Abstract
Brains and blood of Alzheimer’s disease (AD) patients have shown elevated mercury concentrations, but potential involvement of mercury exposure in AD pathogenesis has not been studied at the molecular level. The pathological hallmark of AD brains is deposition of amyloid plaques, consisting mainly of amyloid-β (Aβ) peptides aggregated into amyloid fibrils. Aβ peptide fibrillization is known to be modulated by metal ions such as Cu(II) and Zn(II). Here, we study in vitro the interactions between Aβ peptides and Hg(II) ions by multiple biophysical techniques. Fluorescence spectroscopy and atomic force microscopy (AFM) show that Hg(II) ions have a concentration-dependent inhibiting effect on Aβ fibrillization: at a 1:1 Aβ·Hg(II) ratio only non-fibrillar Aβ aggregates are formed. NMR spectroscopy shows that Hg(II) ions interact with the N-terminal region of Aβ(1–40) with a micromolar affinity, likely via a binding mode similar to that for Cu(II) and Zn(II) ions, i.e., mainly via the histidine residues His6, His13, and His14. Thus, together with Cu(II), Fe(II), Mn(II), Pb(IV), and Zn(II) ions, Hg(II) belongs to a family of metal ions that display residue-specific binding interactions with Aβ peptides and modulate their aggregation processes.
1. Introduction
Alzheimer’s disease (AD) is a progressive, irreversible, and currently incurable neurodegenerative disorder, and the leading cause of dementia worldwide [1,2]. Identifying molecular targets [3] and/or modifiable risk factors related to disease onset and/or early progression is imperative [4,5]. AD risk factors so far identified include advanced age [6,7], genetic mutations associated especially with the AβPP and ApoE genes [8,9,10,11,12,13,14], life style [15,16,17], air pollution including tobacco smoking [18,19,20,21,22], cardio-vascular diseases [23], diabetes [24], traumatic brain injury [25,26], and metal exposure [27,28,29].
The major characteristic AD lesion in the brain is the presence of extracellular amyloid plaques, consisting mainly of amyloid-β (Aβ) peptides aggregated into insoluble fibrils [30] that display the cross-β secondary structure common for many amyloid fibrils [31,32]. The Aβ peptides, produced by two-step enzymatic cleavage of the membrane-bound amyloid-β precursor protein (AβPP), comprise 37–43 residues and are intrinsically disordered in aqueous solution. Aβ peptides have limited solubility in water, as the central and C-terminal Aβ segments are hydrophobic and may fold into a hairpin conformation upon aggregation [33]. The charged N-terminal segment is, however, hydrophilic and interacts readily with cationic molecules and metal ions [34,35,36,37,38]. The Aβ fibrils and plaques are the end-product of an Aβ aggregation process [37,39,40] involving extra- and/or intracellular formation of intermediate, soluble, and likely neurotoxic Aβ oligomers [41,42,43,44] that may transfer from neuron to neuron via exosomes [45,46]. Aβ42 oligomers appear to be the most cell-toxic [43], and oligomer formation appears to be influenced by both hydrophobic and electrostatic effects originating from interactions with, e.g., cellular membranes, metal ions, small molecules, and other proteins [24,37,47,48,49,50,51,52,53,54,55,56].
A second pathological lesion in AD brains is the presence of intracellular neurofibrillary tangles composed of aggregated hyperphosphorylated tau proteins [57,58]. Currently, the most plausible connection between AD and aggregates of Aβ and tau has been formulated in the so-called amyloid cascade hypothesis, but the underlying causative links are not fully understood [39,53,59]. AD brains, furthermore, typically display signs of neuroinflammation [60], increased levels of free oxygen radicals [61,62], and altered concentrations of different metal ions indicative of metal dyshomeostasis [63,64].
The proposed involvement of metal ions in AD pathology [65,66,67] is supported by a number of observations. Accumulation of metal ions such as those of Ca, Cu, Fe, and Zn has been found in AD plaques [68,69,70] and in phosphorylated tau tangles [71]. Cu(II), Fe(II), Mn(II), Pb(IV), and Zn(II) ions are known to bind to specific residues in the Aβ peptides and modulate their aggregation [22,38,65,72,73]. The metal ions are typically coordinated to one or more of the three histidines—His6, His13, and His14—and possibly also to other N-terminal residues such as the negatively charged Asp1 and Glu11 [73]. The metal–peptide interactions are transient, with binding affinities in the nanomolar or micromolar range [74,75,76], allowing formation of dynamic equilibria between different binding modes. The Aβ precursor protein AβPP also binds copper and zinc ions [77], and part of its physiological role might be connected with the regulation of Cu(II) and Zn(II) concentrations in the neuronal synapses, where these ions are released into the synaptic clefts [66] and where Aβ aggregation may be initiated [78]. Elevated Aβ concentrations have been observed in animals and cells exposed to metals such as Ag, As, Cd, Cu, Mn, Pb, and Hg [79,80,81,82,83,84,85,86,87,88,89].
A possible role of Hg in AD pathology is intriguing but also controversial [90]. Increased Hg levels were reported in early studies of brains [91] and blood [92,93] of AD patients [94]. Some later studies have, however, failed to confirm these observations [90,94,95,96]. While Hg is a well-known neurotoxicant, its toxic mechanisms are not fully understood at the molecular level [88,94,97,98,99], and metallic, inorganic, and organic Hg compounds are known to have different toxicity mechanisms and exposure pathways [94,97,98,100].
Few studies have so far investigated the possible influence of Hg(II) and other non-essential metal ions on the molecular events that occur in neurodegenerative diseases, such as aggregation of the Aβ peptide in AD [20,22,29,87,101]. Although Hg(II) ions are known to affect protein aggregation and misfolding in general [102,103,104], we are only aware of one study investigating possible molecular interactions between Hg(II) ions and Aβ peptides [105].
Here, we use solution nuclear magnetic resonance (NMR) and fluorescence spectroscopy, together with solid-state atomic force microscopy (AFM), to study the binding interactions between inorganic Hg(II) ions and Aβ40 and Aβ42 monomers, and the effects of Hg(II) ions on the Aβ amyloid aggregation process and fibril formation from a biophysical perspective.
2. Materials and Methods
2.1. Sample Preparation
Recombinant Aβ42 peptides, with the primary sequence DAEFR5HDSGY10EVHHQ15KLVFF20AEDVG25SNKGA30IIGLM35VGGVV40IA, were purchased lyophilized from rPeptide (Bogart, GA, USA), together with shorter recombinant Aβ16 peptides comprising the first 16 residues in the above sequence. The Aβ16 and Aβ42 peptides were dissolved in 100% hexafluoroisopropanol (HFIP) at concentrations of 100 µM to disassemble preformed peptide aggregates, and then aliquoted into smaller samples of 10–100 µg. The HFIP was evaporated in vacuum and the resulting Aβ films were stored at −80 °C. The peptide concentrations were determined by weight. For the experiments with the Aβ16 and Aβ42 peptides, Hg(II) solutions derived from a Hg(NO3)2 salt were used.
Recombinant unlabeled or uniformly 15N- or 13C–15N-labeled Aβ40 peptides, with a sequence identical to that of Aβ42 (above) but lacking the last two residues, were bought lyophilized from AlexoTech AB (Umeå, Sweden). The peptides were stored at −80 °C until used. The peptide concentration was determined by weight, and the peptide samples were dissolved to monomeric form immediately before each measurement. In brief, the peptides were dissolved in 10 mM sodium hydroxide, pH 12, at a 1 mg/mL concentration, and sonicated in an ice-bath for at least 3 min to avoid having pre-formed aggregates in the peptide solutions. The peptide solution was further diluted in 20 mM sodium phosphate buffer, and all sample preparation steps were performed on ice. For the experiments with the Aβ40 samples, Hg(II) solutions derived from a HgCl2 salt were used.
2.2. Fluorescence Spectroscopy
2.2.1. ThT Fluorescence as a Probe for Aβ Aggregation Kinetics
To monitor the Aβ fibrillization kinetics, 15 µM monomeric Aβ40 peptides were incubated in 20 mM sodium phosphate buffer pH 7.35 at +37 °C in the presence of varying concentrations of HgCl2 (0, 0.2, 0.8, 1.5, 3.0, and 15 µM) and 40 μM Thioflavin T (ThT). ThT is a benzothiazole dye that displays increased fluorescence intensity when bound to amyloid material [106]. The ThT dye was excited at 440 nm, and the fluorescence emission at 480 nm was measured every 3 min in a 96-well plate in a FLUOstar Omega microplate reader (BMG LABTECH, Germany). The Aβ40 samples were incubated under quiescent conditions for two days at +37 °C, and four replicates a la 100 µL per condition were measured. The assay was repeated at least three times. The Aβ aggregation kinetic parameters τ½ and rmax were calculated by sigmoidal curve fitting according to Equation (1) [107] using four replicates per condition:
where F0 is the fluorescence intensity baseline, A is the fluorescence intensity amplitude, rmax is the maximum growth rate, and τ½ is the time when half the Aβ monomer population has been depleted. The aggregation lag time was defined by .
The fibrillization kinetics of Aβ42 was studied using HFIP-treated Aβ42 dissolved at a concentration of 20 µM in 0.02% NH3, and then diluted to a final concentration of 5 µM in a buffered solution containing 20 mM HEPES pH 7.3, 100 mM NaCl, and 5 µM ThT. Aβ42 samples of 500 µL were prepared with different Hg(NO3)2 concentrations (i.e., 0, 5, 7.5, 10, and 15 µM) in a quartz cuvette with 0.5 cm path length, and incubated at +45 °C with high-speed magnetic stirring. The ThT fluorescence emission at 480 nm was measured over time on an LS-55 fluorescence spectrophotometer (PerkinElmer Inc., Waltham, MA, USA) using an excitation wavelength of 440 nm.
2.2.2. Tyrosine Fluorescence Quenching Reporting on Hg(II)·Aβ Binding Affinity
The binding affinity between Aβ16 peptides and Hg(II) ions was evaluated from Cu(II)·Hg(II) binding competition experiments [108]. The Cu(II)·Aβ16 affinity was measured via the quenching effect of Cu(II) ions on the intrinsic fluorescence of Tyr10, the only fluorophore in native Aβ peptides. HFIP-treated Aβ16 was dissolved in 10 mM NaOH at a concentration of 80 µM and divided into 60 µL aliquots that were stored at −20 °C, and then used within the same day to prepare samples containing 8 µM Aβ16 in 100 mM NaCl and 20 mM HEPES pH 7.4, or in 20 mM MES pH 5.0. Titrations with increasing concentrations of CuCl2 were conducted at +25 °C using samples with or without Hg(NO3)2 added in different concentrations, i.e., 50 and 100 µM Hg(II) at both pH 5.0 and pH 7.4. The fluorescence emission intensity at 305 nm (excitation wavelength 270 nm) was recorded using an LS-55 fluorescence spectrophotometer (Perkin-Elmer Inc., Waltham, MA, USA) equipped with a magnetic stirrer. The titrations were carried out by consecutive additions of 1 µL aliquots of 0.3–12 mM stock solutions of CuCl2 to 600 µL Aβ16 peptide solutions in quartz cuvettes with 5 mm path length. After each addition of CuCl2, the solution was stirred for 30 s before recording fluorescence emission spectra.
From the Cu(II) titration curves, recorded in the absence or presence of Hg(II) ions, the apparent dissociation constant (KDapp) of the Cu(II)·Aβ16 complex was determined as the concentration of Cu(II) ions that reduced the Tyr10 fluorescence by 50% (as Hg(II) ions alone have no have no significant effect on the Tyr10 fluorescence intensity: data not shown). The Cu(II)·Aβ16 KDapp values were then plotted versus the concentration of Hg(II) ions present in the samples, and the linear dependence was extrapolated to the point where the KDapp value at zero Hg(II) had been increased by a factor of 2. Assuming competitive binding between Hg(II) and Cu(II) ions to the same Aβ16 binding site, the Hg(II) concentration at this point is approximately equal to the apparent dissociation constant (KD) for the Hg(II)·Aβ16 complex.
2.3. AFM Imaging
Solid-state AFM images were recorded using a ScanAsyst unit (Bruker Corp., Billerica, MA, USA) operating in peak-force mode in air with a sample rate of 1.95 Hz and a resolution of 512 × 512 pixels. At the end of the fluorescence spectroscopy ThT kinetic experiments with Aβ40 samples (above), the aggregated Aβ end products were diluted with distilled water (5 μL sample in 100 μL distilled water) and applied on freshly cleaved mica substrates. After 20 min incubation, the mica substrates were washed three times with distilled water and left to air-dry. This protocol was optimized to obtain images of Aβ fibrils, and might not be optimal for other Aβ aggregates or Hg(II)·Aβ complexes.
2.4. NMR Spectroscopy
Bruker Avance 500 and 700 MHz spectrometers equipped with cryogenic probes were used to record two-dimensional (2D) 1H–15N-HSQC (heteronuclear single quantum coherence) and 2D 1H–13C-HSQC spectra at +5 °C or +25 °C of 84 μM monomeric Aβ(1–40) peptides (either 15N-labeled or 13C-15N-labeled) in 20 mM sodium phosphate buffer at pH 7.35 (90/10 H2O/D2O), either with or without 50 mM sodium dodecyl sulfate (SDS) detergent, and before and after titration with HgCl2. Because NMR spectroscopy is a non-sensitive high-resolution method that requires high protein concentration and pure samples, the chosen Aβ40 concentration was 84 μM. The Aβ40 NMR sample was stable during the experimental time required for the HgCl2 titration series. Excess concentration of SDS (around 50 mM) well above the critical micelle concentration (CMC; around 8 mM) was used as a membrane-mimicking environment. SDS micelles are simple in vitro membrane models with a small size range suitable for NMR spectroscopy [109]. The experiments performed in buffer were recorded at +5 °C due to optimal signal intensity and to limit the peptide aggregation during the experimental time. A temperature of +25 °C was used for the experiments with SDS to avoid precipitation of the SDS detergent. The NMR data were processed with the Topspin version 3.2 software and chemical shifts were referenced to the 1H signal of trimethylsilylpropanoic acid (TSP). The Aβ40 HSQC crosspeak assignment in buffer [110,111,112] and in SDS micelles [113] is known from previous work. Chemical shift differences between the spectra of Aβ40 with and without Hg(II) ions were calculated as the standard weighted average, i.e., Δδ = (((ΔδN/5)2 + (ΔδH)2)/2)1/2 [114,115].
The intensity decrease of the 2D 1H–15N-HSQC crosspeaks is likely caused by two effects, both related to the presence of Hg(II) ions. The first effect is Hg(II)-induced non-amyloid aggregation of Aβ peptides (i.e., precipitation or formation of non-fibrillar amorphous aggregates), which reduces the concentration of Aβ monomers and thus the intensities of all Aβ NMR signals. The second effect decreases more selectively the intensities of the (N-terminal) NMR crosspeaks affected by Hg(II) ion binding, and appears to be caused by chemical exchange on an intermediate time scale. Thus, this second effect reports on the Hg(II)·Aβ binding affinity. To compensate for the first effect, the N-terminal crosspeak intensities were normalized by dividing their signal intensities with the corresponding intensities of the C-terminal crosspeaks, which are affected only by the loss of monomer concentrations. The relative N-terminal intensities reflect only the chemical exchange effect, and were fitted to Equation (2) [116] to yield an estimated apparent dissociation constant (KDapp) for the Hg(II)·Aβ40 complex:
where I0 is the initial fluorescence intensity without Hg(II) ions, I∞ is the steady-state (saturated) intensity at the end of the titration series, [Aβ] is the peptide concentration, [Hg] is the concentration of added Hg(II) ions, and KD is the dissociation constant of the Hg(II)·Aβ complex. The model assumes a single binding site. As no corrections for buffer conditions were made, the calculated dissociation constant should be considered to be apparent.
3. Results
3.1. ThT Fluorescence: Kinetic Effects of Hg(II) Ions on the Aβ40 and Aβ42 Aggregation Processes
The fluorescence intensity of the amyloid-marker molecule ThT was measured when 15 µM Aβ40 samples were incubated for two days, together with different concentrations of HgCl2 during quiescent conditions (Figure 1 and Figure S1). Clear concentration-dependent effects of Hg(II) on the Aβ40 aggregation kinetics were observed (Figure 1). The increased noise level in the kinetic curves following the elongation phase might originate from light scattering effects from large and heterogeneous aggregates. Fitting Equation (1) to the ThT fluorescence curves yielded the kinetic parameters τlag, τ½, rmax, and ThT end-point fluorescence intensity (Figure 1; Table 1). For 15 µM Aβ40 alone, the aggregation lag time (τlag) was approximately 7 h under our experimental conditions, and the aggregation halftime (τ½) was 10 h (Table 1). These kinetic parameters were not much affected by addition of 0.8 µM Hg(II), but clearly increased when 15 µM Aβ40 was incubated in the presence of 1.5 µM or 3 µM Hg(II) (Figure 1). In the presence of 3 µM Hg(II), τlag was around 13 h and τ½ was around 20 h (Table 1). When 15 µM Hg(II) was added, no increase in ThT fluorescence intensity was observed during the entire incubation period, indicating that ThT-active amyloid aggregates did not form. This is consistent with the end-point ThT fluorescence intensity levels, which reflect the amount of ThT-active aggregates at the end of the incubation. These end-point ThT levels strictly decrease with increasing Hg(II) concentrations (Table 1). The maximum amyloid growth rate, rmax, displayed more variation: It first increased from 0.6 to around 0.8 when small amounts (0.8 and 1.5 µM) of Hg(II) were added, and then decreased to 0.3 in the presence of 3 µM Hg(II) ions (Figure 1; Table 1).
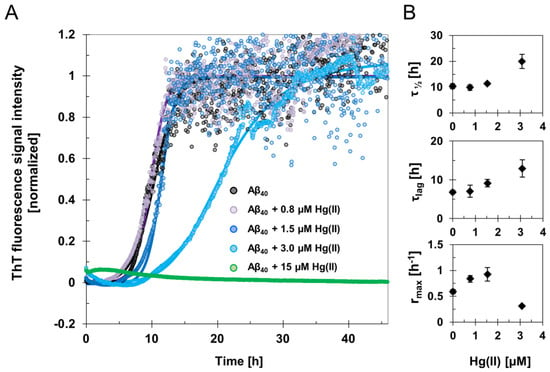
Figure 1.
Amyloid fibril formation monitored by ThT aggregation kinetics assay. (A) ThT fluorescence signal intensity traces of averaged and normalized data from samples with 15 μM Aβ40 peptides in 20 mM sodium phosphate buffer pH 7.4 incubated in the absence and presence of 0.8–15 μM Hg(II) ions at +37 °C under quiescent conditions. The average of four replicates is presented as circles, and the average fit is shown as a solid line for each Hg(II) ion concentration. (B) Phenomenological parameters were extracted from sigmoidal curve fitting of the experimental ThT data in (A) using Equation (1), yielding the aggregation halftime τ½, lag time τlag, and maximum growth rate rmax. The error bars in (B) represent the standard deviation of four replicates.

Table 1.
Kinetic parameters of Aβ40 fibril formation. ThT fluorescence data reflecting Aβ amyloid formation were recorded in the presence of various concentrations of Hg(II) ions. Aggregation halftimes (τ½), lag time (τlag), maximum growth rates (rmax), and ThT end-point fluorescence amplitudes were derived from sigmoidal curve-fitting to Equation (1) and are also presented in Figure 1.
For the Aβ42 peptide incubated under agitation conditions, the ThT kinetics curves again showed a clear dependence on the Hg(II) concentration. Increasing concentrations of Hg(II) ions induce strictly increasing aggregation half times and lag times, and strictly decreasing maximum growth rates and ThT end-point intensities (Figure S2). Although Aβ42 peptides are more prone to aggregate than Aβ40 peptides, similar trends for the effects of Hg(II) ions on Aβ aggregation kinetics were observed for the two peptide versions. Thus, these concentration-dependent effects do not appear to depend on the particular experimental conditions.
3.2. AFM Imaging: Effects of Hg(II) Ions on Aβ40 Aggregate Morphology
AFM images (Figure 2 and Figure S3) were recorded for the aggregation products present at the end of the ThT kinetics experiments. First, 15 µM Aβ40 alone formed typical amyloid fibrils with a height of about 10 nm (Figure 2A and Figure S3A), which is a typical size for Aβ fibrils formed in vitro [42,117]. The aggregates of 15 µM Aβ40 formed in the presence of low concentrations (0.8, 1.5, and 3.0 µM) of HgCl2 displayed shorter fibril-like structures (Figure 2B–D and Figure S3B–D). No amyloid aggregates were observed when the Aβ40 sample was incubated together with equimolar amounts of HgCl2 (Figure 2E and Figure S3E). These results are consistent with the concentration-dependent inhibitory effect of Hg(II) ions on Aβ fibrillization observed with the ThT assays (Figure 1).
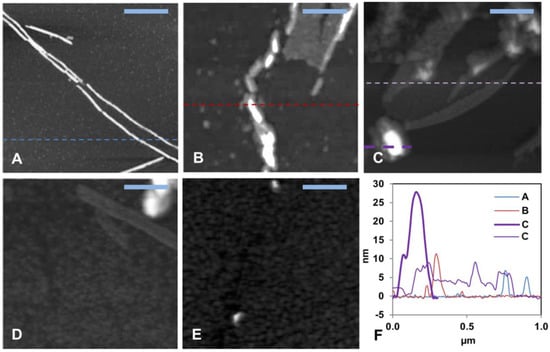
Figure 2.
Solid-state AFM imaging of Aβ40 fibril formation and morphology. (A–D) Topographical images of samples taken from the end of the ThT aggregation experiment (~45 h) in Figure 1. (A) shows 15 μM aggregated Aβ40 peptides alone, (B) Aβ40 + 0.8 μM Hg(II) ions, (C) Aβ40 + 1.5 μM Hg(II) ions, (D) Aβ40 + 3.0 μM Hg(II) ions, (E) Aβ40 + 15 μM Hg(II) ions, and (F) shows cross sectional height information from the images in (A–C). The scale bars represent 0.25 μm.
3.3. NMR Spectroscopy: Molecular Interactions Between Hg(II) Ions and Aβ40 Monomers
High-resolution NMR experiments were conducted to investigate if residue-specific molecular interactions could be observed between Hg(II) ions and monomeric Aβ40 peptides (Figure 3 and Figure 4, Figures S4 and S5). Two-dimensional 1H–15N-HSQC spectra showing the amide crosspeak region for 84 µM monomeric 13C–15N-labeled Aβ40 peptides are presented in Figure 3, recorded either without (Figure 3A) or with 50 mM SDS detergent (Figure 3C), before and after addition of 80 μM Hg(II) ions (Figure 3A) or 30 μM Hg(II) ions (Figure 3C). Addition of Hg(II) ions selectively induces loss of signal intensity mainly for amide crosspeaks corresponding to N-terminal Aβ40 residues (Figure 3A,B), indicating selective Hg(II) binding in this region. These effects are clearly concentration-dependent, as seen in Figures S4 and S5.
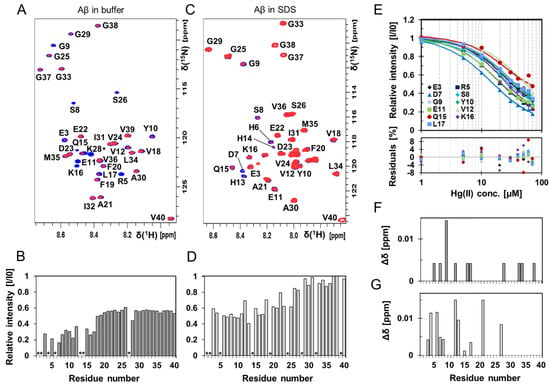
Figure 3.
Two-dimensional (2D) NMR 1H–15N-HSQC experiments showing Aβ40 residue-specific perturbations from Hg(II) ions both in buffer and in the presence of SDS micelles. (A) 700 MHz 1H–15N-HSQC spectra of 84 μM monomeric 13C–15N-labeled Aβ40 peptides alone (blue) and in the presence of 80 μM Hg(II) ions (red) in 20 mM sodium phosphate buffer pH 7.35 at +5 °C. In (B), relative signal intensities determined from the amplitude of the amide crosspeaks in the two spectra in (A) are shown. (C) 500 MHz 1H–15N-HSQC spectra of 84 μM monomeric 15N-labeled Aβ40 peptides (blue) with 30 μM Hg(II) ions (red) in 20 mM sodium phosphate buffer pH 7.35 and 50 mM SDS at +25 °C, and corresponding relative intensities are shown in (D). Residues assigned with a * are not accurately determined or observed because of too fast an exchange with the solvent or due to spectral overlap. (E) Relative intensities from 1H-15N-HSQC spectra (for Aβ40 in buffer solution) for selected residues in the N-terminal part of the Aβ40 peptide were plotted against Hg(II) concentration and fitted globally using Equation (2), assuming one binding site without any buffer correction. Spectra from 84 μM monomeric 13C–15N-labeled Aβ40 peptides were used for the non-quantitative apparent dissociation constant (KDapp*) determination. The estimated apparent dissociation constant was determined to 11 ± 4 μM. Combined chemical shift differences from the spectra in (A,B) are shown in (F,G), respectively.
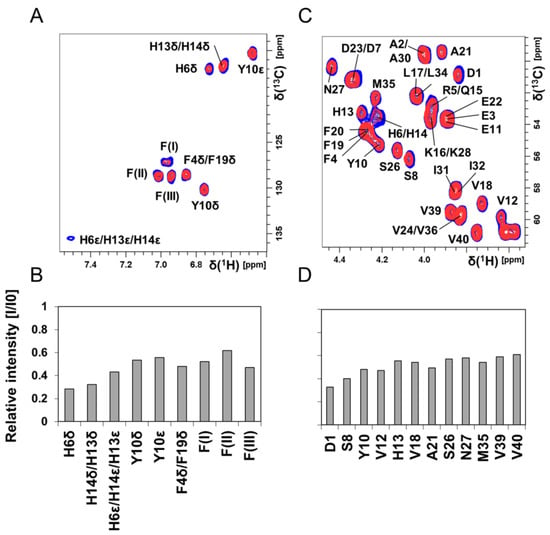
Figure 4.
Two-dimensional (2D) NMR 1H–13C-HSQC experiments showing Aβ40 residue-specific perturbations from Hg(II) ions in buffer; 700 MHz NMR data of 84 μM monomeric 13C-15N-labeled Aβ40 peptides in 20 mM sodium phosphate buffer pH 7.35 at +5 °C are shown in (A–D). (A) 1H–13C-HSQC spectra showing the aromatic region of Aβ peptides (blue) and with 80 μM Hg(II) ions (red). In (B), the relative intensities from the spectra in (A) are shown. Crosspeaks marked with F(I)–F(III) are resonances from phenylalanine residues, without any detailed assignment. (C) 1H–13C-HSQC spectra showing the Cα–H region of Aβ peptides (blue) and with 80 μM Hg(II) ions (red). In (D), the relative intensities from the spectra in (C) are shown. Significant chemical shift changes were not observed. In (D), only crosspeaks without any spectral overlap were included in the evaluation.
Figure 4 shows 2D NMR 1H–13C-HSQC spectra for aromatic (Figure 4A) and Cα–H (Figure 4C) crosspeaks of 84 μM unstructured 13C-15N–Aβ40 peptides in phosphate buffer, before and after addition of 80 μM Hg(II) ions. Again, crosspeaks corresponding to N-terminal Aβ40 residues display the largest intensity loss (Figure 4B,D). Although these NMR observations give no direct information about the metal-binding coordination, the observed loss of NMR signal for specific residues is likely caused by intermediate chemical exchange on the NMR time-scale between a free and a metal-bound state of the Aβ40 peptide, similar to the effect induced by Zn(II) ions [118]. The histidine residues His6, His13, and His14 are markedly affected by the Hg(II) ions (Figure 4B), and might be the main metal-binding ligands. The general loss of Aβ40 crosspeak signal intensity induced by addition of HgCl2 (Figure 3B, Figure 4 and Figure S4D) indicates that Hg(II) ions promote formation of large Aβ aggregates, where some of them are too large to be observed with HSQC NMR and some simply precipitate out of the solution. Thus, the NMR data presented in Figure 3 and Figure 4 mainly stem from interactions between Hg(II) ions and non-aggregated Aβ40 monomers.
Specific binding of Hg(II) ions to the Aβ N-terminal region is observed also when the Aβ40 peptides are bound to SDS micelles (Figure 3C,D), used here as a simple bio-membrane model [109,119]. The 1H–15N-HSQC spectrum for Aβ40 in SDS micelles (Figure 3C) corresponds to a partly α-helical Aβ conformation, where the central (residues 15–24) and C-terminal (residues 29–35) Aβ segments are inserted as α-helices into the micelles [113]. The N-terminal segment is unstructured and located outside the micelle surface, where it can interact with binding agents such as metal ions [116,120]. Addition of Hg(II) ions induces a concentration-dependent intensity loss for N-terminal Aβ40 amide crosspeaks (Figure 3D and Figure S5C), but there is no general loss of amide crosspeak intensity as the Aβ peptides do not aggregate when bound to SDS micelles (at least for Aβ/micelle ratios < 1; cf. Figure 3B,D). Addition of Hg(II) ions induces small changes in the positions of some Aβ40 amide crosspeaks (Figure 3C,G), indicating a structural reorganization of the Aβ40 peptides inside the micelles. This effect might be related to the previously observed increase in helix supercoiling of SDS-bound Aβ induced by Cu(II) ions [116], even though the chemical shifts induced by the Cu(II) and Hg(II) ions are slightly different.
The relative intensities of the Aβ40 1H–15N-HSQC NMR amide crosspeaks corresponding to N-terminal residues were corrected for the general loss of signal intensity caused by Hg(II)-induced aggregation and plotted as a function of added Hg(II) ions (Figure 3E). Fitting Equation (2) globally to all of the curves in Figure 3E produced an apparent Hg(II)·Aβ40 KD value of 11 ± 4 μM.
3.4. Fluorescence Spectroscopy: pH-Dependence of the Hg(II) Binding Affinity to the Aβ16 Monomer
The Aβ16 peptide was used to investigate the pH-dependence of the Hg(II)·Aβ binding affinity, as it is less prone to aggregate during measurements than full-length Aβ peptides. Because Cu(II) ions quench the intrinsic fluorescence of tyrosine residues, while Hg(II) ions do not, the Hg(II)·Aβ16 binding affinity was measured via Cu(II)·Hg(II) competition experiments. Figure 5A,B shows fluorescence quenching data for the Tyr10 residue of 8 µM Aβ16, obtained via stepwise additions of Cu(II) ions in the absence and presence of different concentrations of Hg(II) ions at either pH 7.4 or pH 5.0. The KDapp value for the Cu(II)·Aβ16 complex at pH 7.4 was estimated to be approximately 22 µM in the absence of Hg(II) ions. This value is somewhat higher than the previously reported KDapp values of around 0.5–3 µM, which might be due to different sample preparations and different experimental conditions during the measurements [74,75,76]. When increasing concentrations of Hg(II) ions are present in the pH 7.4 samples, the titration curves are shifted towards weaker Cu(II) affinities, showing that the Hg(II) and Cu(II) ions compete for binding to the Aβ16 peptide (Figure 5A). The KDapp values for the Cu(II)·Aβ16 complex, obtained from the Cu(II) titration series in the presence of 0, 50, and 100 µM Hg(II) ions, are, respectively, 22, 28, and 35 µM. Plotting these KDapp values vs. the Hg(II) concentration (Figure 5A, insert) produced a straight line that was extrapolated to the point where KDapp is two times the KDapp value in absence of Hg(II) ions. At this point, the Hg(II) concentration, which was found to be 170 µM, should be approximately equal to the apparent KD for the Hg(II)·Aβ16 complex.
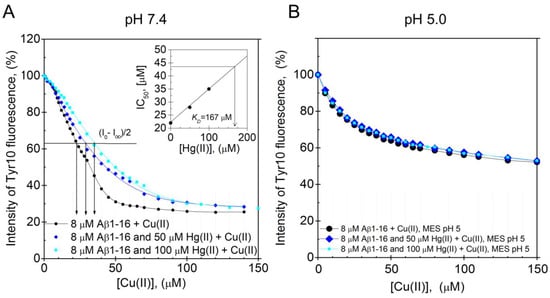
Figure 5.
Dissociation constants for the Aβ·Hg(II) ion complex determined from intrinsic fluorescence quenching. Competitive binding using Tyr10 intrinsic fluorescence quenching experiments were performed by titrating Cu(II) ions onto 8 μM monomeric Aβ16 peptides in 100 mM NaCl + 20 mM HEPES buffer, pH 7.4 (A) or 20 mM MES buffer, pH 5.0 (B) at +25 °C in the absence and presence of 50 and 100 μM Hg(II) ions. A similar binding site for Cu(II) and Hg(II) ions was assumed. In (A), the apparent dissociation constant for the Aβ16·Cu(II) complex was determined to be approximately 22 μM. The relatively high apparent dissociation constant (low affinity) compared to previously reported values [74,75] can possibly be explained by different experimental conditions. The inserted graph in (A) shows the IC50 for Cu(II) as a function of Hg(II) ion concentration. Extrapolation to the point where the IC50 for Cu(II) is increased by a factor of 2 gives an estimated value of 170 μM Hg(II) ions, corresponding to the binding constant. At pH 5.0, the Cu(II) titration series had similar appearances in the absence and presence of Hg(II) ions.
At pH 5.0, addition of up to 100 µM of Hg(II) ions has no influence on the Cu(II) titration curve (Figure 5B), demonstrating that at this lower pH, the binding of Hg(II) ions to Aβ16 is too weak to compete with the binding of Cu(II) ions. This weaker binding of Hg(II) ions to Aβ16 at lower pH is most likely related to protonation of the His residues [121,122,123].
4. Discussion
4.1. The In Vitro Analyses of the Hg(II)·Aβ Complexes and Their Aggregation
Our ThT fluorescence results and AFM images show that Hg(II) ions have a clear and concentration-dependent inhibitory effect on the fibrillization of both Aβ40 (Figure 1 and Figure 2) and Aβ42 (Figure S2) peptides. Instead of forming amyloid fibrils, the Aβ peptides appear to form non-fibrillar amorphous aggregates in the presence of Hg(II) ions. According to the NMR data (Figure 3 and Figure 4), this effect appears to be related to specific binding interactions between Hg(II) ions and the Aβ N-terminal residues. Typical metal-coordinating residues in proteins are cysteines, histidines, and the negatively charged aspartic and glutamic acids. Here, the NMR signals of the Aβ residues His6, His13, and His14 display the most pronounced signal attenuation when Hg(II) ions are added (Figure 4), indicating that the main binding ligands are these three histidines, which coordinate transition metal ions via their imidazole groups. This conclusion is supported by the Hg(II) ions being able to compete with Cu(II) ions for binding to Aβ16 at pH 7.4 (Figure 5A), indicating that similar binding ligands are involved. Cu(II) ions, as well as Fe(II), Mn(II), and Zn(II) ions, have previously been shown to bind to the His residues of Aβ peptides [38,72,75,124,125]. For Pb(IV) ions, both the Tyr10 and the His residues appear to be potential binding ligands [22]. Metal ions such as Ca(II), Cd(II), Cr(III), and Pb(II), on the other hand, do not display residue-specific binding to Aβ monomers [22,126]. The observed His-based binding of Hg(II) ions to Aβ is somewhat surprising, as Hg(II) ions are known to prefer binding ligands such as thiol (−SH) and selenohydryl (−SeH) groups [104,127]. Because Aβ peptides lack Cys and Sec (also known as Se–Cys) residues, it has earlier been suggested that Aβ will not bind Hg(II) ions [128].
The Aβ16, Aβ40, and Aβ42 peptides share the same first 16 residues, and we therefore expect these peptide versions to have similar N-terminal Hg(II) binding modes. The observed binding affinities are, however, not identical. The KDapp value obtained for Hg(II) binding to Aβ40 is 11 ± 4 μM, while for binding to Aβ16 it is around 170 μM. Furthermore, addition of 30 µM Hg(II) ions to the Aβ40 peptides in SDS micelles induces around 50% intensity loss of the N-terminal NMR crosspeak signals (Figure S5C), suggesting an apparent Hg(II)·Aβ40 affinity around 30 µM under those sample conditions. These different results are likely caused by various factors known to influence the binding affinity, such as differences in peptide length, the Aβ aggregation state, and experimental conditions, including buffer type, ionic strength, and pH: As the histidine ligands are very sensitive to protonation around pH 7, small differences in the pH conditions may lead to noticeable differences in binding strength [108,122]. The Hg(II)·Aβ16 affinity measurements were conducted at +25 °C at high salt concentrations (20 mM HEPES pH 7.4 + 100 mM NaCl) on non-aggregated Aβ16 samples (as Aβ16 does not readily aggregate). The Hg(II) affinity measurements to Aβ40 in buffer were conducted at +5 °C at moderate salt concentrations (20 mM sodium phosphate buffer, pH 7.35), on samples that aggregated during the measurements due to the added Hg(II) ions (Figure 3B). The Aβ peptides bound to SDS micelles do not aggregate (at least for Aβ/SDS micelle ratios < 1), but even though previous studies have shown that Aβ peptides positioned in SDS micelles can bind Cu(II) and Zn(II) ions with similar binding affinity as in aqueous solution [116], the binding of Hg(II) ions to SDS-bound Aβ peptides may be somewhat affected by the negatively charged SDS micelles. Although all employed approaches to investigate the Hg(II)·Aβ binding affinity have potential disadvantages, the Cu(II)·Hg(II) competition studies for binding to Aβ16 constitute an indirect method based on the unverified assumption of a shared binding site. Because the NMR experiments measure direct Hg(II)·Aβ40 interactions, we consider them more reliable, and tentatively conclude that the Hg(II)·Aβ binding affinity is in the approximate range of 10–30 µM. Such a conclusion is compatible with the concentration-dependent effects of total fibril inhibition at equimolar concentrations, as observed in the ThT kinetic assay and AFM images (Figure 1 and Figure 2, Figures S2 and S3).
Thus, the apparent Hg(II)·Aβ40 dissociation constant seems to be in a similar range as the 30–60 µM range reported for the Mn(II)·Aβ40 complex [72] and the 1–100 µM range reported for the Zn(II)·Aβ40 complex [75,76,124], but weaker than the 0.5–10 µM range observed for the Cu(II)·Aβ40 complex [72,74,75,76,116,122,124]. When corrected for buffer effects, KD values of 1–50 nM for Aβ·Cu(II) and 0.1–1 µM for Aβ·Zn(II) have been calculated [75,76].
Detailed analyses of Cu(II) and Zn(II) binding to Aβ have shown different effects on peptide aggregation depending on the experimental conditions [38,118,129,130]. The different aggregation pathways might arise from the coordination of multiple Aβ peptides to the same metal ion, which likely is an important factor in Aβ aggregation [130,131]. Although our NMR data indicate that the histidine residues are the main binding ligands to Hg(II) (Figure 4), other N-terminal residues such as Asp1, Glu3, Asp7, Tyr10, and Glu11 are also possible binding partners. When multiple Aβ peptides are involved in coordinating the same metal ion, a multitude of possible binding arrangements are possible, and they are expected to have different effects on the aggregation process. Factors such as pH and salt conditions will influence which binding arrangement is the most favorable, although dynamic equilibria between different arrangements are expected. Investigating such binding modes, and metal binding to Aβ oligomers in general, is an important task for future studies. When a single Aβ peptide coordinates a single metal ion, the peptide appears to adopt a structure unsuitable for fibril formation [118]. Coordinating one metal ion to two or more Aβ peptides usually promotes aggregation, but not necessarily fibrillization [130,131]. In particular, supra-stoichiometric amounts of metal ions often induce rapid formation of amorphous aggregates instead of fibrils [38,132]. Our current results (Figure 1 and Figure 2, Figures S1–S3) show that Hg(II) ions affect Aβ aggregation in a similar fashion as Cu(II) and Zn(II) ions, and prevent fibrillization already at a 1:1 Hg(II)·Aβ ratio. This finding is in agreement with the hypothesis that heavy metals might have a general capacity to induce aggregation of unstructured peptides/proteins [103,104]. As our NMR data indicate that addition of Hg(II) promotes rather than inhibits Aβ aggregation (Figure 3, Figures S4 and S5), Hg(II) ions appear to direct the Aβ aggregation pathway towards unstructured (i.e., non-fibrillar) aggregates. Such unstructured aggregates of Hg(II)·Aβ complexes likely have different electrostatic and hydrophobic properties than Aβ fibrils, which might explain why they are not readily visible in our AFM images (Figure 2 and Figure S3): The incubated Aβ samples were deposited on mica plates using a protocol optimized for imaging of Aβ fibrils.
The Hg(II) ions bind also to Aβ peptides positioned in SDS micelles (Figure 3C,D,G). This is not surprising, as the N-terminal metal-binding Aβ segment is known to be located outside SDS micelles [113], which are considered simplistic membrane models [109]. These results therefore suggest that Hg(II) ions might bind Aβ peptides located in cellular membranes. Such binding could be of biological relevance, as membrane disruption by Aβ oligomers has been suggested to be a neurotoxic mechanism in AD [44,48,133]. It has recently been reported that Hg(II) ions can block membrane channels formed by Aβ42 oligomers [105], which is in line with an earlier observation that membrane leakage of Ca(II) ions induced by Aβ oligomers can be blocked by histidine-binding metal ions and small molecules [134]. Given the inhibitory effects of Hg(II) ions on Aβ fibrillization, it would be interesting to investigate if Hg(II) ions can also modulate the formation of toxic or membrane-disrupting Aβ oligomers.
4.2. Mercury and AD: Clinical Studies and Sources of Exposure
Mercury has, for many decades, been implicated as a risk factor for AD, as elevated Hg levels have been found in early studies of brain and blood of AD patients [10,91,92,93,135,136,137]. Later studies have, however, failed to confirm these higher Hg levels in AD patients [94,95,96]. Because AD patients display altered metal dyshomeostasis [64] that manifests in different ways, including altered plasma/CSF (cerebrospinal fluid) ratios for various metal ions including Hg(II), it has been suggested that AD pathology may involve a compromised blood–CSF barrier [93,137]. On the other hand, the brain metal chemistry is notoriously complex, and studies on beluga whales and humans indicate that Hg(II) and other metal ions accumulate into different brain regions also for non-AD individuals [138,139,140]. One study with a limited number of patients reported elevated Hg concentrations in the two brain regions nucleus basalis of Meynert (NBM) and amygdala [135]. NBM is the major source of cholinergic innervation of the neocortex, and neuronal loss in this region is a well-known pathological feature of both AD and Parkinson’s disease [141].
It is currently unclear if the metal dyshomeostasis observed in AD patients is a cause or effect of the disease progression. Some uncertainties are present in all studies on Hg brain concentrations, due to the difficulties involved in determining metal concentrations in tissue samples and body fluids in general [142], especially for samples stored for a long time in biobanks and, in particular, for ions of Hg [97,143]. Another source of error is the problematic accuracy of AD diagnoses, especially when conducted without reliable biomarkers such as identification of AD plaques via PET scanning or MRI [144,145]. Because symptoms of chronic Hg exposure include personality changes and memory loss, especially for elderly people, Hg poisoning could conceivably be misdiagnosed as AD [10,94,146]. Yet, meta studies support a possible connection between Hg and AD [94]. Studies correlating AD incidence to the number of dental amalgam fillings are often difficult to interpret due to numerous sources of error, and generally show no or little correlation [94]. This suggests that if Hg is a risk factor for AD, then dental amalgam is not the critical source of Hg exposure [147].
Dental amalgam fillings are, nevertheless, the main source of human exposure to metallic Hg [97], which is converted via metabolic oxidation to inorganic Hg(II) ions [148]. Humans are also exposed to fair amounts of lipophilic methyl mercury (MeHg), mainly produced from Hg(II) ions by aquatic microbes [149] and readily bioaccumulates in fish and other marine organisms [97,150]. Less common is exposure to other forms of mercury, such as inorganic Hg2(II) and Hg3(II) polycations and organometallic dimethyl–Hg and ethyl–Hg complexes [97]. During the last decades, industrial release of Hg into the environment has decreased in the Western world but increased in the developing world, mainly due to gold mining and coal burning [128,146,151]. Thus, it still remains relevant to investigate the possible connection between Hg and AD.
4.3. Biological Relevance and Other Molecular Effects on AD Involving Hg Ions
The Hg concentration in the human brain is around 10–50 ng/g (approximately 0.1–0.3 µM) [91,135]. This is significantly lower than the 1–100 µM concentrations of Hg(II) ions used in our in vitro studies. Due to the requirements of the employed spectroscopic techniques, the 10–100 µM Aβ concentrations used in our experiments are also higher than the picomolar–nanomolar Aβ levels typically observed in human brains [152]. Local Aβ concentrations in, e.g., cell membranes may, however, be higher. The higher Aβ concentrations used in our experiments should promote peptide aggregation, but the effect of Hg(II) ions may depend more on the Hg(II)/Aβ ratio than the absolute concentrations. The total inhibition of Aβ fibrillization at 1:1 Hg(II)/Aβ40 ratio (Figure 1 and Figure 2) shows that small amounts of mercury in a critical location can have a large impact on Aβ aggregation. As Hg poisoning correlates with a variety of adverse effects on developing neurites [153], neurotransmission [154], and cognitive function [97,155], the amount of Hg that enters the brain after exposure events clearly has biological impact [10,88,90,94]. While Hg(II) ions do not easily pass the blood–brain barrier (BBB), metallic vapor mercury does [100]. Thus, if metallic vapor mercury passes the BBB and then becomes oxidized to Hg(II), these mercuric ions will be trapped inside the brain. MeHg easily passes across the BBB and the placenta, either by itself or bound to the amino acid cysteine: Such a complex is misrecognized as methionine by transport proteins and therefore freely transported throughout the body [156]. It is currently unclear if Hg is differently deposited in healthy and in AD brains, and if there is co-localization of Hg(II) ions and Aβ peptides in some brain compartments.
Mercury could conceivably affect AD pathology without directly interacting with the Aβ peptides themselves [90,94], for example, via toxic molecular mimicry [157], by promoting the aggregation of the tau fragment R2 [101] and phosphorylation of the tau protein as observed in SHSY5Y neuroblastoma cells [87], or via interactions between other forms of Aβ and Hg than those studied here. MeHg and Hg(II) ions bind to and affect the functions of important intracellular biomolecules with essential thiol (−SH) and selenohydryl (−SeH) groups, such as cysteine, homocysteine, metallothioneins, selenoproteins, glutathione (GSH), tubulin, ion channel proteins, transporters, metabolic enzymes, and N-methyl-D-aspartate (NMDA) receptors, thereby influencing or even damaging various tissues including nerve cells [94,97,153,157,158]. Hg exposure furthermore increased the release of Aβ in SHSY5Y neuroblastoma cells [87], which may promote Aβ aggregation. In Wistar rats exposed to MeHg, the Aβ levels increased in the hippocampus but decreased in the CSF, likely due to impaired Aβ transport [159]. Increased production of AβPP and decreased production of the Aβ-clearing enzyme neprilysin was observed in PC12 cells exposed to Hg(II) and MeHg [89]. In neuroblastoma cells exposed to HgCl2, the increased release of Aβ could be reversed by addition of melatonin [87], which is known to chelate metal ions [160]. Notably, one study using primary endothelial cells from transgenic mice reported increased AβPP expression and sAPPβ secretion in the presence of oxygen radicals [161].
Antioxidants such as GSH and many selenoproteins are known to be blocked by Hg [162], and one important toxic mechanism of various forms of Hg is disruption of the molecular defense against reactive oxygen species (ROS), especially in the mitochondria [98]. This mechanism may be important for both AD and general Hg intoxication. An impaired ROS defense obviously allows for higher concentrations of oxygen radicals, which, as stated above, was reported to promote Aβ production [161]. However, AD pathology appears to involve also cellular and molecular damage directly caused by oxygen radicals, likely formed by Fenton-type reactions with redox-active metal ions such as Cu(II)/Cu(I) and Fe(III)/Fe(II) [61,163,164]. The Hg-induced disruption of the ROS defense [88] likely promotes the ROS-related component of AD pathology [94,98]. This might be particularly relevant for the mitochondria, as they are often affected by Hg exposure [165,166], and as mitochondrial dysfunction is commonly observed in AD brain neurons [62,167,168,169].
Interestingly, AD and Hg intoxication have a common risk factor in the gene encoding the apolipoprotein E (ApoE). Hg exposure typically produces worse outcomes in individuals with the ApoEε4 allele [170,171,172,173], and this allele is linked also to an increased probability of developing AD [8,9,10,11,14]. The underlying reasons for this similar risk factor are unclear, however, and a number of explanations have been proposed [8,174], including the possibility that the beneficial ApoE variants may interact with and promote clearance of the Aβ peptide [175] or Hg ions [9,11]. ApoEε4 proteins might bind metal ions such as Hg(II) less efficiently than other ApoE isoforms, as they have two Arg residues in positions where ApoEε2 proteins have two −SH-containing Cys residues [11]. ApoEε3 has one Arg and one Cys residue. Whether the negative effects of the ApoEε4 allele are in fact related to a possibly reduced capacity for binding and eliminating Hg ions remains to be investigated. Nonetheless, studies investigating the relationship between AD and Hg exposure should benefit from taking into account the ApoE genotype of the studied individuals [173,174,176].
5. Conclusions
Hg(II) ions display specific binding to the N-terminal part of the Aβ peptide, likely coordinated mainly via the Aβ residues His6, His13, and His14, with an apparent Hg(II)·Aβ40 binding affinity in the micromolar range. The Hg(II) ions inhibit Aβ40 and Aβ42 fibrillization in a concentration-dependent manner, and at a 1:1 Hg(II)/Aβ ratio only non-fibrillar Aβ aggregates are formed. The observed molecular interactions support potential involvement of Hg(II) ions in the Aβ amyloid aggregation processes associated with AD pathology.
Supplementary Materials
The following are available online at https://www.mdpi.com/2218-273X/10/1/44/s1: Figure S1: Amyloid fibril formation of Aβ40 peptides monitored by Thioflavin T (ThT) aggregation kinetics assay, Figure S2: Amyloid fibril formation of Aβ42 peptides monitored by Thioflavin T (ThT) aggregation kinetics assay, Figure S3: Solid state AFM imaging of fibril formation and morphology, Figure S4: Two-dimensional (2D) NMR 1H–15N-HSQC titration experiments showing Aβ40 residue-specific perturbations from Hg(II) ions in buffer, Figure S5: Two-dimensional (2D) NMR 1H–15N-HSQC titration experiments showing Aβ40 residue-specific perturbations from Hg(II) ions in SDS micelles.
Author Contributions
Conceptualization, A.G., C.W., P.P., P.M.R., T.S., and S.K.T.S.W.; investigation, A.G., A.N., C.W., J.J., M.F., S.B.S., and S.K.T.S.W.; writing—original draft preparation, P.R., S.B.S., and S.K.T.S.W.; writing—review and editing, A.G., P.P., and T.S.; supervision, A.G., P.P., and S.K.T.S.W.; project administration, S.K.T.S.W.; funding acquisition, A.G., P.P., P.R., and S.K.T.S.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Alzheimer Foundation, the Swedish Research Council, and the Brain Foundation to A.G., from the Magnus Bergvall Foundation to S.K.T.S.W. and P.R., from the Ulla-Carin Lindquist ALS Foundation to P.R., and from the Estonian Ministry of Education and Research (grant IUT 19-8) to P.P.
Acknowledgments
The authors thank Monica Nordberg for helpful discussions.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015—The Global Impact of Dementia; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Alzheimer’s-Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Bjørklund, G.; Aaseth, J.; Dadar, M.; Chirumbolo, S. Molecular Targets in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7032–7044. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Miyata, M.; Smith, J.D. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat. Genet. 1996, 14, 55–61. [Google Scholar] [CrossRef]
- Mutter, J.; Naumann, J.; Sadaghiani, C.; Schneider, R.; Walach, H. Alzheimer disease: Mercury as pathogenetic factor and apolipoprotein E as a moderator. Neuroendocrinol. Lett. 2004, 25, 331–339. [Google Scholar]
- Godfrey, M.E.; Wojcik, D.P.; Krone, C.A. Apolipoprotein E genotyping as a potential biomarker for mercury neurotoxicity. J. Alzheimer’s Dis. 2003, 5, 189–195. [Google Scholar] [CrossRef]
- Gessel, M.M.; Bernstein, S.; Kemper, M.; Teplow, D.B.; Bowers, M.T. Familial Alzheimer’s disease mutations differentially alter amyloid beta-protein oligomerization. ACS Chem. Neurosci. 2012, 3, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Herring, A.; Munster, Y.; Metzdorf, J.; Bolczek, B.; Krussel, S.; Krieter, D.; Yavuz, I.; Karim, F.; Roggendorf, C.; Stang, A.; et al. Late running is not too late against Alzheimer’s pathology. Neurobiol. Dis. 2016, 94, 44–54. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Wang, H.F.; Jiang, T.; Tan, M.S.; Tan, L.; Zhao, Q.F.; Li, J.Q.; Wang, J.; Yu, J.T. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1299–1306. [Google Scholar] [CrossRef]
- Rolandi, E.; Frisoni, G.B.; Cavedo, E. Efficacy of lifestyle interventions on clinical and neuroimaging outcomes in elderly. Ageing Res. Rev. 2016, 25, 1–12. [Google Scholar] [CrossRef]
- Maher, B.A.; Ahmed, I.A.; Karloukovski, V.; MacLaren, D.A.; Foulds, P.G.; Allsop, D.; Mann, D.M.; Torres-Jardon, R.; Calderon-Garciduenas, L. Magnetite pollution nanoparticles in the human brain. Proc. Natl. Acad. Sci. USA 2016, 113, 10797–10801. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Kavanaugh, M.; Block, M.; D’Angiulli, A.; Delgado-Chavez, R.; Torres-Jardon, R.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Osnaya, N.; Villarreal-Calderon, R.; et al. Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J. Alzheimer’s Dis. 2012, 28, 93–107. [Google Scholar] [CrossRef]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W.; Alzheimer’s Disease Neuroimaging Initiative. Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms. Alzheimer’s Dement. 2014, 10, S122–S145. [Google Scholar] [CrossRef]
- Cataldo, J.K.; Prochaska, J.J.; Glantz, S.A. Cigarette smoking is a risk factor for Alzheimer’s Disease: An analysis controlling for tobacco industry affiliation. J. Alzheimer’s Dis. 2010, 19, 465–480. [Google Scholar] [CrossRef]
- Wallin, C.; Sholts, S.B.; Österlund, N.; Luo, J.; Jarvet, J.; Roos, P.M.; Ilag, L.; Gräslund, A.; Wärmländer, S.K.T.S. Alzheimer’s disease and cigarette smoke components: Effects of nicotine, PAHs, and Cd(II), Cr(III), Pb(II), Pb(IV) ions on amyloid-beta peptide aggregation. Sci. Rep. 2017, 7, 14423. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.J.; Zhang, M.; Xu, Z.Q.; Gao, C.Y.; Fang, C.Q.; Yan, J.C.; Zhou, H.D.; Chongqing Ageing Study Group. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology 2011, 76, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Reciprocal Molecular Interactions between the Abeta Peptide Linked to Alzheimer’s Disease and Insulin Linked to Diabetes Mellitus Type II. ACS Chem. Neurosci. 2016, 7, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, T.M.; Thakur, M.K. Traumatic brain injury: A risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 2012, 36, 1376–1381. [Google Scholar] [CrossRef]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Wärmländer, S.K.; Jarvet, J.; Zhao, L.; Jia, X.; Narayana, V.K.; Shankar, S.K.; Olofsson, A.; et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, X.; Yang, J.; Suo, J.; Chen, J.; Liu, X.; Zhao, X. Chronic exposure to aluminum and risk of Alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2016, 610, 200–206. [Google Scholar] [CrossRef]
- Modgil, S.; Lahiri, D.K.; Sharma, V.L.; Anand, A. Role of early life exposure and environment on neurodegeneration: Implications on brain disorders. Transl. Neurodegener. 2014, 3, 9. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Sunde, M.; Blake, C.C. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q. Rev. Biophys. 1998, 31, 1–39. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Abelein, A.; Abrahams, J.P.; Danielsson, J.; Gräslund, A.; Jarvet, J.; Luo, J.; Tiiman, A.; Wärmländer, S.K. The hairpin conformation of the amyloid beta peptide is an important structural motif along the aggregation pathway. J. Biol. Inorg. Chem. 2014, 19, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Wärmländer, S.; Tiiman, A.; Abelein, A.; Luo, J.; Jarvet, J.; Söderberg, K.L.; Danielsson, J.; Gräslund, A. Biophysical studies of the amyloid beta-peptide: Interactions with metal ions and small molecules. Chembiochem 2013, 14, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yu, C.H.; Yu, H.; Borstnar, R.; Kamerlin, S.C.; Gräslund, A.; Abrahams, J.P.; Wärmländer, S.K. Cellular polyamines promote amyloid-beta (Abeta) peptide fibrillation and modulate the aggregation pathways. ACS Chem. Neurosci. 2013, 4, 454–462. [Google Scholar] [CrossRef]
- Luo, J.; Mohammed, I.; Wärmländer, S.K.; Hiruma, Y.; Gräslund, A.; Abrahams, J.P. Endogenous polyamines reduce the toxicity of soluble abeta peptide aggregates associated with Alzheimer’s disease. Biomacromolecules 2014, 15, 1985–1991. [Google Scholar] [CrossRef]
- Owen, M.C.; Gnutt, D.; Gao, M.; Wärmländer, S.K.T.S.; Jarvet, J.; Gräslund, A.; Winter, R.; Ebbinghaus, S.; Strodel, B. Effects of in vivo conditions on amyloid aggregation. Chem. Soc. Rev. 2019, 48, 3946–3996. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-beta peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Sandberg, A.; Luheshi, L.M.; Sollvander, S.; Pereira de Barros, T.; Macao, B.; Knowles, T.P.; Biverstal, H.; Lendel, C.; Ekholm-Petterson, F.; Dubnovitsky, A.; et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [Google Scholar] [CrossRef]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Alzheimer peptides aggregate into transient nanoglobules that nucleate fibrils. Biochemistry 2014, 53, 6302–6308. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-beta oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-beta Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Agholme, L.; Kurudenkandy, F.R.; Granseth, B.; Marcusson, J.; Hallbeck, M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J. Neurosci. 2012, 32, 8767–8777. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjo, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-beta oligomers distinct from amyloid-beta derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef]
- Wärmländer, S.K.T.S.; Österlund, N.; Wallin, C.; Wu, J.; Luo, J.; Tiiman, A.; Jarvet, J.; Gräslund, A. Metal binding to the Amyloid-β peptides in the presence of biomembranes: Potential mechanisms of cell toxicity. J. Biol. Inorg. Chem. 2019, 24, 1189–1196, in press. [Google Scholar]
- Österlund, N.; Kulkarni, Y.S.; Misiaszek, A.D.; Wallin, C.; Krüger, D.M.; Liao, Q.; Mashayekhy Rad, F.; Jarvet, J.; Strodel, B.; Wärmländer, S.K.T.S.; et al. Amyloid-beta Peptide Interactions with Amphiphilic Surfactants: Electrostatic and Hydrophobic Effects. ACS Chem. Neurosci. 2018, 9, 1680–1692. [Google Scholar] [CrossRef]
- Österlund, N.; Moons, R.; Ilag, L.L.; Sobott, F.; Gräslund, A. Native Ion Mobility-Mass Spectrometry Reveals the Formation of beta-Barrel Shaped Amyloid-beta Hexamers in a Membrane-Mimicking Environment. J. Am. Chem. Soc. 2019, 141, 10440–10450. [Google Scholar] [CrossRef]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Human lysozyme inhibits the in vitro aggregation of Abeta peptides, which in vivo are associated with Alzheimer’s disease. Chem. Commun. (Camb.) 2013, 49, 6507–6509. [Google Scholar] [CrossRef]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Non-chaperone proteins can inhibit aggregation and cytotoxicity of Alzheimer amyloid beta peptide. J. Biol. Chem. 2014, 289, 27766–27775. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Cross-interactions between the Alzheimer Disease Amyloid-beta Peptide and Other Amyloid Proteins: A Further Aspect of the Amyloid Cascade Hypothesis. J. Biol. Chem. 2016, 291, 16485–16493. [Google Scholar] [CrossRef] [PubMed]
- Leshem, G.; Richman, M.; Lisniansky, E.; Antman-Passig, M.; Habashi, M.; Gräslund, A.; Wärmländer, S.K.T.S.; Rahimipour, S. Photoactive chlorin e6 is a multifunctional modulator of amyloid-beta aggregation and toxicity via specific interactions with its histidine residues. Chem. Sci. 2019, 10, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Richman, M.; Wilk, S.; Chemerovski, M.; Warmlander, S.K.; Wahlstrom, A.; Graslund, A.; Rahimipour, S. In vitro and mechanistic studies of an antiamyloidogenic self-assembled cyclic D,L-alpha-peptide architecture. J. Am. Chem. Soc. 2013, 135, 3474–3484. [Google Scholar] [CrossRef]
- Luo, J.; Otero, J.M.; Yu, C.H.; Wärmländer, S.K.; Gräslund, A.; Overhand, M.; Abrahams, J.P. Inhibiting and reversing amyloid-beta peptide (1–40) fibril formation with gramicidin S and engineered analogues. Chemistry 2013, 19, 17338–17348. [Google Scholar] [CrossRef]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Cell-to-Cell Transmission of Pathological Tau: A Review. JAMA Neurol. 2019, 76, 101–108. [Google Scholar] [CrossRef]
- Wallin, C.; Hiruma, Y.; Wärmländer, S.K.T.S.; Huvent, I.; Jarvet, J.; Abrahams, J.P.; Gräslund, A.; Lippens, G.; Luo, J. The Neuronal Tau Protein Blocks in Vitro Fibrillation of the Amyloid-beta (Abeta) Peptide at the Oligomeric Stage. J. Am. Chem. Soc. 2018. [Google Scholar] [CrossRef]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.G.; Morris, K.L.; Sada, A.A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of Amyloid-beta in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Duce, J.A.; Bush, A.I.; Adlard, P.A. Role of amyloid-β–metal interactions in Alzheimer’s disease. Future Neurol. 2011, 6, 641–659. [Google Scholar] [CrossRef]
- Wang, Z.X.; Tan, L.; Wang, H.F.; Ma, J.; Liu, J.; Tan, M.S.; Sun, J.H.; Zhu, X.C.; Jiang, T.; Yu, J.T. Serum Iron, Zinc, and Copper Levels in Patients with Alzheimer’s Disease: A Replication Study and Meta-Analyses. J. Alzheimer’s Dis. 2015, 47, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Tiiman, A.; Palumaa, P.; Tougu, V. The missing link in the amyloid cascade of Alzheimer’s disease—Metal ions. Neurochem. Int. 2013, 62, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Lei, P.; Bush, A.I. Metallostasis in Alzheimer’s disease. Free Radic. Biol. Med. 2013, 62, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R. Metals in Alzheimer’s disease: A systemic perspective. Front. Biosci. (Landmark Ed) 2012, 17, 451–472. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Miller, L.M.; Wang, Q.; Telivala, T.P.; Smith, R.J.; Lanzirotti, A.; Miklossy, J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 2006, 155, 30–37. [Google Scholar] [CrossRef]
- Beauchemin, D.; Kisilevsky, R. A method based on ICP-MS for the analysis of Alzheimer’s amyloid plaques. Anal. Chem. 1998, 70, 1026–1029. [Google Scholar] [CrossRef]
- Sayre, L.M.; Perry, G.; Harris, P.L.; Liu, Y.; Schubert, K.A.; Smith, M.A. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: A central role for bound transition metals. J. Neurochem. 2000, 74, 270–279. [Google Scholar] [CrossRef]
- Wallin, C.; Kulkarni, Y.S.; Abelein, A.; Jarvet, J.; Liao, Q.; Strodel, B.; Olsson, L.; Luo, J.; Abrahams, J.P.; Sholts, S.B.; et al. Characterization of Mn(II) ion binding to the amyloid-beta peptide in Alzheimer’s disease. J. Trace Elem. Med. Biol. 2016, 38, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Faller, P. Copper and zinc binding to amyloid-beta: Coordination, dynamics, aggregation, reactivity and metal-ion transfer. ChemBioChem 2009, 10, 2837–2845. [Google Scholar] [CrossRef] [PubMed]
- Conte-Daban, A.; Borghesani, V.; Sayen, S.; Guillon, E.; Journaux, Y.; Gontard, G.; Lisnard, L.; Hureau, C. Link between Affinity and Cu(II) Binding Sites to Amyloid-beta Peptides Evaluated by a New Water-Soluble UV-Visible Ratiometric Dye with a Moderate Cu(II) Affinity. Anal. Chem. 2017, 89, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Faller, P.; Hureau, C. Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-beta peptide. Dalton Trans. 2009, 1080–1094. [Google Scholar] [CrossRef]
- Tõugu, V.; Karafin, A.; Palumaa, P. Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-beta peptide. J. Neurochem. 2008, 104, 1249–1259. [Google Scholar] [CrossRef]
- Wild, K.; August, A.; Pietrzik, C.U.; Kins, S. Structure and Synaptic Function of Metal Binding to the Amyloid Precursor Protein and its Proteolytic Fragments. Front. Mol. Neurosci. 2017, 10, 21. [Google Scholar] [CrossRef]
- Branch, T.; Barahona, M.; Dodson, C.A.; Ying, L. Kinetic Analysis Reveals the Identity of Abeta-Metal Complex Responsible for the Initial Aggregation of Abeta in the Synapse. ACS Chem. Neurosci. 2017, 8, 1970–1979. [Google Scholar] [CrossRef]
- Basha, M.R.; Wei, W.; Bakheet, S.A.; Benitez, N.; Siddiqi, H.K.; Ge, Y.-W.; Lahiri, D.K.; Zawia, N.H. The Fetal Basis of Amyloidogenesis: Exposure to Lead and Latent Overexpression of Amyloid Precursor Protein and β-Amyloid in the Aging Brain. J. Neurosci. 2005, 25, 823. [Google Scholar] [CrossRef]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef]
- Huang, C.-L.; Hsiao, I.L.; Lin, H.-C.; Wang, C.-F.; Huang, Y.-J.; Chuang, C.-Y. Silver nanoparticles affect on gene expression of inflammatory and neurodegenerative responses in mouse brain neural cells. Environ. Res. 2015, 136, 253–263. [Google Scholar] [CrossRef]
- Ashok, A.; Rai, N.K.; Tripathi, S.; Bandyopadhyay, S. Exposure to As-, Cd-, and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci. 2015, 143, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato-Negishi, M. Link between aluminum and the pathogenesis of Alzheimer’s disease: The integration of the aluminum and amyloid cascade hypotheses. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Chen, X.; Li, W.; Han, Y.; Liu, P.; Pi, R. Exposure to metal ions regulates mRNA levels of APP and BACE1 in PC12 cells: Blockage by curcumin. Neurosci. Lett. 2008, 440, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Cheng, D.; Laferla, F.M. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J. Neurochem. 2009, 108, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lv, Y.; Yu, S.; Zhao, H.; Yao, L. The effect of cadmium on Abeta levels in APP/PS1 transgenic mice. Exp. Ther. Med. 2012, 4, 125–130. [Google Scholar] [CrossRef]
- Olivieri, G.; Brack, C.; Muller-Spahn, F.; Stahelin, H.B.; Herrmann, M.; Renard, P.; Brockhaus, M.; Hock, C. Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. J. Neurochem. 2000, 74, 231–236. [Google Scholar] [CrossRef]
- Monnet-Tschudi, F.; Zurich, M.G.; Boschat, C.; Corbaz, A.; Honegger, P. Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases. Rev. Environ. Health 2006, 21, 105–117. [Google Scholar] [CrossRef]
- Song, J.W.; Choi, B.S. Mercury induced the Accumulation of Amyloid Beta (Abeta) in PC12 Cells: The Role of Production and Degradation of Abeta. Toxicol. Res. 2013, 29, 235–240. [Google Scholar] [CrossRef]
- Bjørklund, G.; Tinkov, A.A.; Dadar, M.; Rahman, M.M.; Chirumbolo, S.; Skalny, A.V.; Skalnaya, M.G.; Haley, B.E.; Ajsuvakova, O.P.; Aaseth, J. Insights into the Potential Role of Mercury in Alzheimer’s Disease. J. Mol. Neurosci. 2019, 67, 511–533. [Google Scholar] [CrossRef]
- Ehmann, W.D.; Markesbery, W.R.; Alauddin, M.; Hossain, T.I.; Brubaker, E.H. Brain trace elements in Alzheimer’s disease. Neurotoxicology 1986, 7, 195–206. [Google Scholar]
- Hock, C.; Drasch, G.; Golombowski, S.; Muller-Spahn, F.; Willershausen-Zonnchen, B.; Schwarz, P.; Hock, U.; Growdon, J.H.; Nitsch, R.M. Increased blood mercury levels in patients with Alzheimer’s disease. J. Neural Transm. (Vienna) 1998, 105, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Gerhardsson, L.; Lundh, T.; Minthon, L.; Londos, E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement. Geriatr. Cognit. Disord. 2008, 25, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Mutter, J.; Curth, A.; Naumann, J.; Deth, R.; Walach, H. Does inorganic mercury play a role in Alzheimer’s disease? A systematic review and an integrated molecular mechanism. J. Alzheimer’s Dis. 2010, 22, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.T.; Harry, G.J.; Hayden, K.M.; Szabo, D.T.; Birnbaum, L. Comparison of Metal Levels between Postmortem Brain and Ventricular Fluid in Alzheimer’s Disease and Nondemented Elderly Controls. Toxicol. Sci. 2016, 150, 292–300. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, D.W.; Park, K.S.; Joung, H. Serum trace metal levels in Alzheimer’s disease and normal control groups. Am. J. Alzheimer’s Dis. Other Dement. 2014, 29, 76–83. [Google Scholar] [CrossRef]
- Berlin, M.; Zalups, R.K.; Fowler, B.A. Chapter 46: Mercury. In Handbook on the Toxicology of Metals, 4th ed.; Nordberg, G.F., Fowler, B.A., Nordberg, M., Eds.; Elsevier/Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Carocci, A.; Rovito, N.; Sinicropi, M.S.; Genchi, G. Mercury toxicity and neurodegenerative effects. Rev. Environ. Contam. Toxicol. 2014, 229, 1–18. [Google Scholar] [CrossRef]
- Wu, X.; Cobbina, S.J.; Mao, G.; Xu, H.; Zhang, Z.; Yang, L. A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ. Sci. Pollut. Res. Int. 2016, 23, 8244–8259. [Google Scholar] [CrossRef]
- Aschner, M.; Aschner, J.L. Mercury neurotoxicity: Mechanisms of blood-brain barrier transport. Neurosci. Biobehav. Rev. 1990, 14, 169–176. [Google Scholar] [CrossRef]
- Yang, D.J.; Shi, S.; Zheng, L.F.; Yao, T.M.; Ji, L.N. Mercury(II) promotes the in vitro aggregation of tau fragment corresponding to the second repeat of microtubule-binding domain: Coordination and conformational transition. Biopolymers 2010, 93, 1100–1107. [Google Scholar] [CrossRef]
- Arnhold, F.; Guhrs, K.H.; von Mikecz, A. Amyloid domains in the cell nucleus controlled by nucleoskeletal protein lamin B1 reveal a new pathway of mercury neurotoxicity. PeerJ 2015, 3, e754. [Google Scholar] [CrossRef]
- Sharma, S.K.; Goloubinoff, P.; Christen, P. Heavy metal ions are potent inhibitors of protein folding. Biochem. Biophys. Res. Commun. 2008, 372, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Tamas, M.J.; Sharma, S.K.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 2014, 4, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Meleleo, D.; Notarachille, G.; Mangini, V.; Arnesano, F. Concentration-dependent effects of mercury and lead on Abeta42: Possible implications for Alzheimer’s disease. Eur. Biophys. J. 2019, 48, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, E.; Boland, B.; Walsh, D.M.; Linse, S. Amyloid beta-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 2010, 1, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Alies, B.; Renaglia, E.; Rozga, M.; Bal, W.; Faller, P.; Hureau, C. Cu(II) affinity for the Alzheimer’s peptide: Tyrosine fluorescence studies revisited. Anal. Chem. 2013, 85, 1501–1508. [Google Scholar] [CrossRef]
- Österlund, N.; Luo, J.; Wärmländer, S.K.T.S.; Gräslund, A. Membrane-mimetic systems for biophysical studies of the amyloid-beta peptide. Biochim. Biophys. Acta Proteins Proteom. 2018. [Google Scholar] [CrossRef]
- Danielsson, J.; Andersson, A.; Jarvet, J.; Gräslund, A. 15N relaxation study of the amyloid beta-peptide: Structural propensities and persistence length. Magn. Reson. Chem. 2006, 44, S114–S121. [Google Scholar] [CrossRef]
- Roche, J.; Shen, Y.; Lee, J.H.; Ying, J.; Bax, A. Monomeric Abeta(1-40) and Abeta(1-42) Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry 2016, 55, 762–775. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Matsuzaki, K.; Hoshino, M. Transient formation of intermediate conformational states of amyloid-beta peptide revealed by heteronuclear magnetic resonance spectroscopy. FEBS Lett. 2011, 585, 1097–1102. [Google Scholar] [CrossRef]
- Jarvet, J.; Danielsson, J.; Damberg, P.; Oleszczuk, M.; Gräslund, A. Positioning of the Alzheimer Abeta(1–40) peptide in SDS micelles using NMR and paramagnetic probes. J. Biomol. NMR 2007, 39, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, J.; Wahlström, A.; Danielsson, J.; Markova, N.; Ekblad, C.; Gräslund, A.; Abrahmsen, L.; Karlström, A.E.; Wärmländer, S.K. N-terminal engineering of amyloid-beta-binding Affibody molecules yields improved chemical synthesis and higher binding affinity. Protein Sci. 2010, 19, 2319–2329. [Google Scholar] [CrossRef] [PubMed]
- Tiiman, A.; Luo, J.; Wallin, C.; Olsson, L.; Lindgren, J.; Jarvet, J.; Roos, P.M.; Sholts, S.B.; Rahimipour, S.; Abrahams, J.P.; et al. Specific Binding of Cu(II) Ions to Amyloid-Beta Peptides Bound to Aggregation-Inhibiting Molecules or SDS Micelles Creates Complexes that Generate Radical Oxygen Species. J. Alzheimer’s Dis. 2016, 54, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta 2000, 1502, 16–30. [Google Scholar] [CrossRef]
- Abelein, A.; Gräslund, A.; Danielsson, J. Zinc as chaperone-mimicking agent for retardation of amyloid beta peptide fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, 5407–5412. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein-membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Stewart, K.L.; Radford, S.E. Amyloid plaques beyond Abeta: A survey of the diverse modulators of amyloid aggregation. Biophys. Rev. 2017, 9, 405–419. [Google Scholar] [CrossRef]
- Zhang, S.; Casey, N.; Lee, J.P. Residual structure in the Alzheimer’s disease peptide: Probing the origin of a central hydrophobic cluster. Fold. Des. 1998, 3, 413–422. [Google Scholar] [CrossRef]
- Lindgren, J.; Segerfeldt, P.; Sholts, S.B.; Gräslund, A.; Karlström, A.E.; Wärmländer, S.K. Engineered non-fluorescent Affibody molecules facilitate studies of the amyloid-beta (Abeta) peptide in monomeric form: Low pH was found to reduce Abeta/Cu(II) binding affinity. J. Inorg. Biochem. 2013, 120, 18–23. [Google Scholar] [CrossRef]
- Ghalebani, L.; Wahlström, A.; Danielsson, J.; Wärmländer, S.K.; Gräslund, A. pH-dependence of the specific binding of Cu(II) and Zn(II) ions to the amyloid-beta peptide. Biochem. Biophys. Res. Commun. 2012, 421, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, J.; Pierattelli, R.; Banci, L.; Gräslund, A. High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid beta-peptide. FEBS J. 2007, 274, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Bousejra-ElGarah, F.; Bijani, C.; Coppel, Y.; Faller, P.; Hureau, C. Iron(II) binding to amyloid-beta, the Alzheimer’s peptide. Inorg. Chem. 2011, 50, 9024–9030. [Google Scholar] [CrossRef] [PubMed]
- Brännström, K.; Öhman, A.; Lindhagen-Persson, M.; Olofsson, A. Ca(2+) enhances Abeta polymerization rate and fibrillar stability in a dynamic manner. Biochem. J. 2013, 450, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.; Guo, H.-B.; Parks, J.M.; Gu, B.; Summers, A.O.; Miller, S.M.; Liang, L.; Smith, J.C. Why Mercury Prefers Soft Ligands. J. Phys. Chem. Lett. 2013, 4, 2317–2322. [Google Scholar] [CrossRef]
- Charlet, L.; Chapron, Y.; Faller, P.; Kirsch, R.; Stone, A.T.; Baveye, P.C. Neurodegenerative diseases and exposure to the environmental metals Mn, Pb, and Hg. Coord. Chem. Rev. 2012, 256, 2147–2163. [Google Scholar] [CrossRef]
- Lv, Z.; Condron, M.M.; Teplow, D.B.; Lyubchenko, Y.L. Nanoprobing of the effect of Cu(2+) cations on misfolding, interaction and aggregation of amyloid beta peptide. J. Neuroimmune Pharmacol. 2013, 8, 262–273. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. USA 2010, 107, 9490–9495. [Google Scholar] [CrossRef]
- Wineman-Fisher, V.; Bloch, D.N.; Miller, Y. Challenges in studying the structures of metal-amyloid oligomers related to type 2 diabetes, Parkinson’s disease, and Alzheimer’s disease. Coord. Chem. Rev. 2016, 327, 20–26. [Google Scholar] [CrossRef]
- Tougu, V.; Karafin, A.; Zovo, K.; Chung, R.S.; Howells, C.; West, A.K.; Palumaa, P. Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-beta (1-42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 2009, 110, 1784–1795. [Google Scholar] [CrossRef]
- Serra-Batiste, M.; Ninot-Pedrosa, M.; Bayoumi, M.; Gairi, M.; Maglia, G.; Carulla, N. Abeta42 assembles into specific beta-barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. USA 2016, 113, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Diaz, J.C.; Flora, M. Efficiency of histidine-associating compounds for blocking the alzheimer’s Abeta channel activity and cytotoxicity. Biophys. J. 2008, 95, 4879–4889. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.M.; Markesbery, W.R.; Ehmann, W.D.; Mao, Y.X.; Vance, D.E. Regional brain trace-element studies in Alzheimer’s disease. Neurotoxicology 1988, 9, 1–7. [Google Scholar] [PubMed]
- Basun, H.; Forssell, L.G.; Wetterberg, L.; Winblad, B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J. Neural Transm. Park Dis. Dement. Sect. 1991, 3, 231–258. [Google Scholar] [PubMed]
- Gerhardsson, L.; Lundh, T.; Londos, E.; Minthon, L. Cerebrospinal fluid/plasma quotients of essential and non-essential metals in patients with Alzheimer’s disease. J. Neural Transm. (Vienna) 2011, 118, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Ostertag, S.K.; Stern, G.A.; Wang, F.; Lemes, M.; Chan, H.M. Mercury distribution and speciation in different brain regions of beluga whales (Delphinapterus leucas). Sci. Total Environ. 2013, 456–457, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Kum Jew, S. Inorganic mercury in human astrocytes, oligodendrocytes, corticomotoneurons and the locus ceruleus: Implications for multiple sclerosis, neurodegenerative disorders and gliomas. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2018, 31, 807–819. [Google Scholar] [CrossRef]
- Pamphlett, R.; Kum Jew, S. Different Populations of Human Locus Ceruleus Neurons Contain Heavy Metals or Hyperphosphorylated Tau: Implications for Amyloid-beta and Tau Pathology in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 437–447. [Google Scholar] [CrossRef]
- Liu, A.K.; Chang, R.C.; Pearce, R.K.; Gentleman, S.M. Nucleus basalis of Meynert revisited: Anatomy, history and differential involvement in Alzheimer’s and Parkinson’s disease. Acta Neuropathol. 2015, 129, 527–540. [Google Scholar] [CrossRef]
- Roos, P.M. Ultraclean paired sampling for metal analysis in neurodegenerative disorders. J. Trace Elem. Med. Biol. 2019, 52, 48–52. [Google Scholar] [CrossRef]
- Nuttall, K.L. Interpreting mercury in blood and urine of individual patients. Ann. Clin. Lab. Sci. 2004, 34, 235–250. [Google Scholar] [PubMed]
- Frisoni, G.B.; Boccardi, M.; Barkhof, F.; Blennow, K.; Cappa, S.; Chiotis, K.; Demonet, J.F.; Garibotto, V.; Giannakopoulos, P.; Gietl, A.; et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 2017, 16, 661–676. [Google Scholar] [CrossRef]
- Shim, Y.S.; Roe, C.M.; Buckles, V.D.; Morris, J.C. Clinicopathologic study of Alzheimer’s disease: Alzheimer mimics. J. Alzheimer’s Dis. 2013, 35, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P. Mercury exposure and Alzheimer’s disease in India—An imminent threat? Sci. Total Environ. 2017, 589, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Saxe, S.R.; Wekstein, M.W.; Kryscio, R.J.; Henry, R.G.; Cornett, C.R.; Snowdon, D.A.; Grant, F.T.; Schmitt, F.A.; Donegan, S.J.; Wekstein, D.R.; et al. Alzheimer’s disease, dental amalgam and mercury. J. Am. Dent. Assoc. 1999, 130, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Park, J.D.; Zheng, W. Human exposure and health effects of inorganic and elemental mercury. J. Prev. Med. Public Health 2012, 45, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Lin, H.; Zheng, W.; Tomanicek, S.J.; Johs, A.; Feng, X.; Elias, D.A.; Liang, L.; Gu, B. Oxidation and methylation of dissolved elemental mercury by anaerobic bacteria. Nat. Geosci. 2013, 6, 751–754. [Google Scholar] [CrossRef]
- UNEP. Global Mercury Assessment 2013: Sources, Emissions, Releases and Environmental Transport; UNEP Chemicals Branch: Geneva, Switzerland, 2013. [Google Scholar]
- Streets, D.G.; Devane, M.K.; Lu, Z.; Bond, T.C.; Sunderland, E.M.; Jacob, D.J. All-time releases of mercury to the atmosphere from human activities. Environ. Sci. Technol. 2011, 45, 10485–10491. [Google Scholar] [CrossRef]
- Roberts, K.F.; Elbert, D.L.; Kasten, T.P.; Patterson, B.W.; Sigurdson, W.C.; Connors, R.E.; Ovod, V.; Munsell, L.Y.; Mawuenyega, K.G.; Miller-Thomas, M.M.; et al. Amyloid-beta efflux from the central nervous system into the plasma. Ann. Neurol. 2014, 76, 837–844. [Google Scholar] [CrossRef]
- Leong, C.C.; Syed, N.I.; Lorscheider, F.L. Retrograde degeneration of neurite membrane structural integrity of nerve growth cones following in vitro exposure to mercury. Neuroreport 2001, 12, 733–737. [Google Scholar] [CrossRef]
- Fitsanakis, V.A.; Aschner, M. The importance of glutamate, glycine, and gamma-aminobutyric acid transport and regulation in manganese, mercury and lead neurotoxicity. Toxicol. Appl. Pharmacol. 2005, 204, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.S.; Kwok, K.M.; Chan, P.H.; So, H.K.; Li, A.M.; Ng, P.C.; Fok, T.F. Long term neurocognitive impact of low dose prenatal methylmercury exposure in Hong Kong. Environ. Int. 2013, 54, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Kerper, L.E.; Ballatori, N.; Clarkson, T.W. Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am. J. Physiol. 1992, 262, R761–R765. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 2005, 204, 274–308. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Farkas, S.; Kortbeek, S.; Zhang, F.X.; Chen, L.; Zamponi, G.W.; Syed, N.I. Mercury-induced toxicity of rat cortical neurons is mediated through N-Methyl-D-Aspartate receptors. Mol. Brain 2012, 5, 30. [Google Scholar] [CrossRef]
- Kim, D.K.; Park, J.D.; Choi, B.S. Mercury-induced amyloid-beta (Abeta) accumulation in the brain is mediated by disruption of Abeta transport. J. Toxicol. Sci. 2014, 39, 625–635. [Google Scholar] [CrossRef]
- Limson, J.; Nyokong, T.; Daya, S. The interaction of melatonin and its precursors with aluminium, cadmium, copper, iron, lead, and zinc: An adsorptive voltammetric study. J. Pineal Res. 1998, 24, 15–21. [Google Scholar] [CrossRef]
- Muche, A.; Arendt, T.; Schliebs, R. Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells. PLoS ONE 2017, 12, e0178127. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Sasakura, C.; Yoneda, S. Binding sites for the (Hg-Se) complex on selenoprotein P. Biochim. Biophys. Acta 1998, 1429, 102–112. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.H.; Luz, A.L.; Cao, X.; Maurer, L.L.; Blawas, A.M.; Aballay, A.; Pan, W.K.; Meyer, J.N. Effects of methyl and inorganic mercury exposure on genome homeostasis and mitochondrial function in Caenorhabditis elegans. DNA Repair (Amst) 2017, 52, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.C.; de Paula, E.S.; Pazin, M.; Carneiro, M.F.H.; Grotto, D.; Barbosa, F., Jr.; Dorta, D.J. Niacin prevents mitochondrial oxidative stress caused by sub-chronic exposure to methylmercury. Drug Chem. Toxicol. 2018, 1–7. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Ng, S.; Lin, C.-C.; Hwang, Y.-H.; Hsieh, W.-S.; Liao, H.-F.; Chen, P.-C. Mercury, APOE, and children’s neurodevelopment. Neurotoxicology 2013, 37, 85–92. [Google Scholar] [CrossRef]
- Snoj Tratnik, J.; Falnoga, I.; Trdin, A.; Mazej, D.; Fajon, V.; Miklavcic, A.; Kobal, A.B.; Osredkar, J.; Sesek Briski, A.; Krsnik, M.; et al. Prenatal mercury exposure, neurodevelopment and apolipoprotein E genetic polymorphism. Environ. Res. 2017, 152, 375–385. [Google Scholar] [CrossRef]
- Ng, S.; Lin, C.C.; Jeng, S.F.; Hwang, Y.H.; Hsieh, W.S.; Chen, P.C. Mercury, APOE, and child behavior. Chemosphere 2015, 120, 123–130. [Google Scholar] [CrossRef]
- Wojcik, D.P.; Godfrey, M.E.; Christie, D.; Haley, B.E. Mercury toxicity presenting as chronic fatigue, memory impairment and depression: Diagnosis, treatment, susceptibility, and outcomes in a New Zealand general practice setting (1994–2006). Neuroendocrinol Lett. 2006, 27, 415–423. [Google Scholar]
- Morris, M.C.; Brockman, J.; Schneider, J.A.; Wang, Y.; Bennett, D.A.; Tangney, C.C.; van de Rest, O. Association of Seafood Consumption, Brain Mercury Level, and APOE epsilon4 Status With Brain Neuropathology in Older Adults. JAMA 2016, 315, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Marechal, J.D.; Wärmländer, S.; Gräslund, A.; Peralvarez-Marin, A. In silico analysis of the apolipoprotein E and the amyloid beta peptide interaction: Misfolding induced by frustration of the salt bridge network. PLoS Comput. Biol. 2010, 6, e1000663. [Google Scholar] [CrossRef] [PubMed]
- Napier, M.D.; Poole, C.; Satten, G.A.; Ashley-Koch, A.; Marrie, R.A.; Williamson, D.M. Heavy metals, organic solvents, and multiple sclerosis: An exploratory look at gene-environment interactions. Arch. Environ. Occup. Health 2016, 71, 26–34. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).