schenk, P.M.; Finazzi, G.; Hankamer, B. Improved photobiological H2 production in engineered green algal cells. J. Biol. Chem. 2005, 280, 34170–34177. [Google Scholar] [CrossRef]
- Link, H.; Christodoulou, D.; Sauer, U. Advancing metabolic models with kinetic information. Curr. Opin. Biotechnol. 2014, 29, 8–14. [Google Scholar]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Gerosa, L.; Sauer, U. Regulation and control of metabolic fluxes in microbes. Curr. Opin. Biotechnol. 2011, 22, 566–575. [Google Scholar] [CrossRef]
- Fernie, A.R.; Stitt, M. On the discordance of metabolomics with proteomics and transcriptomics: Coping with increasing complexity in logic, chemistry, and network interactions scientific correspondence. Plant Physiol. 2012, 158, 1139–1145. [Google Scholar] [CrossRef]
- Reaves, M.L.; Rabinowitz, J.D. Metabolomics in systems microbiology. Curr. Opin. Biotechnol. 2011, 22, 17–25. [Google Scholar] [CrossRef]
- Dunn, W.B. Current trends and future requirements for the mass spectrometric investigation of microbial, mammalian and plant metabolomes. Phys. Biol. 2008, 5, 011001. [Google Scholar] [CrossRef]
- Baidoo, E.E.; Benke, P.I.; Keasling, J.D. Mass spectrometry-based microbial metabolomics. Methods Mol. Biol. 2012, 881, 215–278. [Google Scholar] [CrossRef]
- Obata, T.; Fernie, A. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef]
- Halket, J.M.; Waterman, D.; Przyborowska, A.M.; Patel, R.K.; Fraser, P.D.; Bramley, P.M. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J. Exp. Bot 2005, 56, 219–243. [Google Scholar]
- Giavalisco, P.; Li, Y.; Matthes, A.; Eckhardt, A.; Hubberten, H.-M.; Hesse, H.; Segu, S.; Hummel, J.; Köhl, K.; Willmitzer, L. Elemental formula annotation of polar and lipophilic metabolites using 13C, 15N and 34S isotope labelling, in combination with high-resolution mass spectrometry. Plant J. 2011, 68, 364–376. [Google Scholar] [CrossRef]
- Arrivault, S.; Guenther, M.; Ivakov, A.; Feil, R.; Vosloh, D.; Van Dongen, J.T.; Sulpice, R.; Stitt, M. Use of reverse-phase liquid chromatography, linked to tandem mass spectrometry, to profile the Calvin cycle and other metabolic intermediates in Arabidopsis rosettes at different carbon dioxide concentrations. Plant J. 2009, 59, 826–839. [Google Scholar] [CrossRef]
- Tohge, T.; Fernie, A.R. Combining genetic diversity, informatics and metabolomics to facilitate annotation of plant gene function. Nat. Protocols 2010, 5, 1210–1227. [Google Scholar] [CrossRef]
- Jellum, E.; Størseth, P.; Alexander, J.; Helland, P.; Stokke, O.; Teig, E. Application of glass capillary-column gas chromatography-mass spectrometry to the studies of human diseases. J. Chromatogr. A 1976, 126, 487–493. [Google Scholar] [CrossRef]
- Goodman, S.I.; Helland, P.; Stokke, O.; Flatmark, A.; Jellum, E. Organic acid profiles of human tissue biopsies by capillary gas chromatography-mass spectrometry. J. Chromatogr. A 1977, 142, 497–503. [Google Scholar] [CrossRef]
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc 2006, 1, 387–396. [Google Scholar] [CrossRef]
- Little, J.L. Artifacts in trimethylsilyl derivatization reactions and ways to avoid them. J. Chromatogr. A 1999, 844, 1–22. [Google Scholar] [CrossRef]
- Allwood, J.W.; Erban, A.; Koning, S.; Dunn, W.; Luedemann, A.; Lommen, A.; Kay, L.; Löscher, R.; Kopka, J.; Goodacre, R. Inter-laboratory reproducibility of fast gas chromatography-electron impact-time of flight mass spectrometry (GC–EI–TOF/MS) based plant metabolomics. Metabolomics 2009, 5, 479–496. [Google Scholar] [CrossRef]
- Schauer, N.; Steinhauser, D.; Strelkov, S.; Schomburg, D.; Allison, G.; Moritz, T.; Lundgren, K.; Roessner-Tunali, U.; Forbes, M.G.; Willmitzer, L.; et al. GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett. 2005, 579, 1332–1337. [Google Scholar] [CrossRef]
- Wagner, C.; Sefkow, M.; Kopka, J. Construction and application of a mass spectral and retention time index database generated from plant GC/EI-TOF-MS metabolite profiles. Phytochemistry 2003, 62, 887–900. [Google Scholar] [CrossRef]
- Kopka, J. Current challenges and developments in GC-MS based metabolite profiling technology. J. Biotechnol. 2006, 124, 312–322. [Google Scholar] [CrossRef]
- Kováts, E. Gas-chromatographische Charakterisierung organischer Verbindungen. Teil 1: Retentionsindices aliphatischer Halogenide, Alkohole, Aldehyde und Ketone. Helvetica Chimica Acta 1958, 41, 1915–1932. [Google Scholar] [CrossRef]
- Strehmel, N.; Hummel, J.; Erban, A.; Strassburg, K.; Kopka, J. Retention index thresholds for compound matching in GC-MS metabolite profiling. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 871, 182–190. [Google Scholar] [CrossRef]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmüller, E.; Dörmann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. GMDB@DB: The golm metabolome database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar] [CrossRef]
- Erban, A.; Schauer, N.; Fernie, A.R.; Kopka, J. Nonsupervised construction and application of mass spectral and retention time index libraries from time-of-flight gas chromatography-mass spectrometry metabolite profiles. Methods Mol. Biol. 2007, 358, 19–38. [Google Scholar] [CrossRef]
- Luedemann, A.; Strassburg, K.; Erban, A.; Kopka, J. TagFinder for the quantitative analysis of gas chromatography—mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinformatics 2008, 24, 732–737. [Google Scholar] [CrossRef]
- Tohge, T.; Fernie, A.R. Web-based resources for mass-spectrometry-based metabolomics: A user’s guide. Phytochemistry 2009, 70, 450–456. [Google Scholar] [CrossRef]
- Tohge, T.; Mettler, T.; Arrivault, S.; Carroll, A.J.; Stitt, M.; Fernie, A.R. From models to crop species: Caveats and solutions for translational metabolomics. Front. Plant Sci 2011, 2, 61. [Google Scholar]
- Fernie, A.R.; Aharoni, A.; Willmitzer, L.; Stitt, M.; Tohge, T.; Kopka, J.; Carroll, A.J.; Saito, K.; Fraser, P.D.; DeLuca, V. Recommendations for reporting metabolite data. Plant Cell 2011, 23, 2477–2482. [Google Scholar] [CrossRef]
- Bennett, B.D.; Yuan, J.; Kimball, E.H.; Rabinowitz, J.D. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat. Protocols 2008, 3, 1299–1311. [Google Scholar] [CrossRef]
- Villas-Bôas, S. G.; Roessner, U.; Hansen, M. A. E.; Smedsgaard, J.; Nielsen, J. Sampling and sample preparation. In Metabolome Analysis: An Introduction; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 39–82. [Google Scholar]
- Koning, W.d.; Dam, K.V. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 1992, 204, 118–123. [Google Scholar] [CrossRef]
- Villas-Bôas, S.G.; Bruheim, P. Cold glycerol-saline: The promising quenching solution for accurate intracellular metabolite analysis of microbial cells. Anal. Biochem 2007, 370, 87–97. [Google Scholar] [CrossRef]
- Barupal, D.K.; Kind, T.; Kothari, S.L.; Lee do, Y.; Fiehn, O. Hydrocarbon phenotyping of algal species using pyrolysis-gas chromatography mass spectrometry. BMC Biotechnol. 2010, 10, 40. [Google Scholar] [CrossRef]
- Taymaz-Nikerel, H.; de Mey, M.; Ras, C.; ten Pierick, A.; Seifar, R.M.; van Dam, J.C.; Heijnen, J.J.; van Gulik, W.M. Development and application of a differential method for reliable metabolome analysis in Escherichia coli. Anal. Biochem. 2009, 386, 9–19. [Google Scholar] [CrossRef]
- Van Gulik, W.M. Fast sampling for quantitative microbial metabolomics. Curr. Opin. Biotechnol. 2010, 21, 27–34. [Google Scholar] [CrossRef]
- Bolten, C.J.; Kiefer, P.; Letisse, F.; Portais, J.-C.; Wittmann, C. Sampling for metabolome analysis of microorganisms. Anal. Chem. 2007, 79, 3843–3849. [Google Scholar] [CrossRef]
- Canelas, A.B.; Ras, C.; Pierick, A.; Dam, J.C.; Heijnen, J.J.; Gulik, W.M. Leakage-free rapid quenching technique for yeast metabolomics. Metabolomics 2008, 4, 226–239. [Google Scholar] [CrossRef]
- Volmer, M.; Northoff, S.; Scholz, S.; Thute, T.; Buntemeyer, H.; Noll, T. Fast filtration for metabolome sampling of suspended animal cells. Biotechnol. Lett. 2011, 33, 495–502. [Google Scholar] [CrossRef]
- Faijes, M.; Mars, A.E.; Smid, E.J. Comparison of quenching and extraction methodologies for metabolome analysis of Lactobacillus plantarum. Microb Cell. Fact. 2007, 6, 27. [Google Scholar] [CrossRef]
- Link, H.; Anselment, B.; Weuster-Botz, D. Leakage of adenylates during cold methanol/glycerol quenching of Escherichia coli. Metabolomics 2008, 4, 240–247. [Google Scholar] [CrossRef]
- Spura, J.; Reimer, L.C.; Wieloch, P.; Schreiber, K.; Buchinger, S.; Schomburg, D. A method for enzyme quenching in microbial metabolome analysis successfully applied to gram-positive and gram-negative bacteria and yeast. Anal. Biochem. 2009, 394, 192–201. [Google Scholar] [CrossRef]
- Bölling, C.; Fiehn, O. Metabolite profiling of Chlamydomonas reinhardtii under nutrient deprivation. Plant Physiol. 2005, 139, 1995–2005. [Google Scholar] [CrossRef]
- Lee do, Y.; Fiehn, O. High quality metabolomic data for Chlamydomonas reinhardtii. Plant Methods 2008, 4, 7. [Google Scholar] [CrossRef]
- Kempa, S.; Hummel, J.; Schwemmer, T.; Pietzke, M.; Strehmel, N.; Wienkoop, S.; Kopka, J.; Weckwerth, W. An automated GCxGC-TOF-MS protocol for batch-wise extraction and alignment of mass isotopomer matrixes from differential 13C-labelling experiments: A case study for photoautotrophic-mixotrophic grown Chlamydomonas reinhardtii cells. J. Basic Microbiol. 2009, 49, 82–91. [Google Scholar] [CrossRef]
- Renberg, L.; Johansson, A.I.; Shutova, T.; Stenlund, H.; Aksmann, A.; Raven, J.A.; Gardestrom, P.; Moritz, T.; Samuelsson, G. A metabolomic approach to study major metabolite changes during acclimation to limiting CO2 in Chlamydomonas reinhardtii. Plant Physiol. 2010, 154, 187–196. [Google Scholar] [CrossRef]
- Timmins, M.; Zhou, W.; Rupprecht, J.; Lim, L.; Thomas-Hall, S.R.; Doebbe, A.; Kruse, O.; Hankamer, B.; Marx, U.C.; Smith, S.M.; et al. The metabolome of Chlamydomonas reinhardtii following induction of anaerobic H2 production by sulfur depletion. J. Biol. Chem. 2009, 284, 23415–23425. [Google Scholar] [CrossRef]
- Doebbe, A.; Keck, M.; La Russa, M.; Mussgnug, J.H.; Hankamer, B.; Tekce, E.; Niehaus, K.; Kruse, O. The interplay of proton, electron, and metabolite supply for photosynthetic H2 production in Chlamydomonas reinhardtii. J. Biol. Chem. 2010, 285, 30247–30260. [Google Scholar]
- Wienkoop, S.; Weiss, J.; May, P.; Kempa, S.; Irgang, S.; Recuenco-Munoz, L.; Pietzke, M.; Schwemmer, T.; Rupprecht, J.; Egelhofer, V.; et al. Targeted proteomics for Chlamydomonas reinhardtii combined with rapid subcellular protein fractionation, metabolomics and metabolic flux analyses. Mol. Biosyst. 2010, 6, 1018–1031. [Google Scholar] [CrossRef]
- Catalanotti, C.; Dubini, A.; Subramanian, V.; Yang, W.; Magneschi, L.; Mus, F.; Seibert, M.; Posewitz, M.C.; Grossman, A.R. Altered fermentative metabolism in Chlamydomonas reinhardtii mutants lacking pyruvate formate lyase and both pyruvate formate lyase and alcohol dehydrogenase. Plant Cell 2012, 24, 692–707. [Google Scholar] [CrossRef]
- Lee do, Y.; Park, J.J.; Barupal, D.K.; Fiehn, O. System response of metabolic networks in Chlamydomonas reinhardtii to total available ammonium. Mol. Cell. Proteomics 2012, 11, 973–988. [Google Scholar] [CrossRef]
- Mastrobuoni, G.; Irgang, S.; Pietzke, M.; Wenzel, M.; Assmus, H.; Schulze, W.; Kempa, S. Proteome dynamics and early salt stress response of the photosynthetic organism Chlamydomonas reinhardtii. BMC Genomics 2012, 13, 215. [Google Scholar] [CrossRef]
- Valledor, L.; Furuhashi, T.; Hanak, A.M.; Weckwerth, W. Systemic cold stress adaptation of Chlamydomonas reinhardtii. Mol. Cell. Proteomics 2013, 12, 2032–2047. [Google Scholar] [CrossRef]
- Davis, M.C.; Fiehn, O.; Durnford, D.G. Metabolic acclimation to excess light intensity in Chlamydomonas reinhardtii. Plant Cell. Environ. 2013, 36, 1391–1405. [Google Scholar] [CrossRef]
- Lee do, Y.; Fiehn, O. Metabolomic response of Chlamydomonas reinhardtii to the inhibition of target of rapamycin (TOR) by rapamycin. J. Microbiol. Biotechnol. 2013, 23, 923–931. [Google Scholar] [CrossRef]
- Krall, L.; Huege, J.; Catchpole, G.; Steinhauser, D.; Willmitzer, L. Assessment of sampling strategies for gas chromatography-mass spectrometry (GC-MS) based metabolomics of cyanobacteria. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2952–2960. [Google Scholar] [CrossRef]
- Jozefczuk, S.; Klie, S.; Catchpole, G.; Szymanski, J.; Cuadros-Inostroza, A.; Steinhauser, D.; Selbig, J.; Willmitzer, L. Metabolomic and transcriptomic stress response of Escherichia coli. Mol. Syst. Biol. 2010, 6, 364. [Google Scholar]
- Szymanski, J.; Jozefczuk, S.; Nikoloski, Z.; Selbig, J.; Nikiforova, V.; Catchpole, G.; Willmitzer, L. Stability of metabolic correlations under changing environmental conditions in Escherichia coli--a systems approach. PLoS One 2009, 4, e7441. [Google Scholar] [CrossRef]
- Meyer, H.; Liebeke, M.; Lalk, M. A protocol for the investigation of the intracellular Staphylococcus aureus metabolome. Anal. Biochem. 2010, 401, 250–259. [Google Scholar] [CrossRef]
- Shin, M.H.; Lee do, Y.; Liu, K.H.; Fiehn, O.; Kim, K.H. Evaluation of sampling and extraction methodologies for the global metabolic profiling of Saccharophagus degradans. Anal. Chem. 2010, 82, 6660–6666. [Google Scholar] [CrossRef]
- Kim, S.; Lee do, Y.; Wohlgemuth, G.; Park, H.S.; Fiehn, O.; Kim, K.H. Evaluation and optimization of metabolome sample preparation methods for Saccharomyces cerevisiae. Anal. Chem. 2013, 85, 2169–2176. [Google Scholar] [CrossRef]
- Hess, J.L.; Tolbert, N.E. Glycolate pathway in algae. Plant Physiol. 1967, 42, 371–379. [Google Scholar] [CrossRef]
- Brauer, M.J.; Yuan, J.; Bennett, B.D.; Lu, W.; Kimball, E.; Botstein, D.; Rabinowitz, J.D. Conservation of the metabolomic response to starvation across two divergent microbes. Proc. Natl. Acad. Sci. USA. 2006, 103, 19302–19307. [Google Scholar]
- Link, H.; Kochanowski, K.; Sauer, U. Systematic identification of allosteric protein-metabolite interactions that control enzyme activity in vivo. Nat. Biotechnol. 2013, 31, 357–361. [Google Scholar]
- Canelas, A.B.; ten Pierick, A.; Ras, C.; Seifar, R.M.; van Dam, J.C.; van Gulik, W.M.; Heijnen, J.J. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal. Chem. 2009, 81, 7379–7389. [Google Scholar]
- Gullberg, J.; Jonsson, P.; Nordstrom, A.; Sjostrom, M.; Moritz, T. Design of experiments: An efficient strategy to identify factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal. Biochem. 2004, 331, 283–295. [Google Scholar] [CrossRef]
- Magneschi, L.; Catalanotti, C.; Subramanian, V.; Dubini, A.; Yang, W.; Mus, F.; Posewitz, M.C.; Seibert, M.; Perata, P.; Grossman, A.R. A mutant in the ADH1 gene of Chlamydomonas reinhardtii elicits metabolic restructuring during anaerobiosis. Plant Physiol. 2012.
- May, P.; Wienkoop, S.; Kempa, S.; Usadel, B.; Christian, N.; Rupprecht, J.; Weiss, J.; Recuenco-Munoz, L.; Ebenhoh, O.; Weckwerth, W.; et al. Metabolomics- and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics 2008, 179, 157–166. [Google Scholar] [CrossRef]
- Dörmann, P.; Benning, C. Galactolipids rule in seed plants. Trends Plant Sci 2002, 7, 112–118. [Google Scholar] [CrossRef]
- Büscher, J.M.; Czernik, D.; Ewald, J.C.; Sauer, U.; Zamboni, N. Cross-platform comparison of methods for quantitative metabolomics of primary metabolism. Anal. Chem. 2009, 81, 2135–2143. [Google Scholar] [CrossRef]
- Kanani, H.H.; Klapa, M.I. Data correction strategy for metabolomics analysis using gas chromatography-mass spectrometry. Metab. Eng. 2007, 9, 39–51. [Google Scholar] [CrossRef]
- Hajšlová, J.; Zrostlíková, J. Matrix effects in (ultra)trace analysis of pesticide residues in food and biotic matrices. J. Chromatogr. A 2003, 1000, 181–197. [Google Scholar]
- Koek, M.M.; Muilwijk, B.; van Stee, L.L.P.; Hankemeier, T. Higher mass loadability in comprehensive two-dimensional gas chromatography–mass spectrometry for improved analytical performance in metabolomics analysis. J. Chromatogr. A 2008, 1186, 420–429. [Google Scholar]
- Hutschenreuther, A.; Kiontke, A.; Birkenmeier, G.; Birkemeyer, C. Comparison of extraction conditions and normalization approaches for cellular metabolomics of adherent growing cells with GC-MS. Anal. Methods 2012, 4, 1953–1963. [Google Scholar] [CrossRef]
- Harris, E.H. The Chlamydomonas Sourcebook: Introduction to Chlamydomonas and Its Laboratory Use, 2nd ed.; Elsevier/Academic Press: San Diego, CA, USA, 2008; Volume 1. [Google Scholar]
- Teplitski, M.; Chen, H.; Rajamani, S.; Gao, M.; Merighi, M.; Sayre, R.T.; Robinson, J.B.; Rolfe, B.G.; Bauer, W.D. Chlamydomonas reinhardtii secretes compounds that mimic bacterial signals and interfere with quorum sensing regulation in bacteria. Plant Physiol. 2004, 134, 137–146. [Google Scholar] [CrossRef]
- Van den Berg, R.; Hoefsloot, H.; Westerhuis, J.; Smilde, A.; van der Werf, M. Centering, scaling, andtransformations: Improving the biological information content of metabolomics data. BMC Genomics 2006, 7, 142. [Google Scholar] [CrossRef]
- Mashego, M.R.; Wu, L.; Van Dam, J.C.; Ras, C.; Vinke, J.L.; Van Winden, W.A.; Van Gulik, W.M.; Heijnen, J.J. MIRACLE: Mass isotopomer ratio analysis of U-13C-labeled extracts. A new method for accurate quantification of changes in concentrations of intracellular metabolites. Biotechnol. Bioeng. 2004, 85, 620–628. [Google Scholar] [CrossRef]
- Koek, M.M.; Muilwijk, B.; van der Werf, M.J.; Hankemeier, T. Microbial metabolomics with gas chromatography/mass spectrometry. Anal. Chem. 2006, 78, 1272–1281. [Google Scholar] [CrossRef]
- Cleveland, W.S.; Devlin, S.J. Locally Weighted Regression: An Approach to Regression Analysis by Local Fitting. J. Am. Statist. Assoc. 1988, 83, 596–610. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protocol. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Huege, J.; Krall, L.; Steinhauser, M.C.; Giavalisco, P.; Rippka, R.; Tandeau de Marsac, N.; Steinhauser, D. Sample amount alternatives for data adjustment in comparative cyanobacterial metabolomics. Anal. Bioanalyt. Chem. 2011, 399, 3503–3517. [Google Scholar] [CrossRef]
- Ewald, J.C.; Heux, S.p.; Zamboni, N. High-throughput quantitative metabolomics: Workflow for cultivation, quenching, and analysis of yeast in a multiwell format. Anal. Chem 2009, 81, 3623–3629. [Google Scholar] [CrossRef]
- Svensen, Ø.; Frette, Ø.; Erga, S.R. Scattering properties of microalgae: The effect of cell size and cell wall. Appl. Opt. 2007, 46, 5762–5769. [Google Scholar] [CrossRef]
- Horst, C.J.; Fishkind, D.J.; Pazour, G.J.; Witman, G.B. An insertional mutant of Chlamydomonas reinhardtii with defective microtubule positioning. Cell. Motil. Cytoskeleton 1999, 44, 143–154. [Google Scholar] [CrossRef]
- Tredwell, G.D.; Edwards-Jones, B.; Leak, D.J.; Bundy, J.G. The development of metabolomic sampling procedures for Pichia pastoris, and baseline metabolome data. PLoS One 2011, 6, e16286. [Google Scholar]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Kohl, S.M.; Klein, M.S.; Hochrein, J.; Oefner, P.J.; Spang, R.; Gronwald, W. State-of-the art data normalization methods improve NMR-based metabolomic analysis. Metabolomics 2012, 8, 146–160. [Google Scholar] [CrossRef]
- Ejigu, B.A.; Valkenborg, D.; Baggerman, G.; Vanaerschot, M.; Witters, E.; Dujardin, J.C.; Burzykowski, T.; Berg, M. Evaluation of normalization methods to pave the way towards large-scale LC-MS-based metabolomics profiling experiments. OMICS 2013, 17, 473–485. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Huber, W.; von Heydebreck, A.; Sültmann, H.; Poustka, A.; Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002, 18, S96–S104. [Google Scholar]
- Dudoit, S.; Yang, Y.H.; Callow, M.J.; Speed, T.P. Statistical methods for identifying differentiallyexpressed genes in replicated cDNA microarray experiments. Stat. Sinica 2002, 12, 111–139. [Google Scholar]
- Kvalheim, O.M.; Brakstad, F.; Liang, Y. Preprocessing of analytical profiles in the presence of homoscedastic or heteroscedastic noise. Anal. Chem. 1994, 66, 43–51. [Google Scholar] [CrossRef]
- Harris, E.H. The Chlamydomonas Sourcebook: Introduction to Chlamydomonas and Its Laboratory Use; Elsevier Science: San Diego, CA, USA, 2009. [Google Scholar]
- Roessner, U.; Wagner, C.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Simultaneous analysis of metabolites in potato tuber by gas chromatography—mass spectrometry. Plant J. 2000, 23, 131–142. [Google Scholar] [CrossRef]
- Luedemann, A.; Strassburg, K.; Erban, A.; Kopka, J. TagFinder for the quantitative analysis of gas chromatography—mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinformatics 2008, 24, 732–737. [Google Scholar]
- Hummel, J.; Strehmel, N.; Selbig, J.; Walther, D.; Kopka, J. Decision tree supported substructure prediction of metabolites from GC-MS profiles. Metabolomics 2010, 6, 322–333. [Google Scholar] [CrossRef]
- Accord.NET Framework. Available online: http://code.google.com/p/accord/ (accessed on 25 November 2012).
- FSharpChart-Wrapping System.Windows.Forms.DataVisualization.Charting. Available online: http://code.msdn.microsoft.com/windowsdesktop/FSharpChart-b59073f5 (accessed on 19 November 2012).
- Douma, R.D.; de Jonge, L.P.; Jonker, C.T.; Seifar, R.M.; Heijnen, J.J.; van Gulik, W.M. Intracellular metabolite determination in the presence of extracellular abundance: Application to the penicillin biosynthesis pathway in Penicillium chrysogenum. Biotechnol. Bioeng. 2010, 107, 105–115. [Google Scholar] [CrossRef]
- Kronthaler, J.; Gstraunthaler, G.; Heel, C. Optimizing high-throughput metabolomic biomarker screening: A study of quenching solutions to freeze intracellular metabolism in CHO cells. OMICS 2012, 16, 90–97. [Google Scholar] [CrossRef]
- Feist, A.M.; Zielinski, D.C.; Orth, J.D.; Schellenberger, J.; Herrgard, M.J.; Palsson, B.O. Model-driven evaluation of the production potential for growth-coupled products of Escherichia coli. Metab Eng. 2010, 12, 173–186. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Veyel, D.; Erban, A.; Fehrle, I.; Kopka, J.; Schroda, M. Rationales and Approaches for Studying Metabolism in Eukaryotic Microalgae. Metabolites 2014, 4, 184-217. https://doi.org/10.3390/metabo4020184
Veyel D, Erban A, Fehrle I, Kopka J, Schroda M. Rationales and Approaches for Studying Metabolism in Eukaryotic Microalgae. Metabolites. 2014; 4(2):184-217. https://doi.org/10.3390/metabo4020184
Chicago/Turabian StyleVeyel, Daniel, Alexander Erban, Ines Fehrle, Joachim Kopka, and Michael Schroda. 2014. "Rationales and Approaches for Studying Metabolism in Eukaryotic Microalgae" Metabolites 4, no. 2: 184-217. https://doi.org/10.3390/metabo4020184
APA StyleVeyel, D., Erban, A., Fehrle, I., Kopka, J., & Schroda, M. (2014). Rationales and Approaches for Studying Metabolism in Eukaryotic Microalgae. Metabolites, 4(2), 184-217. https://doi.org/10.3390/metabo4020184