In Silico Optimization of Mass Spectrometry Fragmentation Strategies in Metabolomics

 ,
,  , , and
, , and 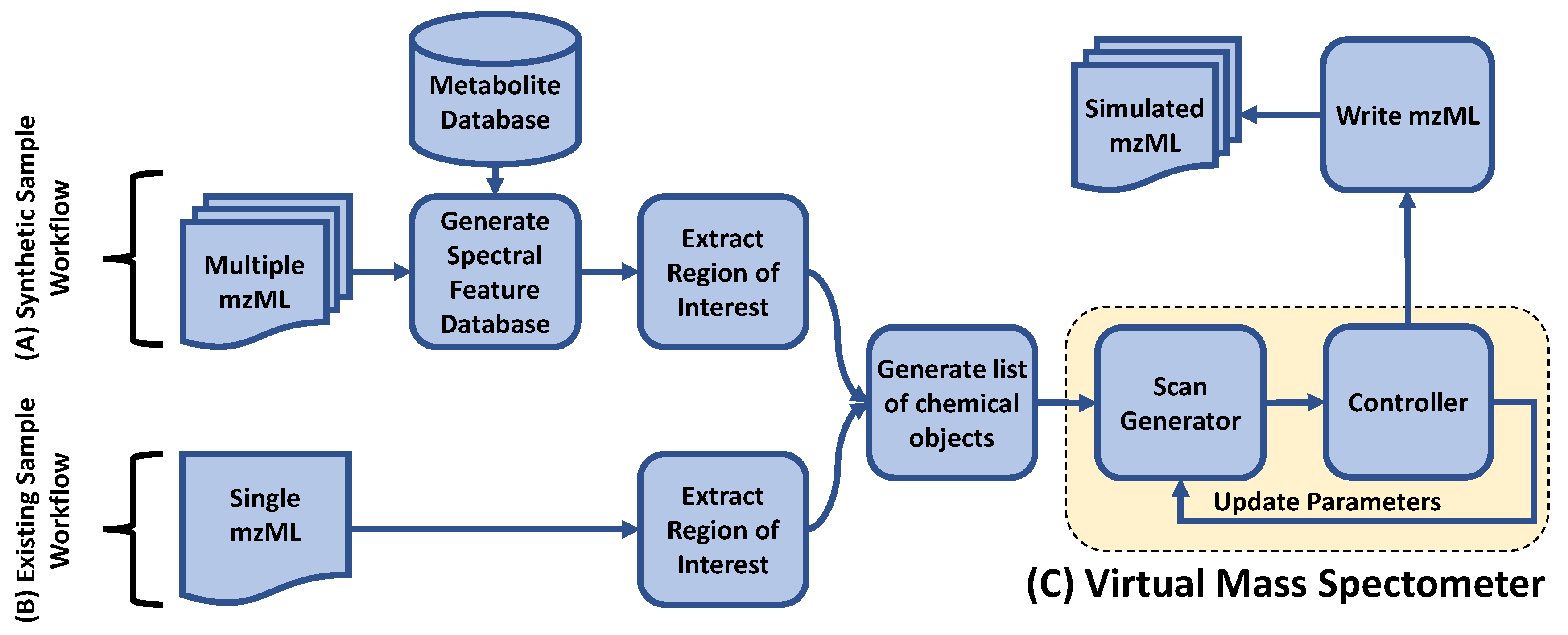{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. LC-MS/MS Materials and Methods
2.1.1. Samples
2.1.2. Liquid Chromatography
2.1.3. Mass Spectrometer Acquisition
2.1.4. Data Transformation
2.2. Computational Methods
2.2.1. Overall Framework
2.2.2. Chemical Objects
2.2.3. Spectral Feature Database
2.2.4. ROI Extraction and Normalization
2.2.5. MS2 Scan Generation
2.2.6. Scan Time
2.3. Controllers
2.3.1. MS1 Controller
2.3.2. Top-N DDA Controller
2.3.3. DsDA Controller
3. Results
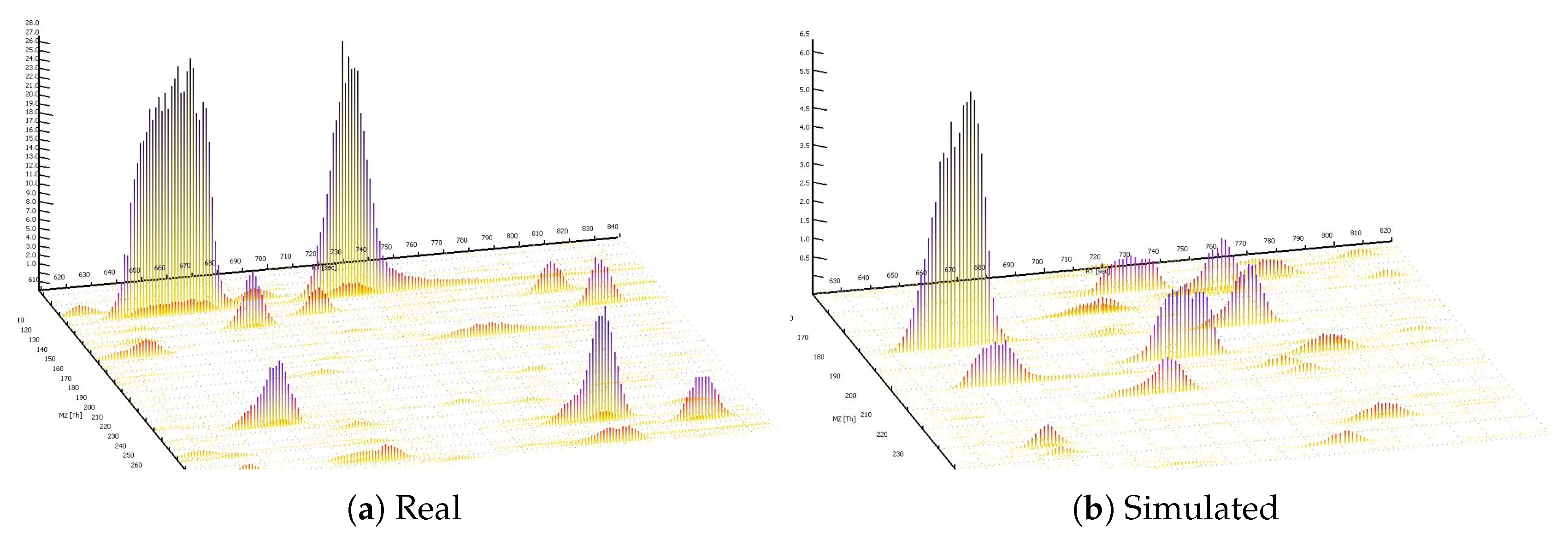
3.1. MS1 Simulations
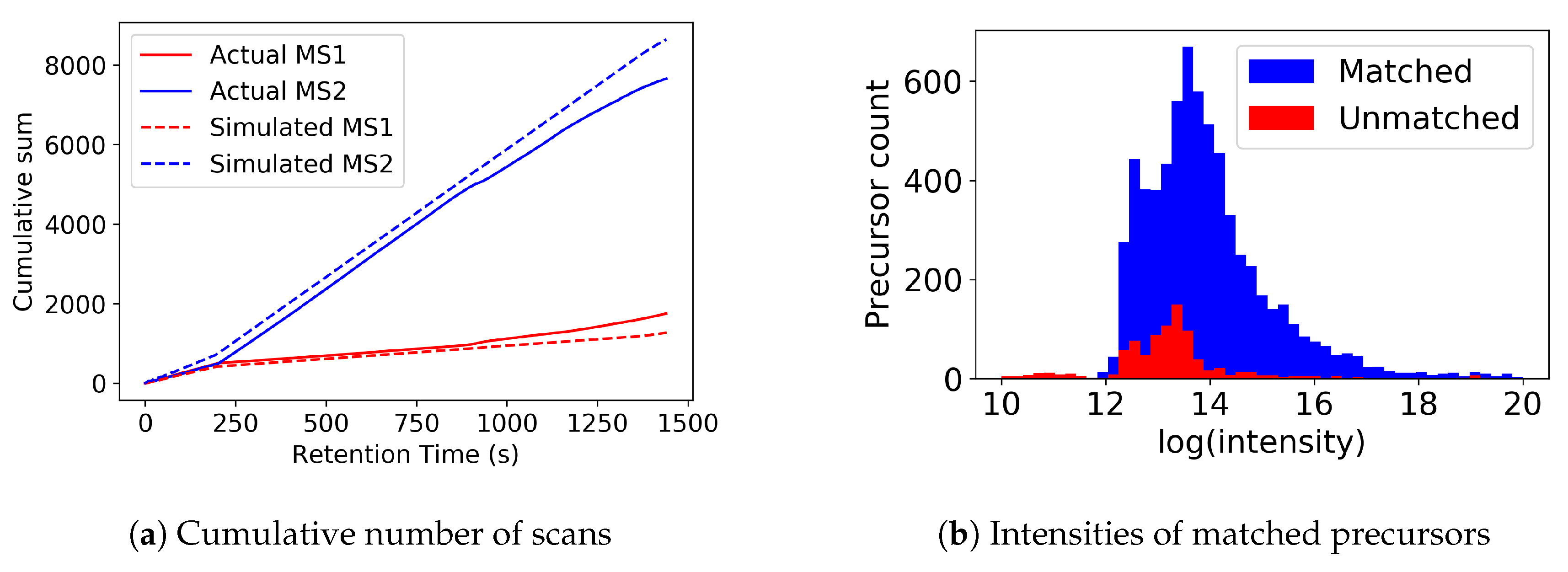
3.2. Top-N Simulations
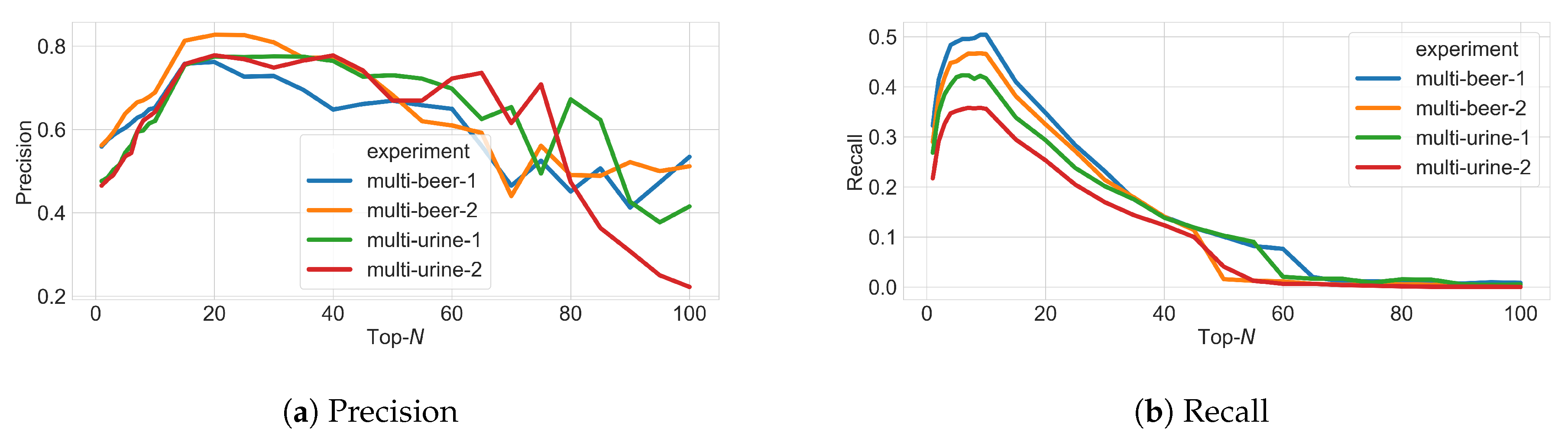
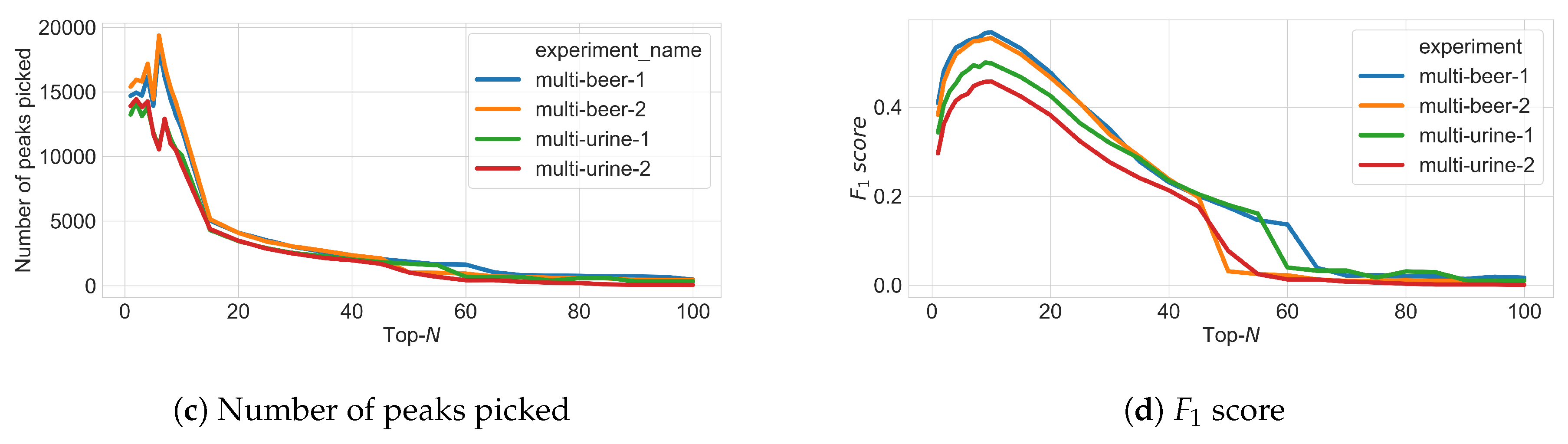
3.3. Varying N in Top-N Simulations
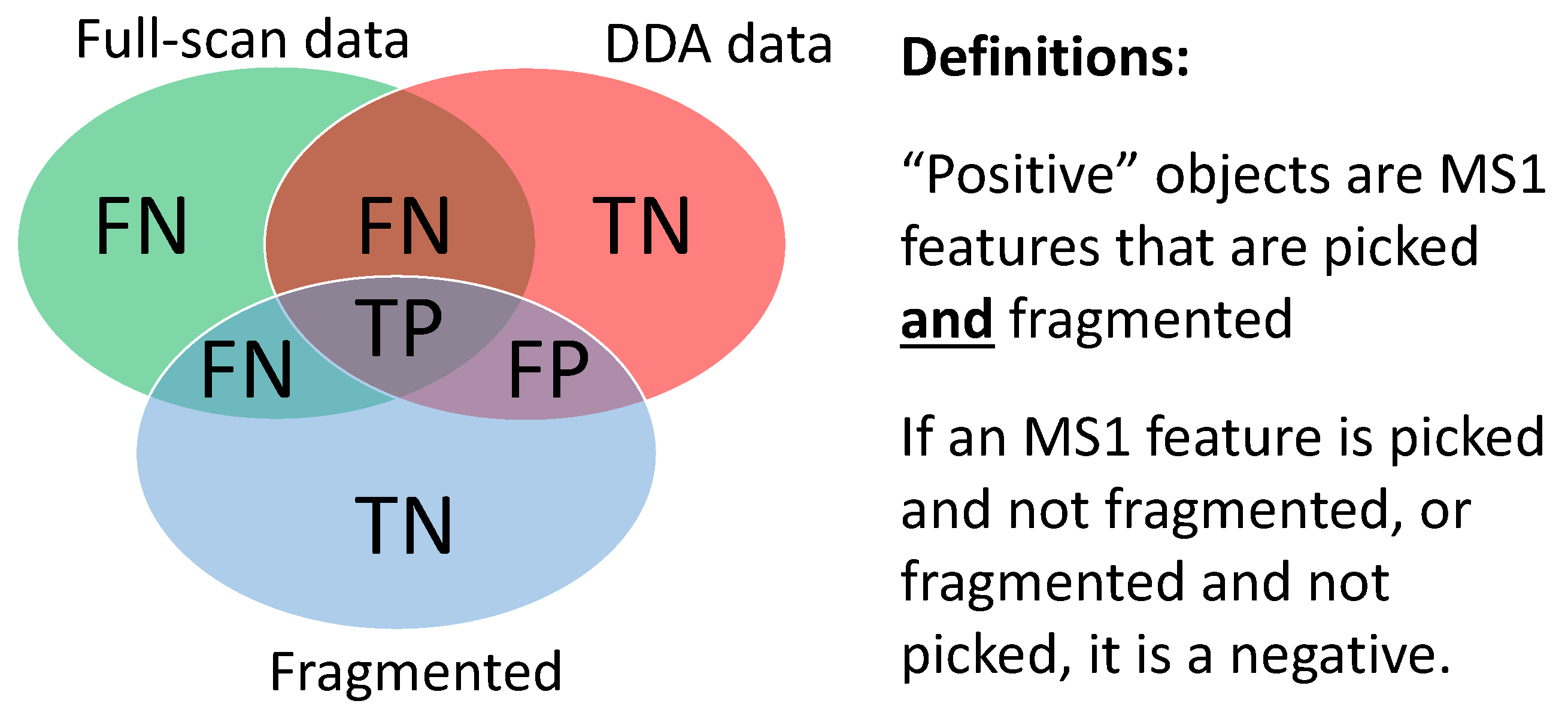
- True Positives (TP): MS1 features from ground truth (found in both fragmentation and full-scan files) that are fragmented above the minimum intensity threshold.
- False Positives (FP): MS1 features not from ground truth (found in fragmentation file but not in full-scan file) that are fragmented above the minimum intensity threshold.
- False Negatives (FN): MS1 features not from ground truth (not found in fragmentation file but found in full-scan file) that are not fragmented or fragmented below the minimum intensity threshold.
- True Negatives (TN): MS1 features not from ground truth (found in fragmentation file but not found in full-scan file) that are not fragmented or fragmented below the minimum intensity threshold.
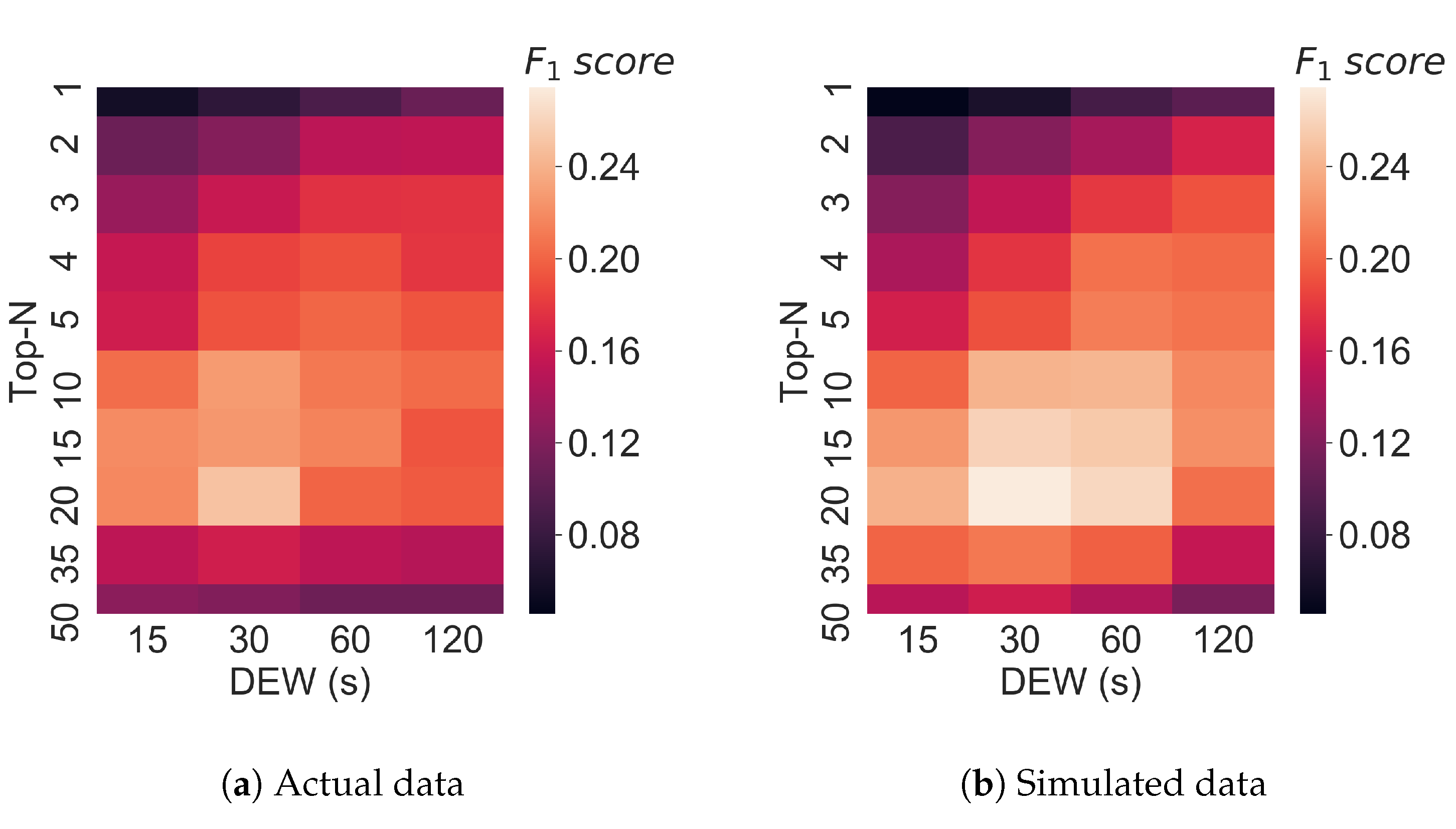
3.4. Varying Multiple Parameters in Top-N Simulations
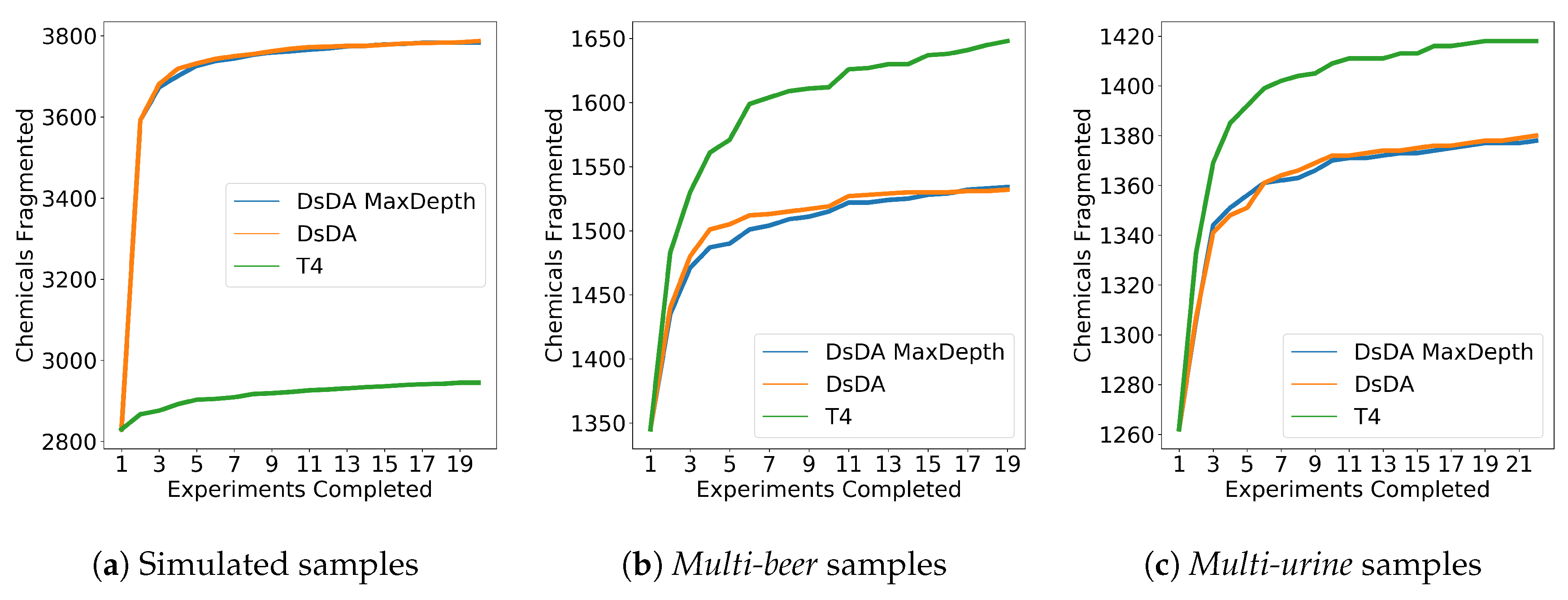
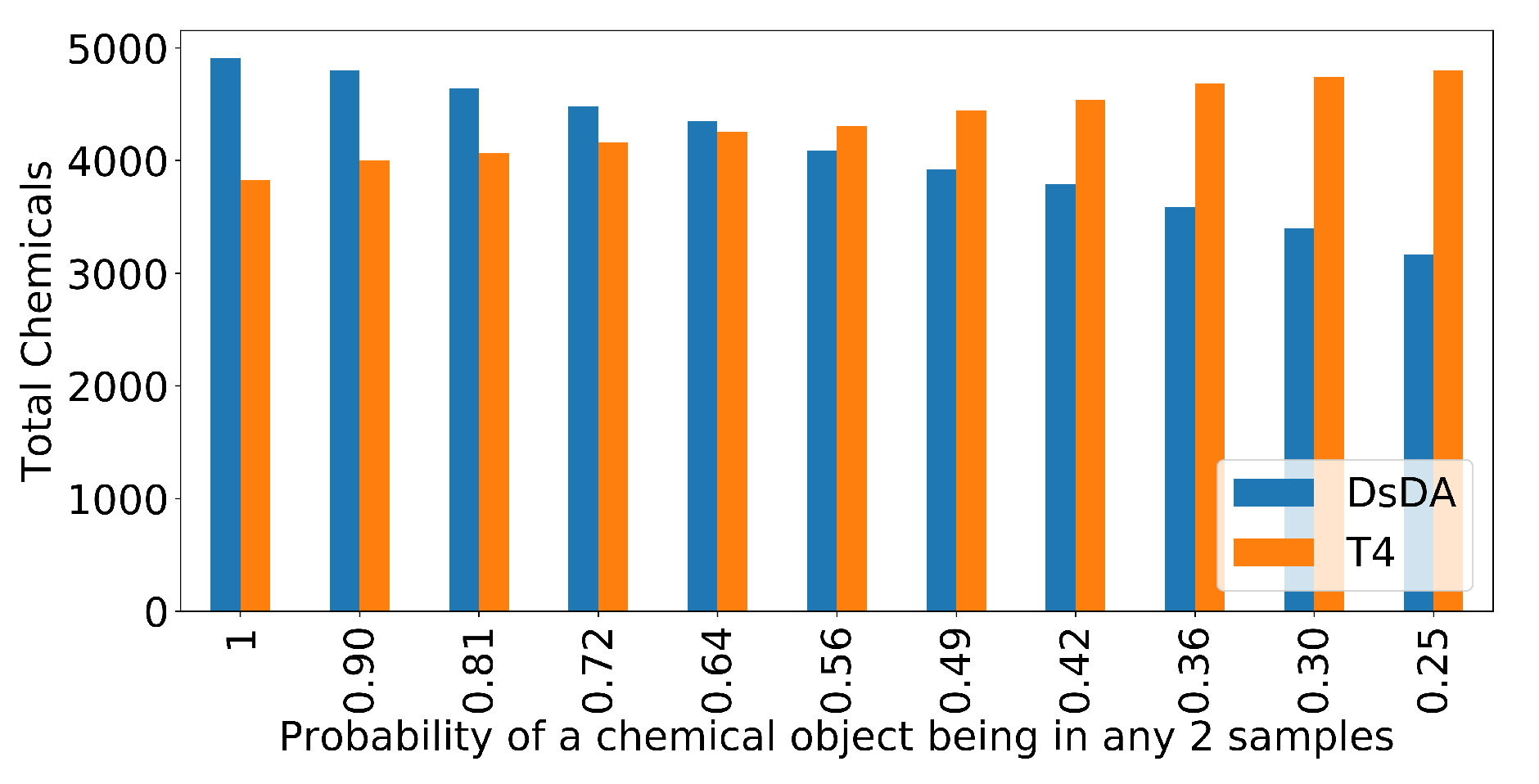
3.5. DsDA Simulations
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Kang, K.B.; Caraballo-Rodríguez, A.M.; Nothias, L.F.; Wandy, J.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; Van Der Hooft, J.J. MolNetEnhancer: Enhanced molecular networks by integrating metabolome mining and annotation tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Walker, S. Nested data independent MS/MS acquisition. Anal. Bioanal. Chem. 2016, 408, 5031–5040. [Google Scholar] [CrossRef] [PubMed]
- Broeckling, C.D.; Hoyes, E.; Richardson, K.; Brown, J.M.; Prenni, J.E. Comprehensive Tandem-Mass-Spectrometry Coverage of Complex Samples Enabled by Data-Set-Dependent Acquisition. Anal. Chem. 2018, 90, 8020–8027. [Google Scholar] [CrossRef] [PubMed]
- Noyce, A.B.; Smith, R.; Dalgleish, J.; Taylor, R.M.; Erb, K.C.; Okuda, N.; Prince, J.T. Mspire-Simulator: LC-MS shotgun proteomic simulator for creating realistic gold standard data. J. Proteome Res. 2013, 12, 5742–5749. [Google Scholar] [CrossRef]
- Smith, R.; Prince, J.T. JAMSS: Proteomics mass spectrometry simulation in Java. Bioinformatics 2014, 31, 791–793. [Google Scholar] [CrossRef][Green Version]
- Schulz-Trieglaff, O.; Pfeifer, N.; Gröpl, C.; Kohlbacher, O.; Reinert, K. LC-MSsim–a simulation software for liquid chromatography mass spectrometry data. BMC Bioinform. 2008, 9, 423. [Google Scholar] [CrossRef]
- Bielow, C.; Aiche, S.; Andreotti, S.; Reinert, K. MSSimulator: Simulation of mass spectrometry data. J. Proteome Res. 2011, 10, 2922–2929. [Google Scholar] [CrossRef][Green Version]
- Awan, M.G.; Saeed, F. MaSS-Simulator: A Highly Configurable Simulator for Generating MS/MS Datasets for Benchmarking of Proteomics Algorithms. Proteomics 2018, 18, e1800206. [Google Scholar] [CrossRef]
- Goldfarb, D.; Wang, W.; Major, M.B. MSAcquisitionSimulator: Data-dependent acquisition simulator for LC-MS shotgun proteomics. Bioinformatics 2015, 32, 1269–1271. [Google Scholar] [CrossRef]
- Bailey, D.J.; Grosse-Coosmann, F.; Doshi, M.; Song, Q.; Canterbury, J.D.; Wan, Q.; Senko, M.W. Real-Time Instrument Control of the Orbitrap Tribrid Mass Spectrometer. 2016. Available online: http://tools.thermofisher.com/content/sfs/posters/PN-64748-Orbitrap-Tribrid-Mass-Spectrometer-ASMS2016-PN64748-EN.pdf (accessed on 8 October 2019).
- Schweppe, D.K.; Eng, J.K.; Bailey, D.; Rad, R.; Yu, Q.; Navarrete-Perea, J.; Huttlin, E.L.; Erickson, B.K.; Paulo, J.A.; Gygi, S.P. Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. bioRxiv 2019. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2017, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Van der Hooft, J.J.J.; Wandy, J.; Young, F.; Padmanabhan, S.; Gerasimidis, K.; Burgess, K.E.V.; Barrett, M.P.; Rogers, S. Unsupervised discovery and comparison of structural families across multiple samples in untargeted metabolomics. Anal. Chem. 2017, 89, 7569–7577. [Google Scholar] [CrossRef] [PubMed]
- Creek, D.J.; Jankevics, A.; Breitling, R.; Watson, D.G.; Barrett, M.P.; Burgess, K.E. Toward global metabolomics analysis with hydrophilic interaction liquid chromatography–mass spectrometry: Improved metabolite identification by retention time prediction. Anal. Chem. 2011, 83, 8703–8710. [Google Scholar] [CrossRef]
- Klein, J.; Zaia, J. psims-A declarative writer for mzML and mzIdentML for Python. Mol. Cell. Proteom. 2019, 18, 571–575. [Google Scholar] [CrossRef]
- Kösters, M.; Leufken, J.; Schulze, S.; Sugimoto, K.; Klein, J.; Zahedi, R.; Hippler, M.; Leidel, S.; Fufezan, C. pymzML v2. 0: Introducing a highly compressed and seekable gzip format. Bioinformatics 2018, 34, 2513–2514. [Google Scholar] [CrossRef]
- Van Rossum, G.; Google, Inc. Python Programming Language. In Proceedings of the USENIX Annual Technical Conference, Santa Clara, CA, USA, 17–22 June 2007; Volume 41, p. 36. [Google Scholar]
- Tautenhahn, R.; Boettcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Van Der Hooft, J.J.J.; Wandy, J.; Barrett, M.P.; Burgess, K.E.; Rogers, S. Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. USA 2016, 113, 13738–13743. [Google Scholar] [CrossRef]
- Griffiths, T.L.; Jordan, M.I.; Tenenbaum, J.B.; Blei, D.M. Hierarchical topic models and the nested Chinese restaurant process. Adv. Neural Inf. Process. Syst. 2004, 17–24. [Google Scholar]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Greiner, R.; Wishart, D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 2015, 11, 98–110. [Google Scholar] [CrossRef]
- Wei, J.N.; Belanger, D.; Adams, R.P.; Sculley, D. Rapid Prediction of Electron–Ionization Mass Spectrometry Using Neural Networks. ACS Cent. Sci. 2019, 5, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Sturm, M.; Kohlbacher, O. TOPPView: An open-source viewer for mass spectrometry data. J. Proteome Res. 2009, 8, 3760–3763. [Google Scholar] [CrossRef]
- Smith, R.; Mathis, A.D.; Ventura, D.; Prince, J.T. Proteomics, lipidomics, metabolomics: A mass spectrometry tutorial from a computer scientist’s point of view. BMC Bioinform. 2014, 15, S9. [Google Scholar] [CrossRef]
- Smith, R.; Ventura, D.; Prince, J.T. LC-MS alignment in theory and practice: A comprehensive algorithmic review. Briefings Bioinform. 2013, 16, 104–117. [Google Scholar] [CrossRef]
Sample Availability: The original 19 multi-beer data from [14] are available from GNPS MassIVE MSV000081119 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=3d3801965ccb4b269a3c8547115c544b), while the original multi-urine data from [14] are available from GNPS MassIVE MSV000081118 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=17813156319b488f9b3351c440ac8d92). The BeerQCB data alongside converted mzML files that can be readily used by ViMMS for the multi-beer and multi-urine data can be found at http://dx.doi.org/10.5525/gla.researchdata.870. |









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wandy, J.; Davies, V.; J. J. van der Hooft, J.; Weidt, S.; Daly, R.; Rogers, S. In Silico Optimization of Mass Spectrometry Fragmentation Strategies in Metabolomics. Metabolites 2019, 9, 219. https://doi.org/10.3390/metabo9100219
Wandy J, Davies V, J. J. van der Hooft J, Weidt S, Daly R, Rogers S. In Silico Optimization of Mass Spectrometry Fragmentation Strategies in Metabolomics. Metabolites. 2019; 9(10):219. https://doi.org/10.3390/metabo9100219
Chicago/Turabian StyleWandy, Joe, Vinny Davies, Justin J. J. van der Hooft, Stefan Weidt, Rónán Daly, and Simon Rogers. 2019. "In Silico Optimization of Mass Spectrometry Fragmentation Strategies in Metabolomics" Metabolites 9, no. 10: 219. https://doi.org/10.3390/metabo9100219
APA StyleWandy, J., Davies, V., J. J. van der Hooft, J., Weidt, S., Daly, R., & Rogers, S. (2019). In Silico Optimization of Mass Spectrometry Fragmentation Strategies in Metabolomics. Metabolites, 9(10), 219. https://doi.org/10.3390/metabo9100219