1. Introduction
The process of transcription is the primary means by which all organisms control their specific gene expression. This is achieved through the binding of sets of proteins known as transcription factors to specific DNA sequences often located within the promoter regions of regulated genes. Identifying those genes can provide valuable information regarding a transcription factor and its biological role in an organism’s physiology.
Advances in DNA sequencing techniques have allowed the determination of genomic sequences for over 370 thousand organisms [
1]. Within these, dozens to hundreds of potential transcription factors can be identified using bioinformatics tools. However, it is beyond our present ability to use this sequence information to accurately predict those DNA sequences recognized by a presumed transcription factor and the target genes it regulates. Simple inferences, e.g., assuming transcription factor autoregulation of its own and opposing operons and recognition of palindromic sequences by homodimeric transcription factors, can provide useful leads as to the specific DNA sequences recognized by these proteins and the types of genes that they regulate [
2]. However, a more thorough understanding typically requires experimentally derived data.
In organisms for which we have tractable genetic tools, an approach involving transcription factor gene disruption, measured changes in global gene expression, and bioinformatic homology studies can be used to obtain information ranging from defining regulatory regions to potential biological roles for relatively uncharacterized transcription factors. Such an approach has proven highly effective in the model organism
Escherichia coli, where information regarding its transcription factors, their consensus binding sequences, and the genes that they regulate, has been obtained [
3].
Unfortunately, genetic tools are not always practical in all organisms for which genomic information is available. Thus, there exists a need for alternative approaches for characterizing transcription factors in these organisms. We have developed an alternative, biochemistry-based approach, using the selection method Restriction Endonuclease Protection, Selection, and Amplification (REPSA), massively parallel sequencing, and bioinformatics to determine a consensus binding sequence and thereby identify possible genes regulated by these transcription factors. We have successfully used this approach to investigate three putative TetR-family transcription factors, TTHA0167, TTHA0101, and TTHA0973, in the extreme thermophile
Thermus thermophilus HB8 [
4,
5,
6]. Here, we describe our investigations into the DNA-binding specificity and genomic targets of TTHB023, a putative TetR-related transcriptional repressor protein in
T. thermophilus HB8. This study further improves the use of REPSA in identifying consensus binding sequences and provides further insights into potential transcription factor functions in an important model organism [
7].
3. Discussion
In this paper, we report our biochemistry-based investigation on the
T. thermophilus HB8 transcription factor TTHB023. Using REPSA selection, massively parallel sequencing, and MEME motif elicitation, we found that TTHB023 preferentially binds the consensus sequence, 5′–
(a/c)Y(g/t)A(A/C)YGryCR(g/t)T(c/a)R(g/t)–3′. In comparison, Agari et al. functionally identified TTHB023-regulated genes based on their magnitude change in expression when comparing mRNAs isolated from wild-type and Δ
pfmR strains [
18]. Comparing the nucleotide sequences upstream of 30 candidate genes, they found three,
TTHA0750,
TTHA0987, and
TTHB023, containing similar pseudopalindromic sequences in their upstream regions. These sequences allowed them to derive a predicted consensus TTHB023-binding site, 5′–TA
CCGACCGNTNGGTN–3′. Notably, there is a significant degree of overlap between our consensus sequence and that derived by Agari et al. (see underlining, above), suggesting that they both encompass bona fide TTHB023 recognition sequences. However, given the considerable difference in sample size (992 versus 3) and statistical significance (E-values 7.2 × 10
−1942 versus 9.3 × 10
−6) between the two studies, the consensus we report is likely to be a more accurate representation of the spectrum of DNA-binding sites preferentially recognized by TTHB023.
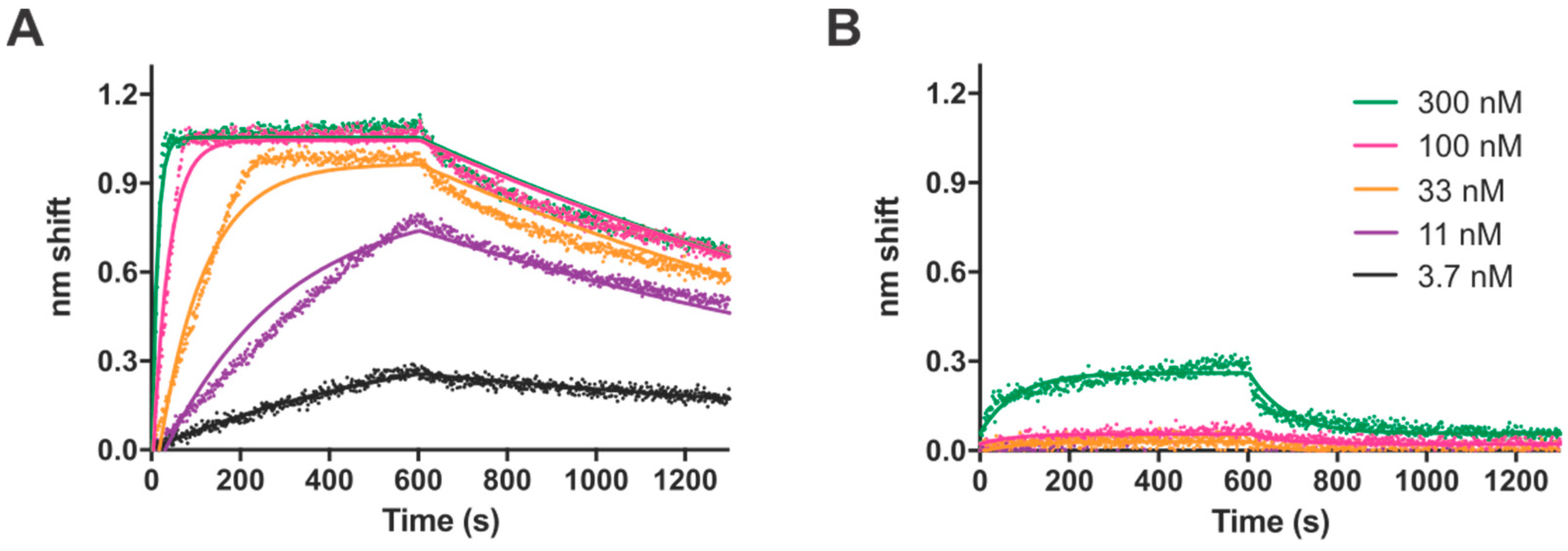
Having defined a consensus recognition sequence, we sought to characterize the TTHB023-DNA interaction biophysically through the use of BLI. DNA probes containing either the consensus sequences or selected point mutations were analyzed, to better ascertain the importance of each nucleotide within the consensus. We found a dissociation constant of 2 nM for the consensus sequence and a range of 4.5 to 279 nM for the different point mutants. Notably, these dissociation constants closely mirrored the statistical significance of each nucleotide position from the MEME-derived position-weight matrix, as represented in the TTHB023 palindromic sequence logo. Thus, these data validate the sequences obtained by our REPSA-based approach as being those DNAs with high-affinity TTHB023 binding sites. Agari et al. performed a much more limited biophysical analysis of TTHB023-DNA binding, choosing to only investigate TTHB023 binding to its own gene’s upstream region using the technique surface plasmon resonance [
18]. Most important, they found similar
kon (5.6 ± 1.1 × 10
5 M
−1·s
−1),
koff (4.3 ± 0.3 × 10
−3 s
−1), and K
D (7.9 ± 1.4 × 10
−9 M) values to what we observed, giving additional credence that the high-affinity binding we both found may be an accurate representation of TTHB023-binding parameters under physiological conditions.
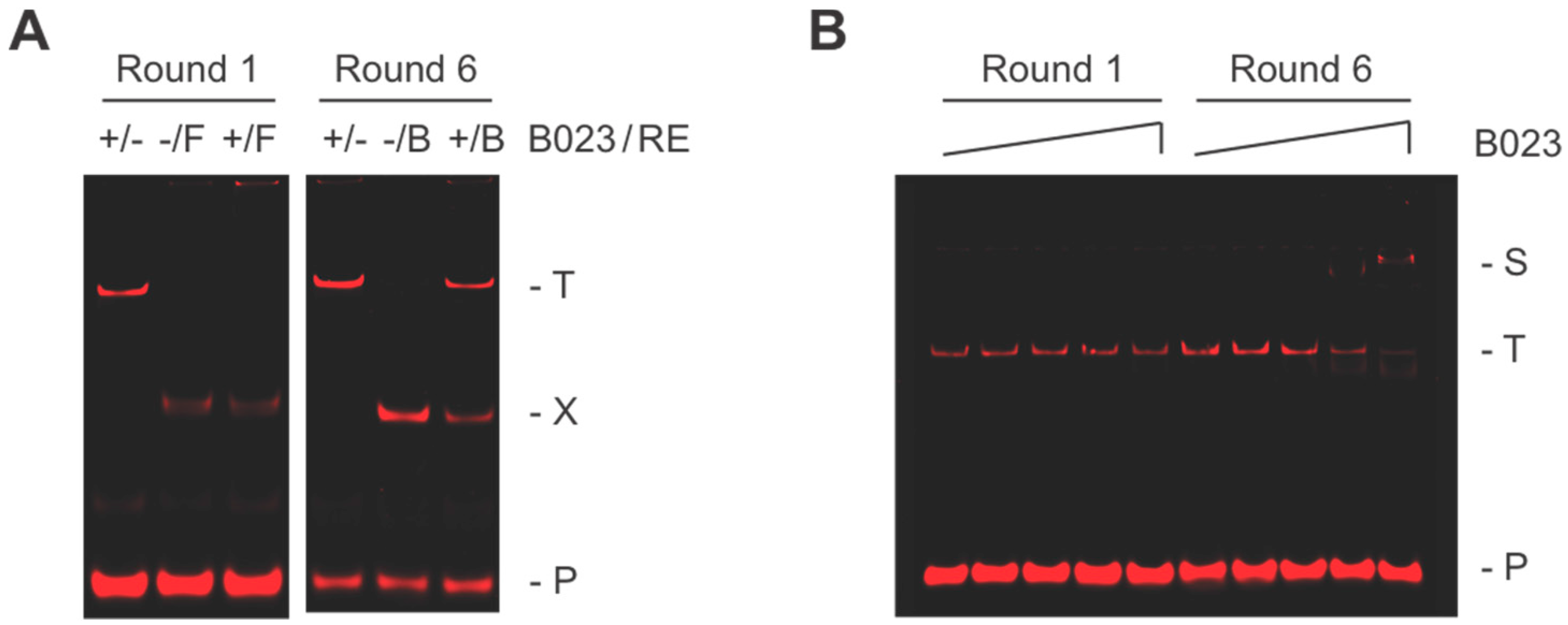
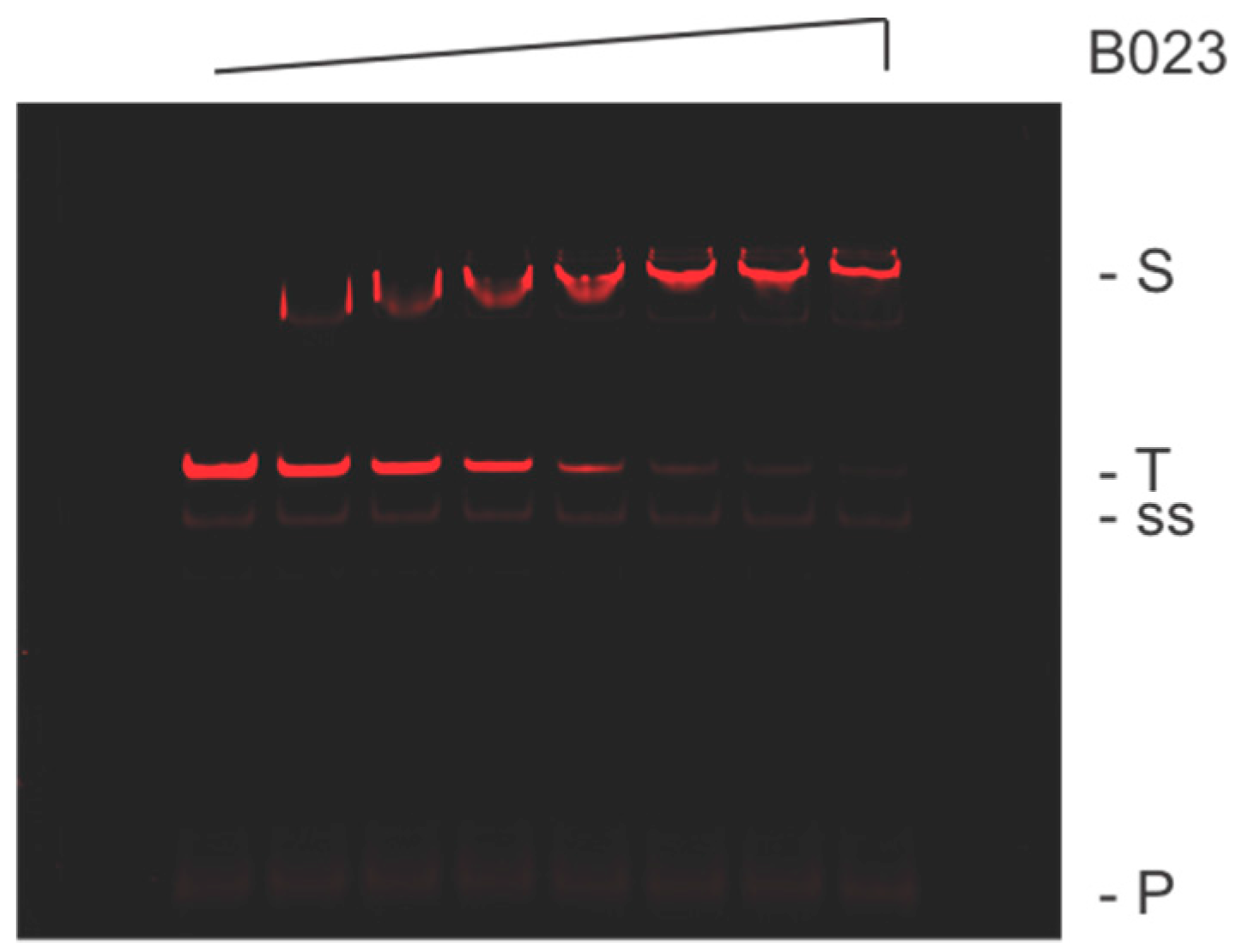
In our biochemistry-based approach, we use a consensus sequence and bioinformatics to identify potential protein binding sites within an organism’s genome. With TTHB023, we initially obtained 96 motif occurrences, which were then reduced to nine based on their homology with the consensus sequence (
p-value ≤ 5.10 × 10
−6) and location within an identifiable promoter region. These nine included two bi-directional promoter regions (
TTHA1315/
16,
TTHA1605/
06) and two promoters (
TTHA0987, TTHB023) containing two TTHB023 binding sites each. Interestingly, four of these sites overlapped with those identified by Agari et al. (
Figure 5, indicated by underlining) [
18]. Our sites were validated using BLI, which found that all but the
TTHA1605/06 shared site exhibited high-affinity TTHB023 binding (K
D from 0.3 to 7.4 nM) comparable to that observed with the consensus sequence. These data strongly suggest that TTHB023 could bind and regulate these promoters in vivo. Special note should be made regarding the location of TTHB023 binding within the promoter region. In many cases (e.g.,
TTHA0987, TTHA0750, TTHB023), the TTHB023-binding sites encompassed their mapped start sites of transcription (+1 site). Such would be expected for a transcriptional repressor that hinders the process of promoter-bound RNA polymerase transitioning to a productive transcriptional state rather than blocking RNA polymerase-promoter access. Thus, TTHB023 may behave more like the QacR regulator in
Staphylococcus aureus than a typical TetR-family transcriptional repressor [
19,
20].
Given that TTHB023 binding sites typically overlapped with core promoter elements (−35 and −10 boxes, +1 site) and that TTHB023 is structurally related to the TetR transcriptional repressor protein, it is quite likely that TTHB023 functions as a transcriptional repressor. Agari et al. used in vitro transcription assays to validate TTHB023 function on the promoters they identified [
18]. In accordance with a role as a transcriptional repressor, they found decreasing transcription with increasing TTHB023 concentration. However, rather high (100 to 500 nM) TTHB023 concentrations were necessary in order to observe appreciable inhibition on most promoters, and similar transcription suppression was observed on a promoter (
TTHA0973) not thought to have a TTHB023 binding site. These findings demonstrate that while validation of transcription factor function on a promoter is important, methods like in vitro transcription have their limitations.
We used available microarray data from wild-type and Δ
pfmR strains of
T. thermophilus HB8 to identify those genes whose expression is most affected by the loss of TTHB023 under normal growth conditions [
13]. We found three of the genes we identified,
TTHB023,
TTHA0750, and
TTHA0987, to be among the top 20 genes that exhibited the greatest change in transcript levels between wild-type and deletion strains.
TTHB023 exhibited a significant (365-fold) decrease in transcripts, as expected, given that this was the gene disrupted in the Δ
pfmR strains.
TTHA0750 and
TTHA0987 both exhibited moderate (3.6 to 3.9-fold) increases in expression, consistent with TTHB023 serving as a transcriptional repressor on their promoters. Further analysis of downstream genes being affected by
TTHB023 depletion found others in the operon
TTHB023–
TTHB014 that were also upregulated, albeit at only moderate levels (1.9 to 3.6-fold). Such is not entirely unexpected, given that prokaryotic transcriptional repressors often suppress the expression of their operons, as part of a negative-feedback regulation loop [
2,
21]. Examining the reported roles of the
TTHB023–
TTHB014 gene products, one finds that many are enzymes potentially involved in fatty acid metabolism. Agari et al. took this information one further, stating that these enzymes may be involved in phenylacetic acid degradation and fatty acid degradation and biosynthesis [
18]. Thus, they refer to TTHB023 as the regulator of
phenylacetic acid and
fatty acid
metabolism, or PfmR.
In our analysis of the
T. thermophilus HB8 genome, we found two gene pairs,
TTHA1315/16 and
TTHA1606/06, that contained potential TTHB023 binding sites within their promoter regions but were not identified by Agari et al. [
18]. Such is understandable as their expression was not appreciably affected when
TTHB023 was deleted. We do not believe that
TTHA1606/06 is an actual target of TTHB023, given its relatively weak binding affinity. However,
TTHA1315/16 does possess an intermediate affinity TTHB023 binding affinity site, making these genes potential targets. One complication with standard genetic approaches for identifying targets of transcription factors is that not all genes have simple regulation programs, i.e., are regulated by a single transcription factor. Many are combinatorially regulated, involving both transcriptional repressors and activators [
21]. Thus, depletion of a single transcription factor may have, at best, only moderate effects. In the case of
TTHA1315/16, we believe multiple transcriptional repressors may be involved in their regulation. In fact, we had previously shown that these genes are likely targets of another
T. thermophilus HB8 TetR-family transcriptional repressor, TTHA0167, otherwise known as SbtR [
4,
22]. We found that the consensus sequence for TTHA0167 is 5′–TGACYrnnyRGTCA–3′, which can be accommodated by the central 14 nucleotides of the TTHB023 consensus sequence in many cases. We have performed BLI measurements with TTHA0167 on the TTHB023 binding sites found in the
TTHA1315/16 and
TTHA1606/06 promoter regions and found evidence for high-affinity binding, with K
D values in the 1–2 nM range (
Table 6). These data strongly suggest that the
TTHA1315 and
TTHA1316 genes may be regulated by both the TTHA0167 and TTHB023 transcriptional repressors, requiring multiple effectors to be present to permit their maximal expression in vivo. This co-ordinate regulation may also help explain why previous investigators did not report
TTHA1315 and
TTHA1316 being regulated by TTHA0167 in vivo, even though they well recognized TTHA0167 binding to these gene promoters in genomic selection experiments [
22]. We were able to confirm this using publicly available microarray data and GEO2R, finding that
TTHA1315 and
TTHA1316 are not among the top 250 genes whose expression is affected by
TTHA0167 deletion (
Table S3). Taken together, these examples point to an inherent strength of a biochemistry-focused approach for transcription factor discovery, especially in cases involving complicated regulatory schemes.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}