The Methoxylated, Highly Conjugated C40 Carotenoids, Spirilloxanthin and Anhydrorhodovibrin, Can Be Separated Using High Performance Liquid Chromatography with Safe and Environmentally Friendly Solvents


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
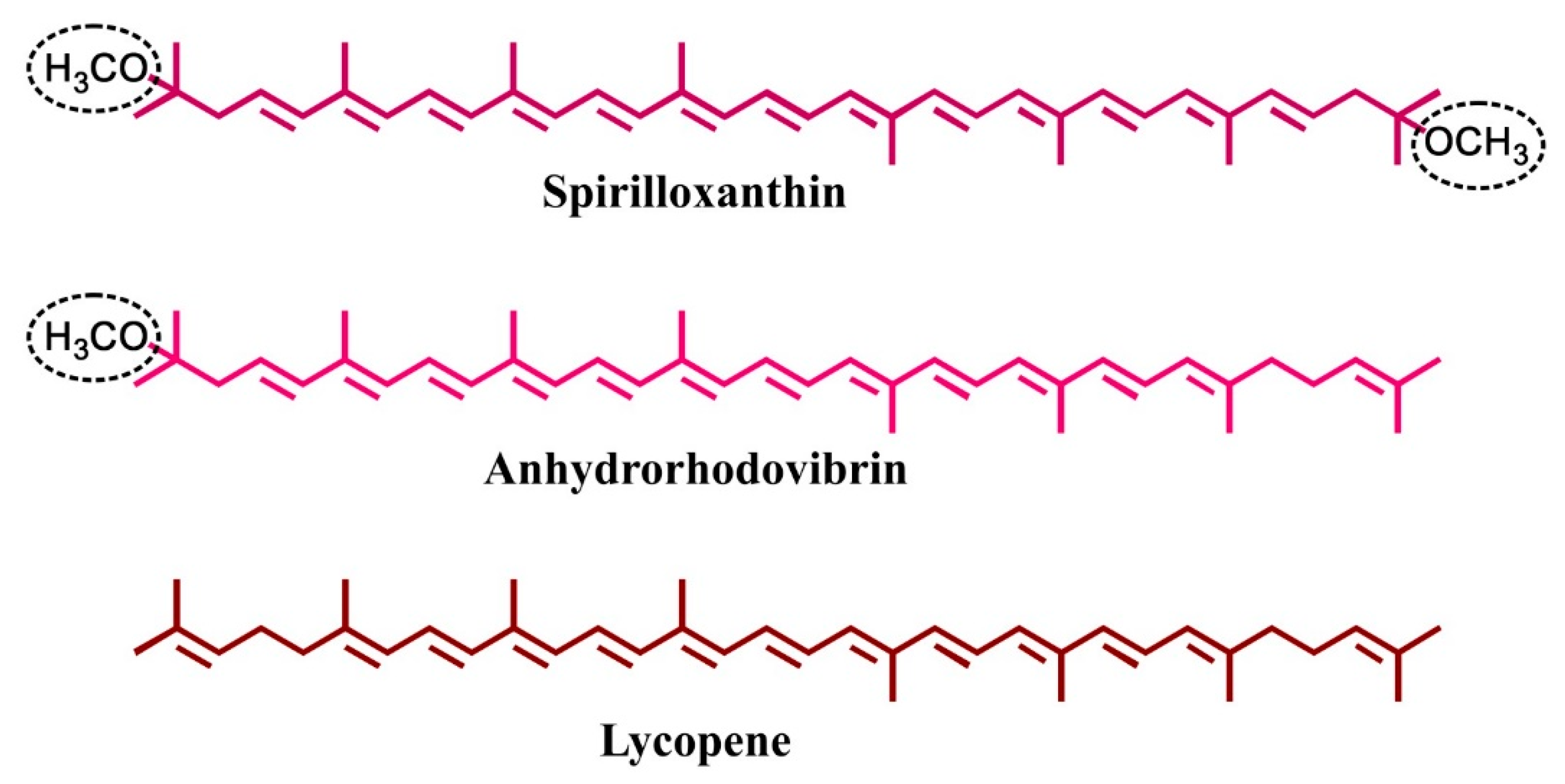
1. Introduction
2. Results
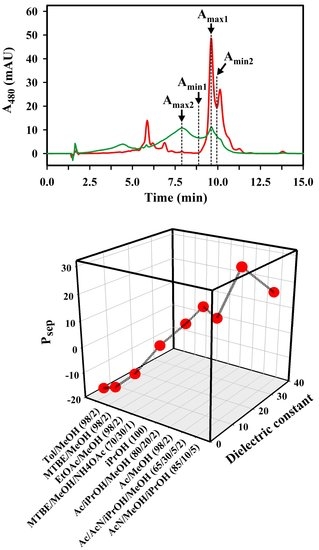
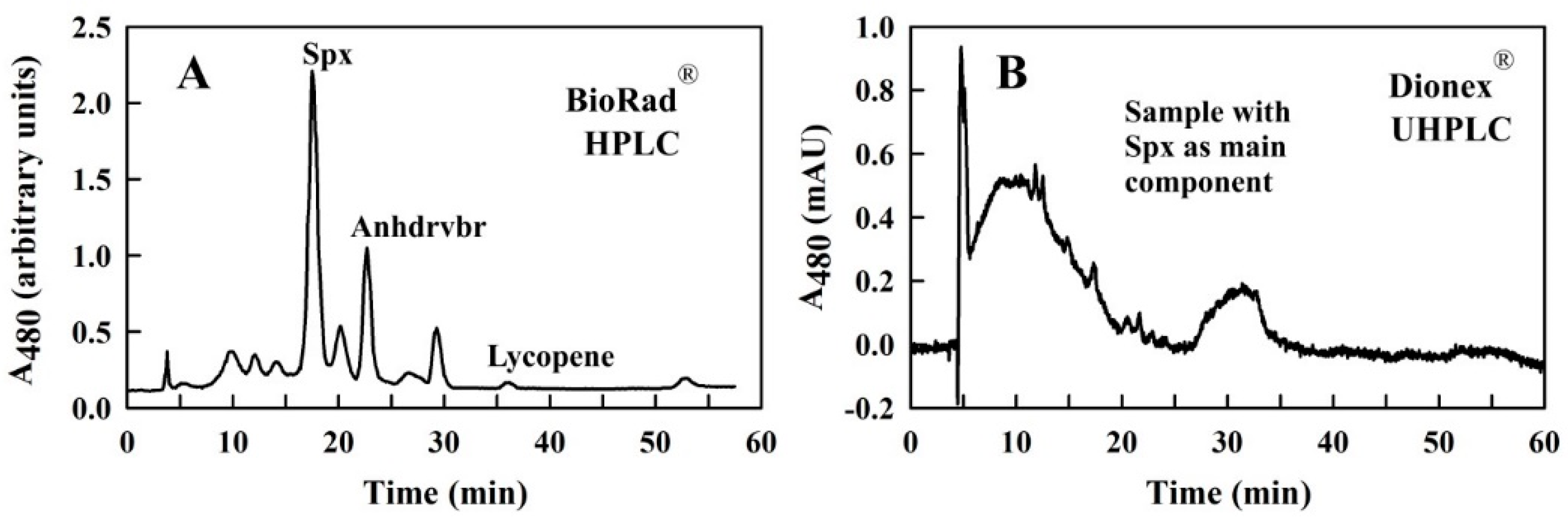
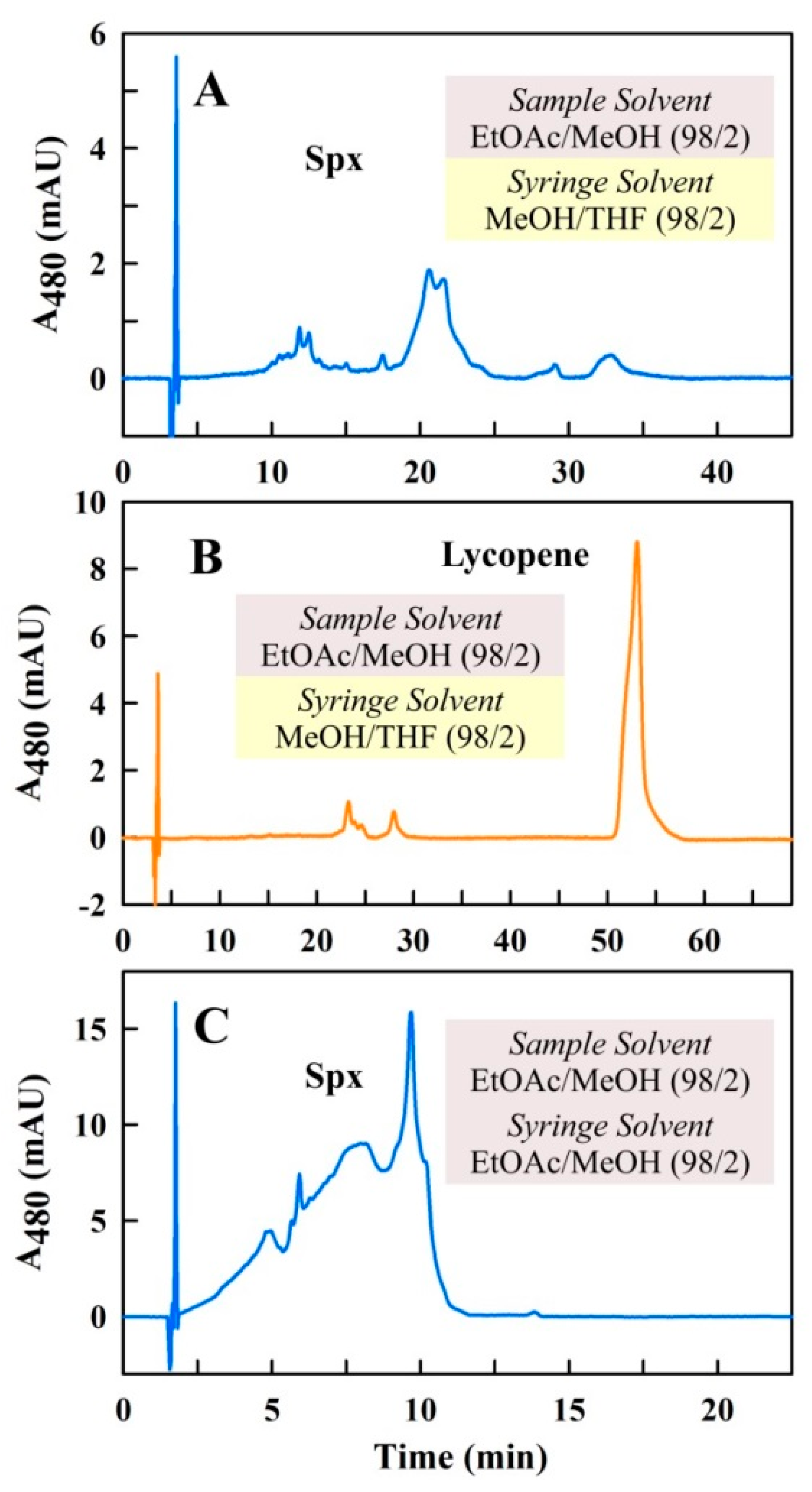
2.1. Initial Considerations for the Separation of Spx Using a Modern UHPLC-System
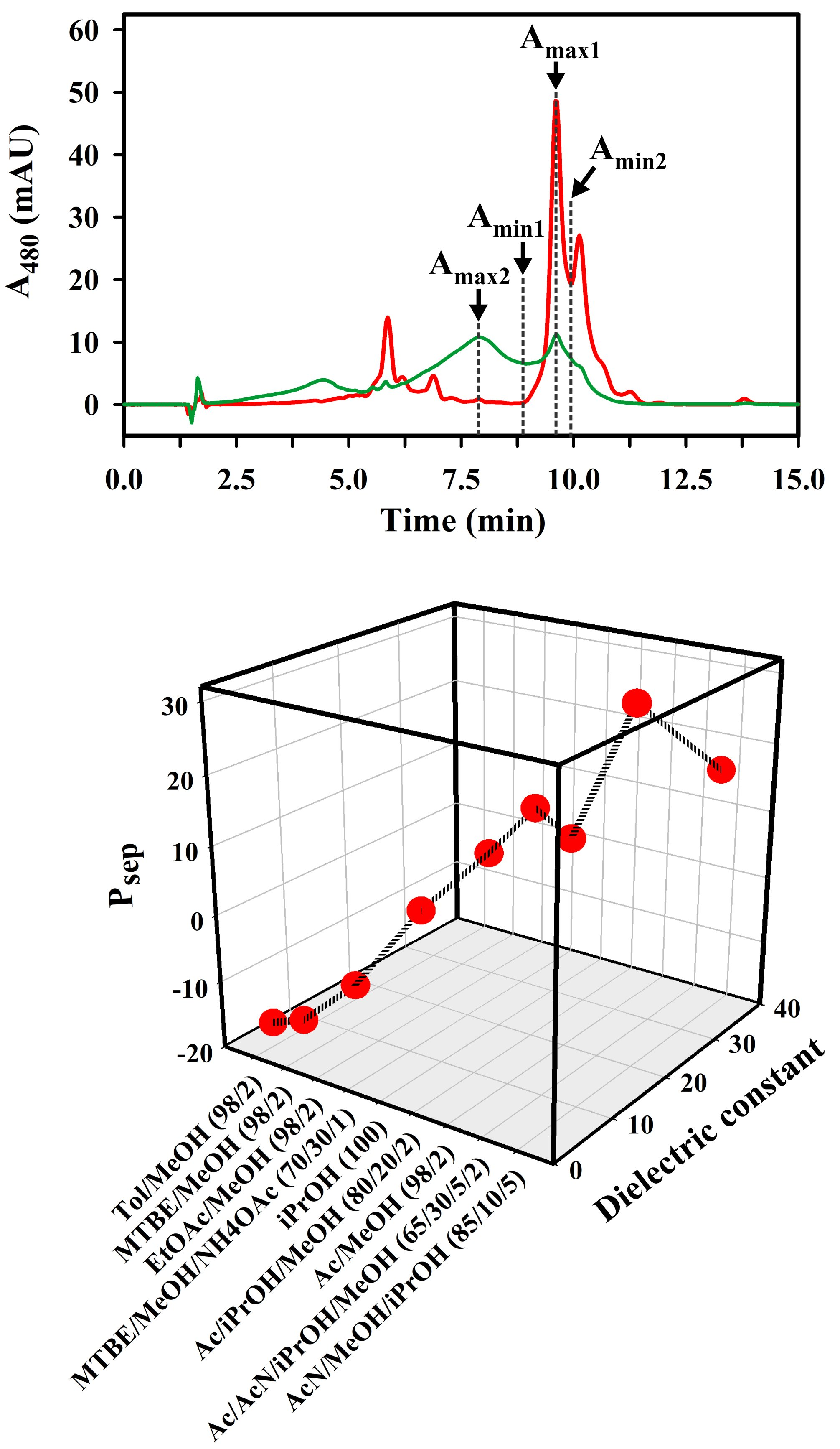
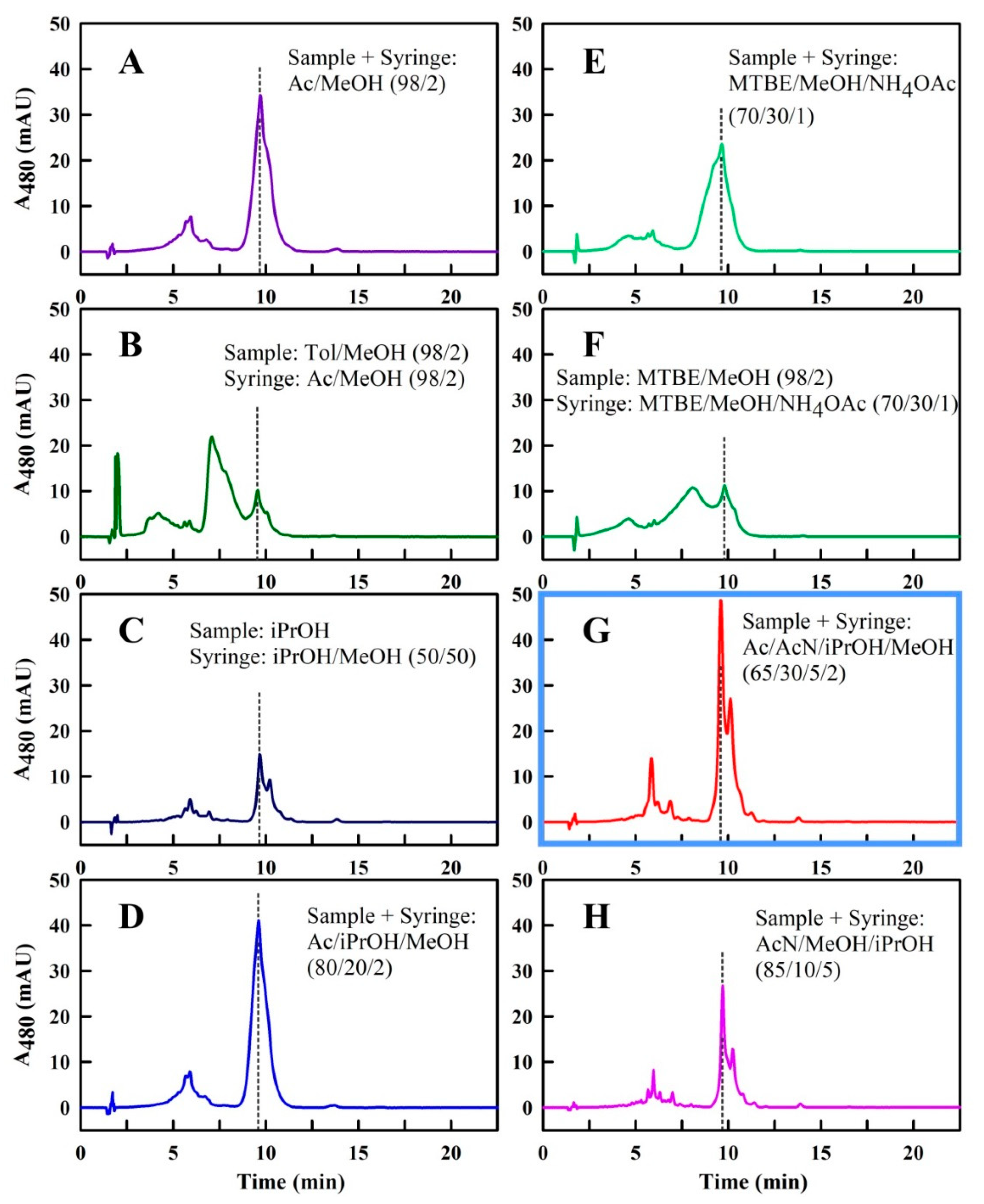
2.2. Optimization of the Solvent System for Use as Sample and Syringe Solvent for Spx UHPLC Runs
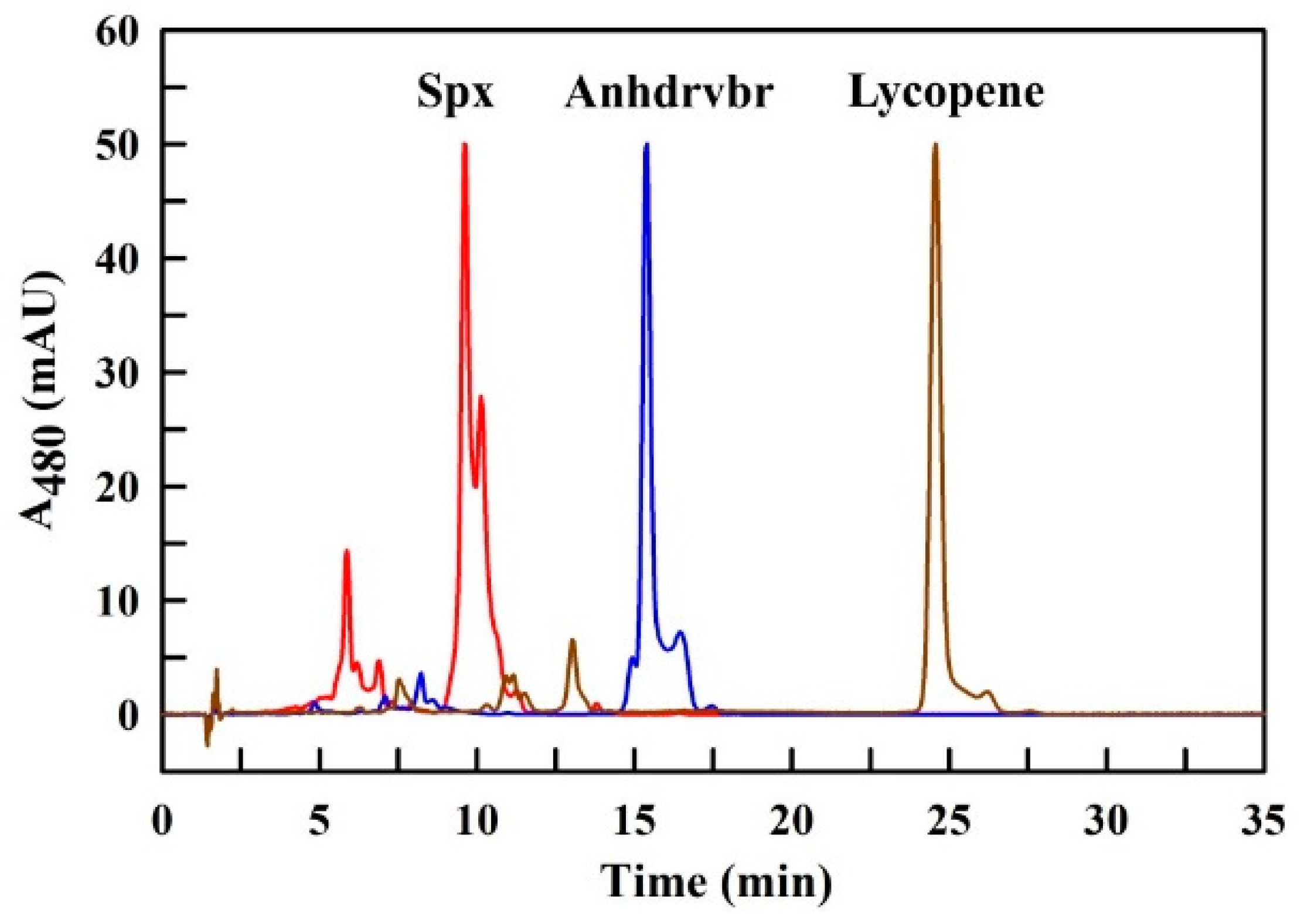
3. Discussion
3.1. Quantitative Analysis of the Solvent Delivery System
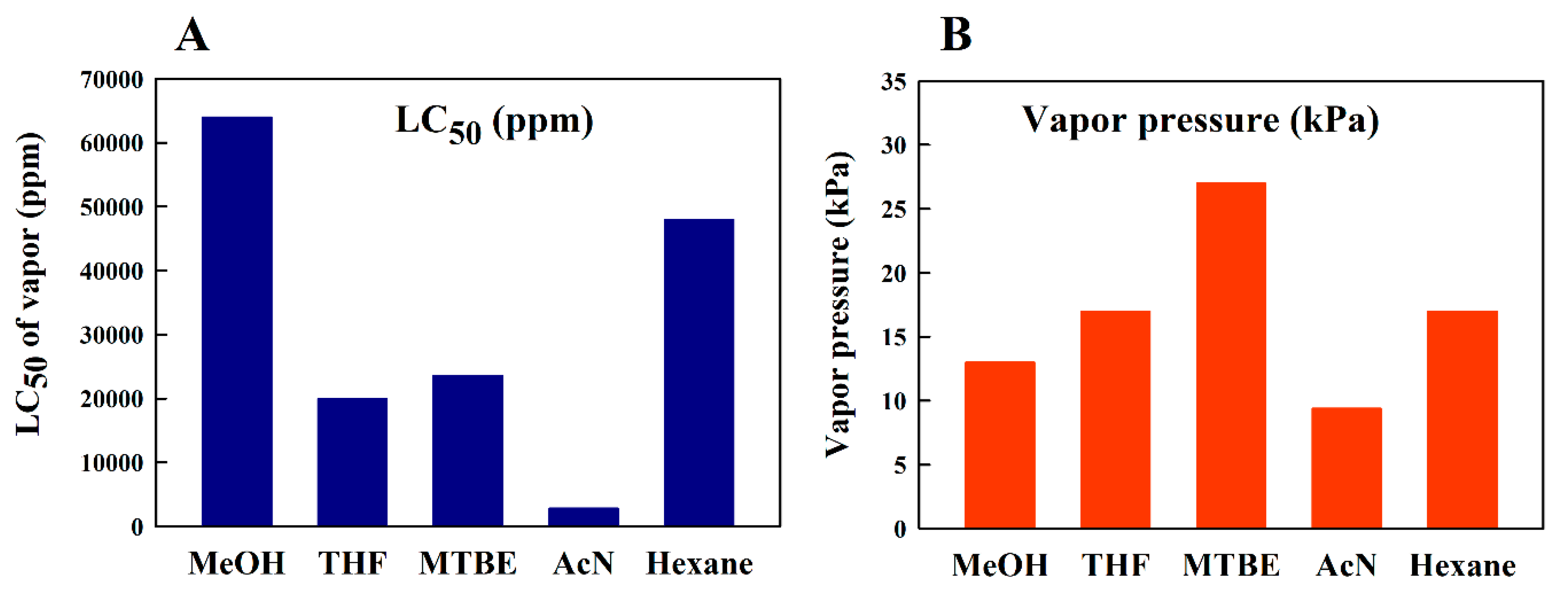
3.2. Safety Considerations
4. Materials and Methods
4.1. Chemicals and Carotenoid Sample Preparation
4.2. Carotenoid Sample Preparation for HPLC Runs
4.3. UHPLC Setup and Settings
4.4. Calculation of Dav
4.5. Comparison of Solvent Toxicities
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, X.-D. Lycopene metabolism and its biological significance. Am. J. Clin. Nutr. 2012, 96, 1214S–1222S. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, R.; Du, J.H.; Liu, T.; Wu, S.S.; Liu, X.H. Lutein, zeaxanthin and meso-zeaxanthin supplementation associated with macular pigment optical density. Nutrients 2016, 8, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Dannert, C.; Umeno, D.; Arnold, F.H. Molecular breeding of carotenoid biosynthetic pathways. Nat. Biotechnol. 2000, 18, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Autenrieth, C.; Ghosh, R. Random mutagenesis and overexpression of rhodopin-3,4-desaturase allows the production of highly conjugated carotenoids in Rhodospirillum rubrum. Arch. Biochem. Biophys. 2015, 572, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.; Takaichi, S.; Steiger, S.; Wang, Z.-Y.; Sandmann, G. Novel hydroxycarotenoids with improved antioxidative properties produced by gene combination in Escherichia coli. Nat. Biotechnol. 2000, 18, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Takaichi, S.; Shimada, K.; Ishidsu, J. Monocyclic cross-conjugated carotenal from an aerobic photosynthetic bacterium, Erythrobacter longus. Phytochemistry 1988, 27, 3605–3609. [Google Scholar] [CrossRef]
- Takaichi, S.; Shimada, K. Characterization of carotenoids in photosynthetic bacteria. Meth. Enzymol. 1992, 213, 374–385. [Google Scholar]
- Takaichi, S.; Sandmann, G.; Schnurr, G.; Satomi, Y.; Suzuki, A.; Misawa, N. The carotenoid 7,8-dihydro-end group can be cyclized by the lycopene cyclases from the bacterium Erwinia uredovora and the higher plant Capsicum annuum. Eur. J. Biochem. 1996, 241, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Takaichi, S.; Maoka, T.; Sasikala, Ch.; Ramana, C.V.; Shimada, K. Genus specific unusual carotenoids in purple bacteria, Phaeospirillum and Roseospira: Structures and biosyntheses. Curr. Microbiol. 2011, 63, 75–80. [Google Scholar] [CrossRef]
- Schwerzmann, R.U.; Bachofen, R. Carotenoid profiles in pigment-protein complexes of Rhodospirillum rubrum. Plant Cell Physiol. 1989, 30, 497–504. [Google Scholar] [CrossRef]
- Naruse, M.; Hashimoto, H.; Kuki, M.; Koyama, Y.J. Triplet excitation of precursors of spirilloxanthin bound to the chromatophores of Rhodospirillum rubrum as detected by transient Raman spectroscopy. J. Mol. Struct. 1991, 242, 15–26. [Google Scholar] [CrossRef]
- Kuki, M.; Naruse, M.; Kakuno, T.; Koyama, Y. Resonance Raman evidence for 15-cis to all-trans photoisomerization of spirilloxanthin bound to a reduced form of the reaction center of Rhodospirillum rubrum S1. Photochem. Photobiol. 1995, 62, 502–508. [Google Scholar] [CrossRef]
- Qian, P.; Saiki, K.; Mizoguchi, T.; Hara, K.; Sashima, T.; Fuji, R.; Koyama, Y. Time-dependent changes in the carotenoid composition and preferential binding of spirilloxanthin to the reaction center and anhydrorhodovibrin to the LH1 antenna complex in Rhodobium marinum. Photochem. Photobiol. 2001, 74, 444–452. [Google Scholar] [CrossRef]
- Connor, A.E.; Britton, G. HPLC analysis of the pigments of purple photosynthetic bacteria. In Current Research in Photosynthesis; Baltscheffsky, M., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1990; Volume 2, pp. 53–56. ISBN 978-94-010-6716-4. [Google Scholar]
- Komori, M.; Ghosh, R.; Takaichi, S.; Hu, Y.; Mizoguchi, T.; Koyama, Y.; Kuki, M. A null lesion in the rhodopin 3,4-desaturase of Rhodosprillum rubrum unmasks a cryptic branch of the carotenoid biosynthetic pathway. Biochemistry 1998, 37, 8987–8994. [Google Scholar] [CrossRef] [PubMed]
- Steiger, S.; Astier, C.; Sandmann, G. Substrate specificity of the expressed carotenoid 3,4-desaturase from Rubrivivax gelatinosus reveals the detailed reaction sequence to spheroidene and spirilloxanthin. Biochem. J. 2000, 349, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Pinta, V.; Ouchane, S.; Picaud, M.; Takaichi, S.; Astier, C.; Reiss-Husson, F. Characterization of unusual hydroxy- and ketocarotenoids in Rubrivivax gelatinosus: Involvement of enzyme CrtF or CrtA. Arch. Microbiol. 2003, 179, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.Q.; Liu, C.; Ernst, H.; Krinsky, N.I.; Russell, R.M.; Wang, X.-D. The biochemical characterization of ferret carotene-9′, 10′-monooxygenase catalyzing cleavage of carotenoids in vitro and in vivo. J. Biol. Chem. 2006, 281, 19327–19338. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Quadro, L. Reverse-phase high-performance liquid chromatography (HPLC) analysis of retinol and retinyl esters in mouse serum and tissues. Meth. Mol. Biol. 2010, 652, 263–275. [Google Scholar]
- Chi, S.C.; Mothersole, D.J.; Dilbeck, P.; Niedzwiedzki, D.M.; Zhang, H.; Qian, P.; Vasilev, C.; Grayson, K.J.; Jackson, P.J.; Martin, E.C.; et al. Assembly of functional photosystem complexes in Rhodobacter sphaeroides incorporating carotenoids from the spirilloxanthin pathway. Biochem. Biophys. Acta 2015, 1847, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedzki, D.M.; Dilbeck, P.L.; Tang, Q.; Mothersole, D.J.; Martin, E.C.; Bocian, D.F.; Holten, D.; Hunter, C.N. Functional characteristics of spirilloxanthin and keto-bearing analogues in light-harvesting LH2 complexes from Rhodobacter sphaeroides with a genetically modified carotenoid synthesis pathway. Biochim. Biophys. Acta 2015, 1847, 640–655. [Google Scholar] [CrossRef]
- Lindal, T.-R.; Liaaen-Jensen, S. Bacterial carotenoids 56.* On the spirilloxanthin stereoisomeric set. Acta Chem. Scand. 1997, 51, 1128–1131. [Google Scholar] [CrossRef]
- Wang, G.-S.; Grammel, H.; Abou-Aisha, K.; Ghosh, R. High-level production of the industrial product lycopene by the photosynthetic bacterium Rhodospirillum rubrum. Appl. Env. Microbiol. 2012, 78, 7205–7215. [Google Scholar] [CrossRef] [PubMed]
- van Breemen, R.B.; Huang, C.-R.; Tan, Y.; Sander, L.C.; Schilling, A.B. Liquid chromatography/mass spectrometry of carotenoids using atmospheric pressure chemical ionization. J. Mass Spectr. 1996, 31, 975–981. [Google Scholar] [CrossRef]
- Kaiser, P.; Surmann, P.; Vallentin, G.; Fuhrmann, H. A small-scale method for quantitation of carotenoids in bacteria and yeasts. J. Microbiol. Meth. 2007, 70, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Albermann, C. High versus low level expression of the lycopene biosynthesis genes from Pantoea ananatis in Escherichia coli. Biotechnol. Lett. 2011, 33, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Fleshman, M.K.; Riedl, K.M.; Novotny, J.A.; Schwartz, S.J.; Harrison, E.H. An LC/MS method for d8-carotene and d4-retinyl esters: β-carotene absorption and its conversion to vitamin A in humans. J. Lipid Res. 2012, 53, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.; Geyer, R.; Surmann, P.; Fuhrmann, H. LC-MS method for screening unknown microbial carotenoids and isoprenoid quinones. J. Microbiol. Meth. 2012, 88, 28–34. [Google Scholar] [CrossRef]
- Takehara, M.; Nishimura, M.; Kuwa, T.; Inoue, Y.; Kitamura, C.; Kumagai, T.; Honda, M. Characterization and thermal isomerization of (all-E)-lycopene. J. Agric. Food Chem. 2014, 62, 264–269. [Google Scholar] [CrossRef]
- Melendez-Martinez, A.J.; Paulino, M.; Stinco, C.M.; Mapelli-Brahm, P.; Wang, X.-D. Study of the time-course of cis/trans (Z/E) isomerization of lycopene, phytoene, and phytofluene from tomato. J. Agric. Food Chem. 2014, 62, 12399–12406. [Google Scholar] [CrossRef]
- Mihaylova, D.; Vrancheva, R.; Petkova, N.; Ognyanov, M.; Desseva, I.; Ivanov, I.; Popova, M.; Popova, A. Carotenoids, tocopherols, organic acids, charbohydrate and mineral content in different medicinal plant extracts. Z. Naturforsch. C 2018, 73, 439–448. [Google Scholar] [CrossRef]
- Bóna-Lovász, J.; Bóna, A.; Ederer, M.; Sawodny, O.; Ghosh, R. A rapid method for the extraction and analysis of carotenoids and other hydrophobic substances suitable for systems biology studies with photosynthetic bacteria. Metabolites 2013, 3, 912–930. [Google Scholar] [CrossRef] [PubMed]
- DDBST Dortmund Data Bank. Pure Component Properties. Available online: http://www.ddbst.com/en/EED/PCP/PCPindex.php (accessed on 23 November 2018).
- TOXNET Toxicology Data Network. NIH U.S. National Library of Medicine. Available online: https://toxnet.nlm.nih.gov (accessed on 23 November 2018).
- PubChem, National Center for Biotechnology Information. PubChem Compound Database. Available online: https://pubchem.ncbi.nlm.nih.gov/search/ (accessed on 27 November 2018).
- Cohen-Bazire, G.; Sistrom, W.R.; Stanier, R.Y. Kinetic studies of pigment synthesis by non-sulfur purple bacteria. J. Cell Comp. Physiol. 1956, 49, 25–68. [Google Scholar] [CrossRef]
- Sistrom, W.R. A requirement for sodium in the growth of Rhodopseudomonas sphaeroides. J. Gen. Microbiol. 1960, 22, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Hauser, H.; Bachofen, R. Reversible dissociation of B873 light-harvesting complex from Rhodospirillum rubrum G9+. Biochemistry 1988, 27, 1004–1014. [Google Scholar] [CrossRef]
- Weast, R.C.; Astle, M.J.; Beyer, W.H. (Eds.) CRC Handbook of Chemistry and Physics, 64th ed.; CRC Press: Boca Raton, FL, USA, 1984; pp. E50–E53. ISBN 978-0849304637. [Google Scholar]
- University of Washington. Dielectric Chart. Available online: https://depts.washington.edu/eooptic/linkfiles/dielectric_chart%5B1%5D.pdf (accessed on 3 December 2018).
- American Chemical Society, Division of Organic Chemistry. Common Solvents Used in Organic Chemistry: Table of Properties. Available online: https://www.organicdivision.org/wp-content/uploads/2016/12/organic_solvents.html (accessed on 3 December 2018).
- Monument Chemical. Technical Product Information. Methyl t-Butyl Ether (MTBE). Available online: https://monumentchemical.com/uploads/files/TDS/MTBE%20-%20TDS.pdf (accessed on 3 December 2018).








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Autenrieth, C.; Ghosh, R. The Methoxylated, Highly Conjugated C40 Carotenoids, Spirilloxanthin and Anhydrorhodovibrin, Can Be Separated Using High Performance Liquid Chromatography with Safe and Environmentally Friendly Solvents. Metabolites 2019, 9, 20. https://doi.org/10.3390/metabo9020020
Autenrieth C, Ghosh R. The Methoxylated, Highly Conjugated C40 Carotenoids, Spirilloxanthin and Anhydrorhodovibrin, Can Be Separated Using High Performance Liquid Chromatography with Safe and Environmentally Friendly Solvents. Metabolites. 2019; 9(2):20. https://doi.org/10.3390/metabo9020020
Chicago/Turabian StyleAutenrieth, Caroline, and Robin Ghosh. 2019. "The Methoxylated, Highly Conjugated C40 Carotenoids, Spirilloxanthin and Anhydrorhodovibrin, Can Be Separated Using High Performance Liquid Chromatography with Safe and Environmentally Friendly Solvents" Metabolites 9, no. 2: 20. https://doi.org/10.3390/metabo9020020
APA StyleAutenrieth, C., & Ghosh, R. (2019). The Methoxylated, Highly Conjugated C40 Carotenoids, Spirilloxanthin and Anhydrorhodovibrin, Can Be Separated Using High Performance Liquid Chromatography with Safe and Environmentally Friendly Solvents. Metabolites, 9(2), 20. https://doi.org/10.3390/metabo9020020