Robust Prediction of Single and Multiple Point Protein Mutations Stability Changes

 , , and
, , and
Abstract
:1. Introduction
2. Methods
2.1. Protein Datasets
2.2. Protein Mutation Prediction Methodology
2.2.1. Single Point Mutations
2.2.2. Multiple Point Mutations
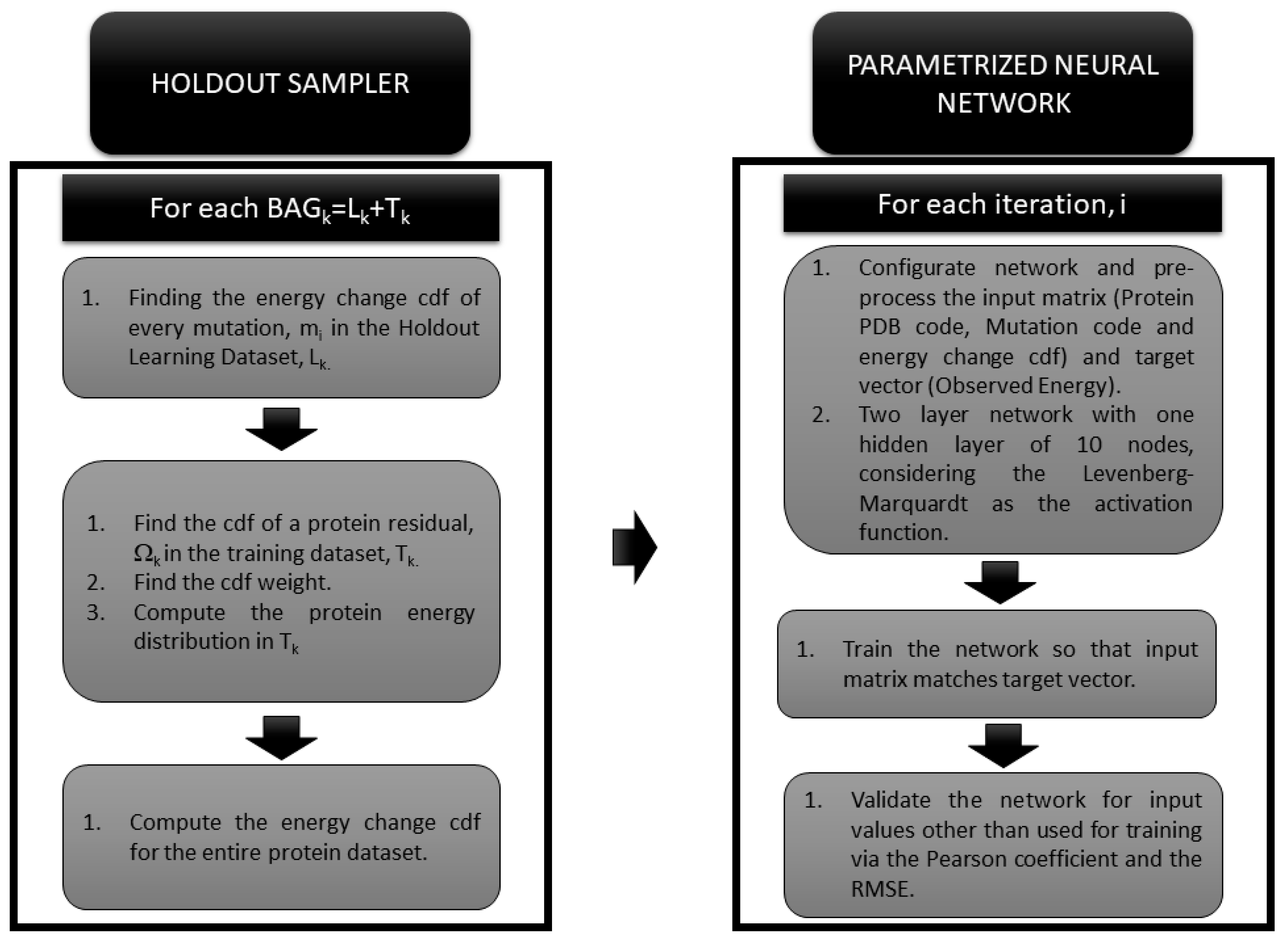
2.3. The Holdout Sampler-Based Uncertainty Predictor
2.4. The Neural Network Based Predictor
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Daggett, V.; Fersht, A.R. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 2003, 28, 18–25. [Google Scholar] [CrossRef]
- Casadio, R.; Compiani, M.; Fariselli, P.; Vivarelli, F. Predicting free energy contributions to the conformational stability of folded proteins from the residue sequence with radial basis function networks. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995, 3, 81–88. [Google Scholar]
- Kumar, M.; Bava, K.; Gromiha, M.; Prabakaran, P.; Kitajima, K.; Uedaira, H.; Sarai, A. ProTherm and Pronit: Thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic. Acids Res. 2005, 34, D204–D206. [Google Scholar] [CrossRef] [Green Version]
- Risch, N.J. Searching for genetic determinants in the new millennium. Nature 2000, 405, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting the effects of amino-acid substitutions on protein function. Annu. Rev. Genom. Hum. Genet. 2006, 7, 61–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, R.; Schwaneberg, U.; Roccatano, D. Computer-aided Protein Directed Evolution: A review of web servers, databases and other computational tools for protein engineering. Comput. Struct. Biotech. J. 2012, 2, e201209008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.I.; Bolon, D.N.A.; Tawfik, D.S. Quantifying and understanding the fitness effects of protein mutations: Laboratory versus nature. Protein Sci. 2016, 25, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Gnad, F.; Baucom, A.; Mukhyala, K.; Manning, G.; Zhang, Z. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genom. 2013, 14, S7. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, E.; Fariselli, P.; Casadio, R. A neural network-based method for predicting protein stability changes upon single point mutations. Bioinformatics 2004, 20, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of Protein Stability Changes for Single-Site Mutations Using Support Vector Machines. Proteins 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Lee, C. Testing homology modeling on mutant proteins: Predicting structural and thermodynamic effects in the ala98-val mutants of t4 lysozyme. Fold. Des. 1995, 1, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002, 11, 2714–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sippl, M.J. Knowledge based potentials for proteins. Curr. Opin. Stuct. Biol. 1995, 5, 229–235. [Google Scholar] [CrossRef]
- Prevost, M.; Wodak, S.; Tidor, B.; Karplus, M. Contribution of the hydrophobic effect to protein stability: Analysis based on simulations of the Ile-96-Ala mutation in barnase. Proc. Natl. Acad. Sci. USA 1991, 88, 10880–10884. [Google Scholar] [CrossRef] [Green Version]
- Topham, C.M.; Srinivasan, N.; Blunell, T.L. Prediction of the stability of protein mutants based on structural environment-dependent amino-acids substitution and propensity tables. Protein Eng. 1997, 10, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhou, Y. Quantifying the effect of burial of amino-acid residues on protein stability. Proteins 2004, 54, 315–322. [Google Scholar] [CrossRef]
- Gillis, D.; Rooman, M. Predicting protein stability changes upon mutation using database-derived potentials: Solvent accesibility determines the importance of local versus non-local interactions along the sequence. J. Mol. Biol. 1997, 272, 276–290. [Google Scholar] [CrossRef] [Green Version]
- Carter, C.W.; LeFebvre, B.C.; Cammer, S.A.; Torpsha, A.; Edgell, M.H. Four body potentials reveal protein specific correlations to stability changes caused by hydrophobic core mutations. J. Mol. Biol. 2001, 311, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Takano, K.; Ota, M.; Ogasahara, K.; Yamagata, Y.; Nishikawa, K.; Yutani, K. Experimental verification of the stability profile of mutant protein [spmp) data using mutant human lysozymes. Protein Eng. 1999, 12, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Domingues, H.; Peters, J.; Schneider, K.H.; Apeler, H.; Sebald, W.; Oschkinat, H.; Serrano, L. Improving the refolding yield of interleukin-4 through the optimization of local interactions. J. Biotechnol. 2000, 84, 217–230. [Google Scholar] [CrossRef]
- Funahashi, J.; Takano, K.; Yutani, K. Are the parameters of various stabilization factors estimated from mutant human lysozymes compatible with other proteins? Protein Eng. 2001, 14, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radestock, S.; Gohlke, H. Exploiting the Link between Protein Rigidity and Thermostability for Data Driven Protein Engineering. Eng. Life Sci. 2008, 8, 507–522. [Google Scholar] [CrossRef]
- Jacobs, D.; Rader, A.; Thorpe, M.; Kuhn, L. Protein Flexibility Predictions Using Graph Theory. Proteins 2001, 44, 150–165. [Google Scholar] [CrossRef]
- Fox, N.; Jagodzinski, F.; Li, Y.; Streinu, I. KINARI-Web: A server for protein rigidity analysis. Nucleic Acids Res. 2011, 39, W177–W183. [Google Scholar] [CrossRef]
- Jagodzinski, F.; Hardy, J.; Streinu, I. Using rigidity analysis to probe mutation-induced structural chagnes in proteins. J. Bioinf. Comput. Biol. 2012, 10, 1242010. [Google Scholar] [CrossRef]
- Jagodzinski, F.; Akbal-Delibas, B.; Haspel, N. An evolutionary Conservation & Rigidity Analysis Machine Learning Approach for Detecting Critical Protein Residues. In Proceedings of the ACM International Conference on Bioinformatics, Computational Biology and Biomedical Informatics (ACM-BCB), Washington, DC, USA, 22–25 September 2013; pp. 780–786. [Google Scholar]
- Dehghanpoor, R.; Ricks, E.; Hursh, K.; Gunderson, S.; Farhoodi, R.; Haspel, N.; Hutchinson, B.; Jagodzinski, F. Predicting the Effect of Single and Multiple Mutations on Protein Structural Stability. Molecules 2018, 23, 251. [Google Scholar] [CrossRef] [Green Version]
- Worth, C.; Preissner, R.; Blundell, L. SDM—A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [CrossRef] [Green Version]
- Brender, J.R.; Zhang, Y. Predicting the effect of mutations on protein-protein binding interactions through structure-based interface profiles. PLoS Comput. Biol. 2015, 11, e1004494. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Xing, P.; Shi, G.; Ji, Z.; Zou, Q. Fast prediction of protein methylation sites using a squence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Tang, J.; Zou, Q. PhosPred-RF: A novel Sequence Based Predictor for Phosphorylation Sites using Sequential Information Only. IEEE Trans. Nanobiosci. 2017, 16, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Duan, Y.; Zou, Q. HPSLPred: An Ensemble Multi-Label Classifier for Human Protein Subcellular Location Prediction with Imbalanced Source. Proteomics 2017, 17, 1700262. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Yarlagadda, R.; Reed, C.C. Structure Based Thermostability Prediction Models for Protein Single Point Mutations with Machine Learning Tools. PLoS ONE 2015, 10, e0138022. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Fang, J. PROTS-RF: A robust model for predicting mutation-induced protein stability changes. PLoS ONE 2012, 7, e47247. [Google Scholar] [CrossRef]
- Wolpert, D.H. Stacked generalization. Neural Netw. 1992, 5, 241–259. [Google Scholar] [CrossRef]
- Breiman, L. Stacked regressions. Mach. Learn. 1996, 24, 49–64. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, M.; Tibshirani, R. Combining estimates in regression and classification. J. Am. Stat. Assoc. 1996, 91, 1641–1650. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L.; Fernández-Muñiz, Z.; Breysse, D. The uncertainty analysis in linear and nonlinear regression revisited: Application to concrete strength estimation. Inverse Probl. Sci. Eng. 2018. [Google Scholar] [CrossRef]
- Fernández-Muñiz, Z.; Hassan, K.; Fernández-Martínez, J.L. Data kit inversion and uncertainty analysis. J. Appl. Geophys. 2019, 161, 228. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L.; Cernea, A.; deAndrés-Galiana, E.J.; Fernández-Ovies, F.J.; Fernández-Muñiz, Z.; Alvarez-Machancoses, O.; Saligan, L.; Sonis, S.T. Sampling Defective Pathways in Phenotype Prediction Problems via the Holdout Sampler. Bioinformatics and Biomedical Engineering. In Proceedings of the International Conference on Bioinformatics and Biomedical Engineering IWBBIO 2018, Granada, Spain, 25–27 April 2018; pp. 24–32. [Google Scholar]
- Abdulla, B.K.; Gromiha, M.M.; Uedaira, H.; Kitajima, K.; Sarai, A. ProTherm, version 4.0: Thermodynamic database for proteins and mutants. Nucleic Acids Res. 2004, 32, D120. [Google Scholar]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Efron, B.; Tibshirani, R. An Introduction to the Bootstrap, 1st ed.; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Jain, A.K.; Mao, J.; Mohiuddin, K.M. Artificial Neural Networks: A tutorial. Computer 1996, 29, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, P.D. Advanced Methods in Neural Computing; John Willey & Sons, Inc.: New York, NY, USA, 1993. [Google Scholar]
- Moré, J.J. The Levenberg-Marquardt algorithm: Implementation and theory. Numer. Anal. 1978, 630, 105–116. [Google Scholar]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. Predict SNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [Green Version]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Pokala, N.; Handel, T.M. Energy functions for protein design: Adjustment with protein–protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J. Mol. Biol. 2005, 347, 203–227. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Tai, D.; Middaugh, C.R.; Zhang, Y.; Fang, J. Prots: A fragment based protein thermo-stability potential. Proteins Struct. Funct. Bioinform. 2012, 80, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef] [Green Version]
- Farhoodi, R.; Shelbourne, M.; Hsieh, R.; Haspel, N.; Hutchinson, B.; Jagodzinski, F. ACM. Predicting the Effect of Point Mutations on Protein Structural Stability. Comput. Biology Health Inform. 2017. [Google Scholar] [CrossRef]
- Wainreb, G.; Wolf, L.; Ashkenazy, H.; Dehouck, Y.; Ben-Tal, N. Protein stability: A single recorded mutation aids in predicting the effects of other mutations in the same amino acid site. Bioinformatics 2011, 27, 3286–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witvliet, D.K.; Strokach, A.; Giraldo-Forero, A.F.; Teyra, J.; Colak, R.; Kim, P.M. ELASPIC web-server: Proteome-wide structure-based prediction of mutation effects on protein stability and binding affinity. Bioinformatics 2016, 32, 1589–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frappier, V.; Chartier, M.; Najmanovich, R.J. ENCoM server: Exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res. 2015, 43, W295–W400. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2013, 30, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, O.; Fernández-Martínez, J.L. The importance of Biological Invariance in Drug Design. Biomed. J. Sci. Tech. Res. 2019, 18, 13211–13212. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Highest Reported Pearson Coefficient (R) | Reference |
|---|---|---|
| Holdout-NN Method | 0.77 | |
| Regression with RF | 0.66 | Li et al. [36] |
| MUpro | 0.48 | Cheng et al. [10] |
| I-Mutant 2.0 | 0.54 | Capriotti et al. [51] |
| LSE | 0.16 | Jia et al. [35] |
| FoldX | 0.50 | Schymkowitz et al. [52] |
| EGAD | 0.60 | Pokala et al. [53] |
| PROTS | 0.40 | Li et al. [54] |
| PopMuSiC-2.0 | 0.62 | Dehouck et al. [55] |
| Prethemut | 0.72 | Farhoodi et al. [56] |
| ProMaya | 0.74 | Wainreb et al. [57] |
| ELASPIC | 0.77 | Witvliet et al. [58] |
| SDM2 | 0.52 | Pandurangan et al. [31] |
| ENCoM | 0.44 | Frappier et al. [59] |
| DynaMut | 0.67 | Rodrigues et al. [50] |
| mCSM | 0.76 | Pires et al. [60] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Machancoses, Ó.; De Andrés-Galiana, E.J.; Fernández-Martínez, J.L.; Kloczkowski, A. Robust Prediction of Single and Multiple Point Protein Mutations Stability Changes. Biomolecules 2020, 10, 67. https://doi.org/10.3390/biom10010067
Álvarez-Machancoses Ó, De Andrés-Galiana EJ, Fernández-Martínez JL, Kloczkowski A. Robust Prediction of Single and Multiple Point Protein Mutations Stability Changes. Biomolecules. 2020; 10(1):67. https://doi.org/10.3390/biom10010067
Chicago/Turabian StyleÁlvarez-Machancoses, Óscar, Enrique J. De Andrés-Galiana, Juan Luis Fernández-Martínez, and Andrzej Kloczkowski. 2020. "Robust Prediction of Single and Multiple Point Protein Mutations Stability Changes" Biomolecules 10, no. 1: 67. https://doi.org/10.3390/biom10010067