
Application of Metabolomics in Drug Resistant Breast Cancer Research


Abstract
:1. Introduction
2. Breast Cancer
2.1. Breast Cancer Biology and Therapeutic Options
2.2. Drug Resistance in Breast Cancer
3. Metabolomics as a Promising New Tool in Breast Cancer Research
3.1. Current Metabolomics Technologies
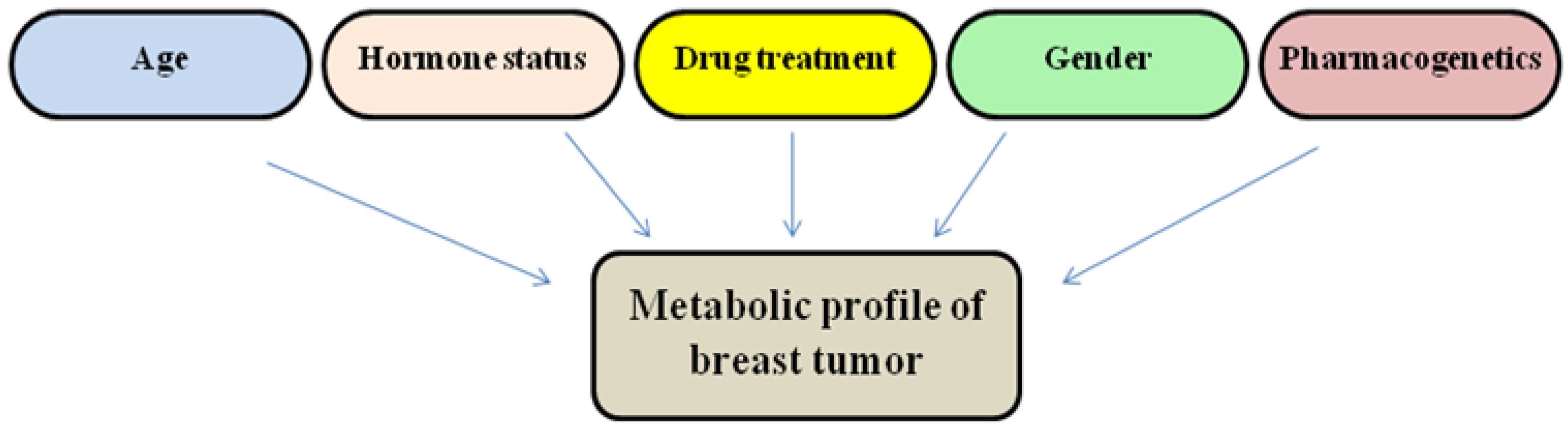
3.2. Metabolites as Powerful Biomarkers of Breast Cancer
3.3. Uncovering New Therapeutic Targets through Metabolomics

{kind=link}
| Biological materials | Approach | Specific treatment | Metabolic pathways identified | Reference |
|---|---|---|---|---|
| ER+ and ER- tumor tissues | GC-MS | None | Increase in glutamate, xanthine, beta-alanine in the ER- disease | [88] |
| MCF7 (ER+) | GC-MS | adriamycin | Increase in glycerol metabolism and decrease in glutathione biosynthesis | [106] |
| MDA-MB-231 (ER-) | NMR | hypoxia | Increase in glutamate, valine, and leucine and decrease in proline, creatine, alanine | [107] |
| MCF7 (ER+) | NMR | ascididemin | Increase in citrate, gluconate and polyunsaturated fatty acids and decrease in glycerophospho-choline and -ethanolamine | [108] |
| serum: early and metastatic breast cancer | NMR | None | Increase in histidine, acetoacetate, glycerol, pyruvate, glycoproteins (N-acetyl), mannose, glutamate and phenylalanine and decrease in alanine | [89] |
| MCF7 (ER+) and MDA-MB-231 (ER-) | NMR | curcumin +/- docetaxel (dose- and time-response) | Changes in glutathione metabolism, lipid metabolism, and glucose utilization - some biphasic changes depending on exposure | [109] |
| MCF7 (ER+) and MDA-MB-231 (ER-) | LC-MS | resveratrol | Increased amino acid and arachidonic acid in both cell lines | [110] |
| serum: recurrent and non-recurrent breast cancer | NMR & GC-MS | None | Changes in amino acids metabolism (glutamic acid, histidine, proline and tyrosine), glycolysis (lactate), phospholipid metabolism (choline) and fatty acid metabolism (nonanedioic acid) | [83] |
| urine: early-/late-stage breast cancer and normal | NMR | None | Changes in metabolites relating to energy metabolism, amino acids, and gut microbial metabolism | [111] |
4. Current Challenges in Metabolomics-Based Breast Cancer Research
4.1. Metabolomics Complements Other “Omics” Disciplines in a Systems Biology Approach towards Precision Medicine

4.2. Targeting Metabolic Pathways in Cancer
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Pinedo, C.; Mjiyad, El.N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Exploiting tumor metabolism: Challenges for clinical translation. J. Clin. Invest. 2013, 123, 3648–3651. [Google Scholar]
- Tyers, M.; Mann, M. From genomics to proteomics. Nature 2003, 422, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Cadoo, K.A.; Fornier, M.N.; Morris, P.G. Biological subtypes of breast cancer: Current concepts and implications for recurrence patterns. Q. J. Nucl. Med. Mol. Imaging 2013, 57, 312–321. [Google Scholar] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar]
- Peintinger, F. Using molecular profiles to tailor treatment in breast cancer: Are they ready for prime time? Curr. Opin. Obstet. Gynecol. 2014, 26, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; Carroll, J.S. Approaches for assessing and discovering protein interactions in cancer. Mol. Cancer Res. 2013, 11, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Invest. 2013, 123, 3652–3658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.H. Metabolomics in noninvasive breast cancer. Clin. Chim. Acta 2013, 424, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Ressom, H.W.; Wang, A.; Xuan, J.; Liu, M.C.; Gehan, E.A.; Wang, Y. The properties of high-dimensional data spaces: Implications for exploring gene and protein expression data. Nat. Rev. Cancer 2008, 8, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Shajahan, A.N.; Riggins, R.B.; Cho, Y.; Crawford, A.; Xuan, J.; Wang, Y.; Zwart, A.; Nehra, R.; Liu, M.C. Gene network signaling in hormone responsiveness modifies apoptosis and autophagy in breast cancer cells. J. Steroid Biochem. Mol. Biol. 2009, 114, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.J.; Eapen, S.; Gorin, B.; Adler, P. The economic burden of metastatic breast cancer: A U.S. managed care perspective. Breast Cancer Res. Treat 2012, 134, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Schnipper, L. Clinical implications of tumor-cell heterogeneity. N. Engl. J. Med. 1986, 314, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar]
- Matsen, C.B.; Neumayer, L.A. Breast cancer: A review for the general surgeon. JAMA Surg. 2013, 148, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef]
- Jorns, J.M.; Healy, P.; Zhao, L. Review of estrogen receptor, progesterone receptor, and HER-2/neu immunohistochemistry impacts on treatment for a small subset of breast cancer patients transferring care to another institution. Arch. Pathol. Lab. Med. 2013, 137, 1660–1663. [Google Scholar] [CrossRef] [PubMed]
- Allegra, J.C.; Barlock, A.; Huff, K.K.; Lippman, M.E. Changes in multiple or sequential estrogen receptor determinations in breast cancer. Cancer 1980, 45, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Liesenfeld, D.B.; Habermann, N.; Owen, R.W.; Scalbert, A.; Ulrich, C.M. Review of mass spectrometry-based metabolomics in cancer research. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 2182–2201. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Chatterjee, N.; Ershler, W.B.; Brawley, O.W. Estrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology, and End Results database. Breast Cancer Res. Treat. 2002, 76, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Skaar, T.; Baumann, K.; Leonessa, F.; James, M.; Lippman, J.; Thompson, E.W.; Freter, C.; Brunner, N. Hormonal carcinogenesis in breast cancer: Cellular and molecular studies of malignant progression. Breast Cancer Res. Treat. 1994, 31, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.; Costantino, J.; Redmond, C.; Poisson, R.; Bowman, D.; Couture, J.; Dimitrov, N.V.; Wolmark, N.; Wickerham, D.L.; Fisher, E.R. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N. Engl. J. Med. 1989, 320, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Howell, A. Selective oestrogen receptor downregulator. Eur. J. Cancer 2002, 38 (Suppl. 6), S61–S62. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Robertson, J.F.; Quaresma, A.J.; Aschermannova, A.; Mauriac, L.; Kleeberg, U.R.; Vergote, I.; Erikstein, B.; Webster, A.; Morris, C. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J. Clin. Oncol. 2002, 20, 3396–3403. [Google Scholar] [CrossRef] [PubMed]
- Buzdar, A.; Jonat, W.; Howell, A.; Jones, S.E.; Blomqvist, C.; Vogel, C.L.; Eiermann, W.; Wolter, J.M.; Azab, M.; Webster, A.; et al. Anastrozole, a potent and selective aromatase inhibitor, versus megestrol acetate in postmenopausal women with advanced breast cancer: results of overview analysis of two phase III trials. Arimidex Study Group. J. Clin. Oncol. 1996, 14, 2000–2011. [Google Scholar] [PubMed]
- Gershanovich, M.; Chaudri, H.A.; Campos, D.; Lurie, H.; Bonaventura, A.; Jeffrey, M.; Buzzi, F.; Bodrogi, I.; Ludwig, H.; Reichardt, P.; et al. Letrozole, a new oral aromatase inhibitor: randomised trial comparing 2.5 mg daily, 0.5 mg daily and aminoglutethimide in postmenopausal women with advanced breast cancer. Letrozole International Trial Group (AR/BC3). Ann. Oncol. 1998, 9, 639–645. [Google Scholar] [CrossRef]
- Howell, A.; Buzdar, A. Are aromatase inhibitors superior to antiestrogens? J. Steroid Biochem. Mol. Biol. 2005, 93, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Mouridsen, H.; Gershanovich, M.; Sun, Y.; Perez-Carrion, R.; Boni, C.; Monnier, A.; Apffelstaedt, J.; Smith, R.; Sleeboom, H.P.; Janicke, F.; et al. Superior efficacy of letrozole versus tamoxifen as first-line therapy for postmenopausal women with advanced breast cancer: results of a phase III study of the International Letrozole Breast Cancer Group. J. Clin. Oncol. 2001, 19, 2596–2606. [Google Scholar]
- Baselga, J.; Cortes, J.; Kim, S.B.; Im, S.A.; Hegg, R.; Im, Y.H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 2012, 366, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Krajewski, K.; Cakar, B.; Ma, C.X. Targeted therapy for breast cancer. Am. J. Pathol. 2013, 183, 1096–1112. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Skaar, T.C.; Bouker, K.B.; Davis, N.; Lee, Y.R; Welch, J.N.; Leonessa, F. Molecular and pharmacological aspects of antiestrogen resistance. J. Steroid Biochem. Mol. Biol. 2001, 76, 71–84. [Google Scholar]
- Clarke, R.; Liu, M.C.; Bouker, K.B.; Gu, Z.; Lee, R.Y.; Zhu, Y.; Skaar, T.C.; Gomez, B.; O’Brien, K.; Wang, Y.; et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003, 22, 7316–7339. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.L.; Li, G.; Loh, S.L.; Sung, W.K.; Liu, E.T. Cellular reprogramming by the conjoint action of ERalpha, FOXA1, and GATA3 to a ligand-inducible growth state. Mol. Syst. Biol. 2011, 7, 526. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Piccart, M.; Sotiriou, C. The use of gene-expression profiling to better understand the clinical heterogeneity of estrogen receptor positive breast cancers and tamoxifen response. Crit. Rev. Oncol. Hematol. 2007, 61, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; D’Santos, C.; Serandour, A.A.; Ali, H.R.; Brown, G.D.; Atkins, A.; Rueda, O.M.; Holmes, K.A.; Theodorou, V.; Robinson, JL; et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013, 3, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E., Jr.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Criscitiello, C.; Azim, H.A., Jr.; Schouten, P.C.; Linn, S.C.; Sotiriou, C. Understanding the biology of triple-negative breast cancer. Ann. Oncol. 2012, 23 (Suppl. 6), VI13–VI18. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Signaling in control of cell growth and metabolism. Cold Spring Harb. Perspect. Biol. 2012, 4, a006783. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Shajahan, A.N.; Wang, Y.; Tyson, J.J.; Riggins, R.B.; Weiner, L.M.; Bauman, W.T.; Xuan, J.; Zhang, B.; Facey, C.; et al. Endoplasmic reticulum stress, the unfolded protein response, and gene network modeling in antiestrogen resistant breast cancer. Horm. Mol. Biol. Clin. Investig. 2011, 5, 35–44. [Google Scholar] [PubMed]
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012, 72, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Riggins, R.B.; Bouton, A.H.; Liu, M.C.; Clarke, R. Antiestrogens, aromatase inhibitors, and apoptosis in breast cancer. Vitam. Horm. 2005, 71, 201–237. [Google Scholar] [PubMed]
- Bouker, K.B.; Skaar, T.C.; Fernandez, D.R.; O’Brien, K.A.; Riggins, R.B.; Cao, D.; Clarke, R. interferon regulatory factor-1 mediates the proapoptotic but not cell cycle arrest effects of the steroidal antiestrogen ICI 182,780 (faslodex, fulvestrant). Cancer Res. 2004, 64, 4030–4039. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Shajahan, A.N.; Warri, A.; Jin, L.; Hilakivi-Clarke, L.A.; Clarke, R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012, 72, 3337–3349. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.C.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Clarke, R. Co-inhibition of BCL-W and BCL2 restores antiestrogen sensitivity through BECN1 and promotes an autophagy-associated necrosis. PLoS One 2010, 5, e8604. [Google Scholar] [CrossRef] [PubMed]
- Gomez, B.P.; Riggins, R.B.; Shajahan, A.N.; Klimach, U.; Wang, A.; Crawford, A.C.; Zhu, Y.; Zwart, A.; Wang, M.; Clarke, R. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 2007, 21, 4013–4027. [Google Scholar] [CrossRef] [PubMed]
- Nehra, R.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Crawford, A.C.; Clarke, R. BCL2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells. FASEB J. 2010, 24, 2040–2055. [Google Scholar] [CrossRef] [PubMed]
- Riggins, R.B.; Zwart, A.; Nehra, R.; Clarke, R. The nuclear factor kappa B inhibitor parthenolide restores ICI 182,780 (Faslodex; fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol. Cancer Ther. 2005, 4, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shajahan, A.N.; Wang, A.; Decker, M.; Minshall, R.D.; Liu, M.C.; Clarke, R. Caveolin-1 tyrosine phosphorylation enhances paclitaxel-mediated cytotoxicity. J. Biol. Chem. 2007, 282, 5934–5943. [Google Scholar] [CrossRef] [PubMed]
- Shajahan, A.N.; Dobbin, Z.C.; Hickman, F.E.; Dakshanamurthy, S.; Clarke, R. Tyrosine-phosphorylated caveolin-1 (Tyr-14) increases sensitivity to paclitaxel by inhibiting BCL2 and BCLxL proteins via c-Jun N-terminal kinase (JNK). J. Biol. Chem. 2012, 287, 17682–17692. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Balko, J.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; Gonzalez-Angulo, A.M.; Mills, G.B.; Miller, W.R.; Wu, H.; et al. A gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistance. Clin. Cancer Res. 2011, 17, 2024–2034. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sergio, C.M.; Loi, S.; Inman, C.K.; Anderson, L.R.; Alles, M.C.; Pinese, M.; Caldon, C.E.; Schutte, J.; Gardiner-Garden, M.; et al. Identification of functional networks of estrogen- and c-Myc-responsive genes and their relationship to response to tamoxifen therapy in breast cancer. PLoS One 2008, 3, e2987. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, Y.; Hua, H.; Li, M.; Luo, T.; Xu, L.; Wang, R.; Liu, D.; Zhang, Y.; Jiang, Y. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J. Cell Mol. Med. 2009, 13, 3888–3897. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Shajahan-Haq, A.N.; Cook, K.L.; Schwartz-Roberts, J.L.; Eltayeb, A.E.; Demas, D.M.; Warri, A.M.; Facey, C.O.; Hilakivi-Clarke, L.A.; Clarke, R. MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol. Cancer 2014, 13, 239. [Google Scholar] [CrossRef] [PubMed]
- Deyati, A.; Younesi, E.; Hofmann-Apitius, M.; Novac, N. Challenges and opportunities for oncology biomarker discovery. Drug Discov. Today 2013, 18, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Armitage, E.G.; Barbas, C. Metabolomics in cancer biomarker discovery: Current trends and future perspectives. J. Pharm. Biomed. Anal. 2014, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Xu, G. Metabolomics for tumor marker discovery and identification based on chromatography-mass spectrometry. Expert Rev. Mol. Diagn. 2013, 13, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Chawla, K. Oncometabolomics in cancer research. Expert Rev. Proteomics 2013, 10, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.L.; Westbrook, J.A.; Brown, J.E. Omic-profiling in breast cancer metastasis to bone: implications for mechanisms, biomarkers and treatment. Cancer Treat. Rev. 2014, 40, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Duarte, I.F.; Gil, A.M. Metabolic signatures of cancer unveiled by NMR spectroscopy of human biofluids. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 62, 51–74. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Dwivedi, D.K.; Jagannathan, N.R. High-resolution NMR spectroscopy of human body fluids and tissues in relation to prostate cancer. NMR Biomed. 2014, 27, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D.; Reeves, R.; Resar, L.S.; Hill, H.H., Jr. Metabolomics of colorectal cancer: past and current analytical platforms. Anal. Bioanal. Chem. 2013, 405, 5013–5030. [Google Scholar] [CrossRef] [PubMed]
- Blekherman, G.; Laubenbacher, R.; Cortes, D.F.; Mendes, P.; Torti, F.M.; Akman, S.; Torti, S.V.; Shulaev, V. Bioinformatics tools for cancer metabolomics. Metabolomics 2011, 7, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Gonzalez, F.J. Challenges and opportunities of metabolomics. J. Cell Physiol. 2012, 227, 2975–2981. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Xiao, J.F.; Tuli, L.; Ressom, H.W. LC-MS-based metabolomics. Mol. Biosyst. 2012, 8, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Mamas, M.; Dunn, W.B.; Neyses, L.; Goodacre, R. The role of metabolites and metabolomics in clinically applicable biomarkers of disease. Arch. Toxicol. 2011, 85, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, K.A.; Styczynski, M.P. Applications of metabolomics in cancer research. J. Carcinog. 2013, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Vander Heiden, M.G.; Cantley, L.C. Rewiring of glycolysis in cancer cell metabolism. Cell Cycle 2010, 9, 4253. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Pinedo, C.; El, M.N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G. Exploiting tumor metabolism: Challenges for clinical translation. J. Clin. Invest. 2013, 123, 3648–3651. [Google Scholar] [CrossRef] [PubMed]
- Asiago, V.M.; Alvarado, L.Z.; Shanaiah, N.; Gowda, G.A.; Owusu-Sarfo, K.; Ballas, R.A.; Raftery, D. Early detection of recurrent breast cancer using metabolite profiling. Cancer Res. 2010, 70, 8309–8318. [Google Scholar] [CrossRef] [PubMed]
- Cadoo, K.A.; Fornier, M.N.; Morris, P.G. Biological subtypes of breast cancer: current concepts and implications for recurrence patterns. Q. J. Nucl. Med. Mol. Imaging. 2013, 57, 312–321. [Google Scholar] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [PubMed]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Weljie, A.M.; Bondareva, A.; Zang, P.; Jirik, F.R. 1H NMR metabolomics identification of markers of hypoxia-induced metabolic shifts in a breast cancer model system. J. Biomol. NMR 2011, 49, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Brockmoller, S.F.; Muller, B.M.; Barupal, D.K.; Richter-Ehrenstein, C.; Kleine-Tebbe, A.; Griffin, J.L.; Oresic, M.; Dietel, M.; Denkert, C.; et al. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: Alterations in glutamine and beta-alanine metabolism. J. Proteomics 2013, 94, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Jobard, E.; Pontoizeau, C.; Blaise, B.J.; Bachelot, T.; Elena-Herrmann, B.; Tredan, O. A serum nuclear magnetic resonance-based metabolomic signature of advanced metastatic human breast cancer. Cancer Lett. 2014, 343, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zhou, B.; Su, M.; Baxter, S.; Zheng, X.; Zhao, X.; Yen, Y.; Jia, W. Mass spectrometry-based quantitative metabolomics revealed a distinct lipid profile in breast cancer patients. Int. J. Mol. Sci. 2013, 14, 8047–8061. [Google Scholar] [CrossRef] [PubMed]
- Bayet-Robert, M.; Morvan, D. Metabolomics reveals metabolic targets and biphasic responses in breast cancer cells treated by curcumin alone and in association with docetaxel. PLoS One 2013, 8, e57971. [Google Scholar] [CrossRef] [PubMed]
- Brauer, H.A.; Makowski, L.; Hoadley, K.A.; Casbas-Hernandez, P.; Lang, L.J.; Roman-Perez, E.; D’Arcy, M.; Freemerman, A.J.; Perou, C.M.; Troester, M.A. Impact of tumor microenvironment and epithelial phenotypes on metabolism in breast cancer. Clin. Cancer Res. 2013, 19, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Mandujano-Tinoco, E.A.; Gallardo-Perez, J.C.; Marin-Hernandez, A.; Moreno-Sanchez, R.; Rodriguez-Enriquez, S. Anti-mitochondrial therapy in human breast cancer multi-cellular spheroids. Biochim. Biophys. Acta 2013, 1833, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Liu, L.; Zhang, J.; Bowers, J.; Gowda, G.A.; Seeger, H.; Fehm, T.; Neubauer, H.J.; Vogel, U.; Clare, S.E.; et al. Metabolomics approach for predicting response to neoadjuvant chemotherapy for breast cancer. Mol. Oncol. 2013, 7, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J. Remodeling of central metabolism in invasive breast cancer compared to normal breast tissue—a GC-TOFMS based metabolomics study. BMC Genomics 2012. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Soga, T.; Pollard, P.J.; Adam, J. The emerging role of fumarate as an oncometabolite. Front. Oncol. 2012, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Claudino, W.M.; Goncalves, P.H.; Di, L.A.; Philip, P.A.; Sarkar, F.H. Metabolomics in cancer: A bench-to-bedside intersection. Crit. Rev. Oncol. Hematol. 2012, 84, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, A.; Lewensohn, R. Metabolomics: Moving to the clinic. J. Neuroimmune Pharmacol. 2010, 5, 4–17. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, T.M. Recent advances in metabolomics in oncology. Bioanalysis 2012, 4, 431–451. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.I.; Cravatt, B.F.; Nomura, D.K. Global profiling strategies for mapping dysregulated metabolic pathways in cancer. Cell Metab. 2012, 16, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.C.; Chan, H.L. Targeting proteomics to investigate metastasis-associated mitochondrial proteins. J. Bioenerg. Biomembr. 2012, 44, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, P.; Yang, Y.; Wang, F.; Qin, H. Metabolomics in the fields of oncology: A review of recent research. Mol. Biol. Rep. 2012, 39, 7505–7511. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Li, M.; Zha, W.; Zhao, Q.; Gu, R.; Liu, L.; Shi, J.; Zhou, J.; Zhou, F.; Wu, X.; et al. Metabolomic approach to evaluating adriamycin pharmacodynamics and resistance in breast cancer cells. Metabolomics 2013, 9, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.L.; Kuo, T.C.; Ho, T.J.; Harn, Y.C.; Wang, S.Y.; Fu, W.M.; Kuo, C.H.; Tseng, Y.J. Metabolomic Dynamic Analysis of Hypoxia in MDA-MB-231 and the Comparison with Inferred Metabolites from Transcriptomics Data. Cancers 2013, 5, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Morvan, D. Functional metabolomics uncovers metabolic alterations associated to severe oxidative stress in MCF7 breast cancer cells exposed to ascididemin. Mar. Drugs 2013, 11, 3846–3860. [Google Scholar] [CrossRef] [PubMed]
- Bayet-Robert, M.; Morvan, D. Metabolomics reveals metabolic targets and biphasic responses in breast cancer cells treated by curcumin alone and in association with docetaxel. PLoS One 2013, 8, e57971. [Google Scholar] [CrossRef] [PubMed]
- Jager, W.; Gruber, A.; Giessrigl, B.; Krupitza, G.; Szekeres, T.; Sonntag, D. Metabolomic analysis of resveratrol-induced effects in the human breast cancer cell lines MCF-7 and MDA-MB-231. OMICS 2011, 15, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Slupsky, C.M.; Steed, H.; Wells, T.H.; Dabbs, K.; Schepansky, A.; Capstick, V.; Faught, W.; Sawyer, M.B. Urine metabolite analysis offers potential early diagnosis of ovarian and breast cancers. Clin. Cancer Res. 2010, 16, 5835–5841. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Wilson, I.D.; Lindon, J.C. Pharmacometabonomics as an effector for personalized medicine. Pharmacogenomics 2011, 12, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.T.; Mendelsohn, J.; Mills, G.B. Bioinformatics and systems biology. Mol. Oncol. 2012, 6, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Bucher, E.; Hilvo, M.; Salek, R.; Oresic, M.; Griffin, J.; Brockmoller, S.; Klauschen, F.; Loibl, S.; Barupal, D.K.; et al. Metabolomics of human breast cancer: New approaches for tumor typing and biomarker discovery. Genome Med. 2012, 4, 37. [Google Scholar] [PubMed]
- Peintinger, F. Using molecular profiles to tailor treatment in breast cancer: Are they ready for prime time? Curr. Opin. Obstet. Gynecol. 2014, 26, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.; Flores, M. A personal view on systems medicine and the emergence of proactive P4 medicine: Predictive, preventive, personalized and participatory. N. Biotechnol. 2012, 29, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.; Auffray, C. Participatory medicine: A driving force for revolutionizing healthcare. Genome Med. 2013, 5, 110. [Google Scholar] [PubMed]
- Hood, L. Systems biology and p4 medicine: Past, present, and future. Rambam Maimonides Med. J. 2013, 4, e0012. [Google Scholar] [CrossRef] [PubMed]
- Cesario, A.; Auffray, C.; Russo, P.; Hood, L. P4 Medicine Needs P4 Education. Curr. Pharm. Des. 2014, 20, 6071–6072. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Huang, Y.; Yan, G.; Cen, X.; Zhao, Y.L. Metabolomics: A revolution for novel cancer marker identification. Comb. Chem. High Throughput Screen. 2012, 15, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Byun, J.; Pennathur, S. Analytical approaches to metabolomics and applications to systems biology. Semin. Nephrol. 2010, 30, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Casado-Vela, J.; Cebrian, A.; Gomez del Pulgar, M.T.; Lacal, J.C. Approaches for the study of cancer: Towards the integration of genomics, proteomics and metabolomics. Clin. Transl. Oncol. 2011, 13, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.W.; Lorkiewicz, P.K.; Sellers, K.; Moseley, H.N.; Higashi, R.M.; Lane, A.N. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol. Ther. 2012, 133, 366–391. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.F.; Zhou, B.; Ressom, H.W. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. Trends Analyt. Chem. 2012, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lin, S.C.; Chen, K.F.; Lai, Y.Y.; Tsai, S.J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 2008, 283, 28106–28114. [Google Scholar] [CrossRef] [PubMed]
- Fanciulli, M.; Bruno, T.; Giovannelli, A.; Gentile, F.P.; Di, P.M.; Rubiu, O.; Floridi, A. Energy metabolism of human LoVo colon carcinoma cells: Correlation to drug resistance and influence of lonidamine. Clin. Cancer Res. 2000, 6, 1590–1597. [Google Scholar] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Horecker, B.L. The pentose phosphate pathway. J. Biol. Chem. 2002, 277, 47965–47971. [Google Scholar] [CrossRef] [PubMed]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef] [PubMed]
- Backos, D.S.; Franklin, C.C.; Reigan, P. The role of glutathione in brain tumor drug resistance. Biochem. Pharmacol. 2012, 83, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zur, H.A.; Coy, J.F.; Lochelt, M. Transketolase-like protein 1 (TKTL1) is required for rapid cell growth and full viability of human tumor cells. Int. J. Cancer 2009, 124, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, F.; Rosa, R.; Vitale, M.; D’Ambrosio, C.; Succoio, M.; Formisano, L.; Nappi, L.; Romano, M.F.; Scaloni, A.; Tortora, G.; et al. Increased anaerobic metabolism is a distinctive signature in a colorectal cancer cellular model of resistance to antiepidermal growth factor receptor antibody. Proteomics 2013, 13, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Mancuso, A.; Bui, T.V.; Tong, X.; Gruber, J.J.; Swider, C.R.; Sanchez, P.V.; Lum, J.J.; Sayed, N.; Melo, J.V.; et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene 2010, 29, 2962–2972. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, S.; Li, Y.; Tang, Z.; Kong, W. Hexokinase 2 overexpression promotes the proliferation and survival of laryngeal squamous cell carcinoma. Tumor Biol. 2013, 35, 3743–3753. [Google Scholar] [CrossRef]
- Gershon, T.R.; Crowther, A.J.; Tikunov, A.; Garcia, I.; Annis, R.; Yuan, H.; Miller, C.R.; Macdonald, J.; Olson, J.; Deshmukh, M. Hexokinase-2-mediated aerobic glycolysis is integral to cerebellar neurogenesis and pathogenesis of medulloblastoma. Cancer Metab. 2013. [Google Scholar] [CrossRef]
- Puzone, R.; Savarino, G.; Salvi, S.; Dal Bello, M.G.; Barletta, G.; Genova, C.; Rijavec, E.; Sini, C.; Esposito, A.I.; Ratto, G.B.; et al. Glyceraldehyde-3-phosphate dehydrogenase gene over expression correlates with poor prognosis in non small cell lung cancer patients. Mol. Cancer 2013, 12, 97. [Google Scholar] [CrossRef] [PubMed]
- Jacquin, M.A.; Chiche, J.; Zunino, B.; Beneteau, M.; Meynet, O.; Pradelli, L.A.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.E. GAPDH binds to active Akt, leading to Bcl-xL increase and escape from caspase-independent cell death. Cell Death Differ. 2013, 20, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Semenza, G.L. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget 2011, 2, 551–556. [Google Scholar] [PubMed]
- Hussien, R.; Brooks, G.A. Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol. Genomics 2011, 43, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Blenis, J. Appetite for destruction: the inhibition of glycolysis as a therapy for tuberous sclerosis complex-related tumors. BMC Biol. 2011, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Jung, W.H.; Koo, J.S. Differential expression of enzymes associated with serine/glycine metabolism in different breast cancer subtypes. PLoS One 2014, 9, e101004. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shajahan-Haq, A.N.; Cheema, M.S.; Clarke, R. Application of Metabolomics in Drug Resistant Breast Cancer Research. Metabolites 2015, 5, 100-118. https://doi.org/10.3390/metabo5010100
Shajahan-Haq AN, Cheema MS, Clarke R. Application of Metabolomics in Drug Resistant Breast Cancer Research. Metabolites. 2015; 5(1):100-118. https://doi.org/10.3390/metabo5010100
Chicago/Turabian StyleShajahan-Haq, Ayesha N., Mehar S. Cheema, and Robert Clarke. 2015. "Application of Metabolomics in Drug Resistant Breast Cancer Research" Metabolites 5, no. 1: 100-118. https://doi.org/10.3390/metabo5010100
APA StyleShajahan-Haq, A. N., Cheema, M. S., & Clarke, R. (2015). Application of Metabolomics in Drug Resistant Breast Cancer Research. Metabolites, 5(1), 100-118. https://doi.org/10.3390/metabo5010100