Role of Glutathione in Cancer: From Mechanisms to Therapies

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. ROS Generation and Signaling
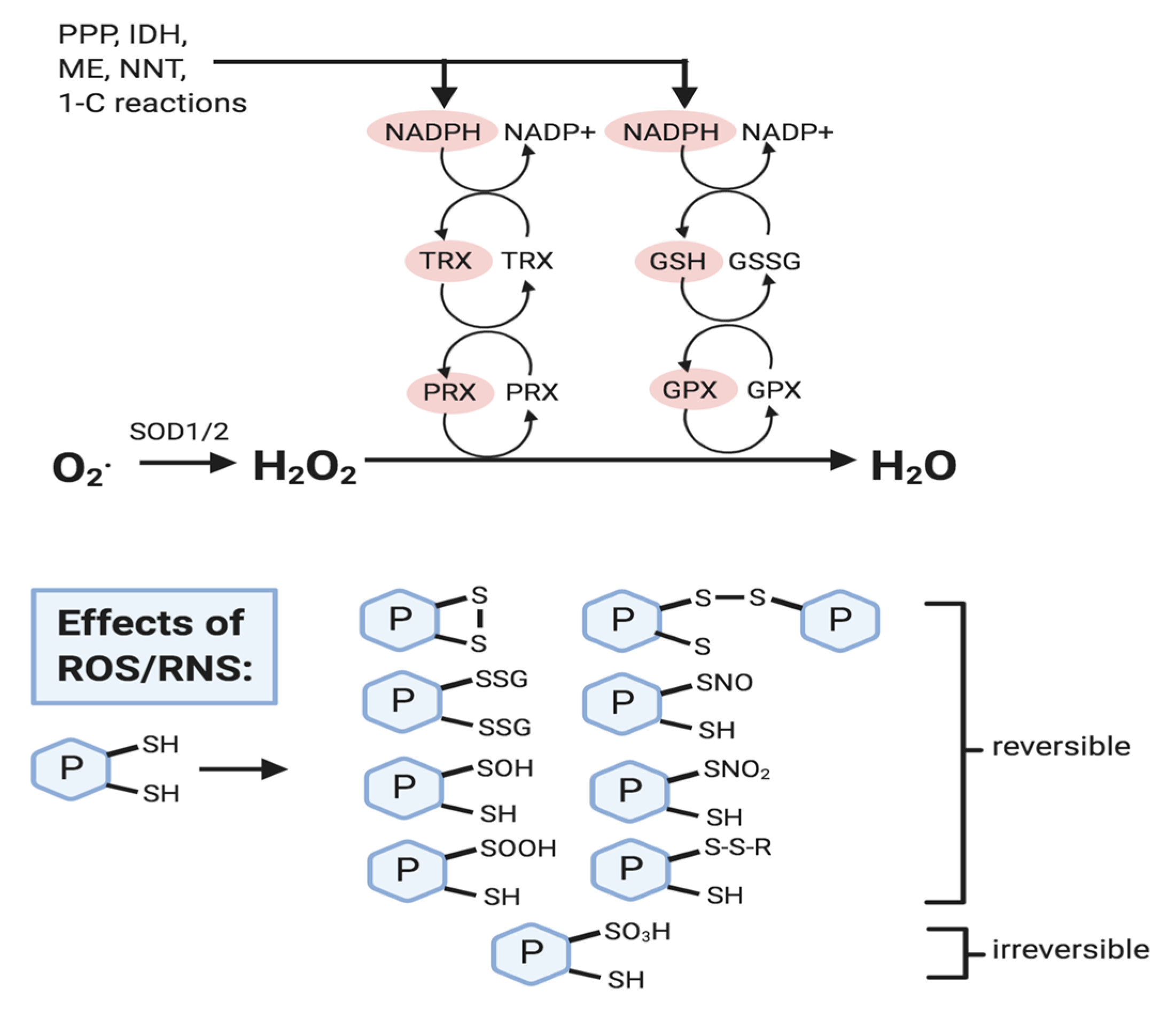
2.1. ROS Control Mechanisms: Striking a Fine Balance
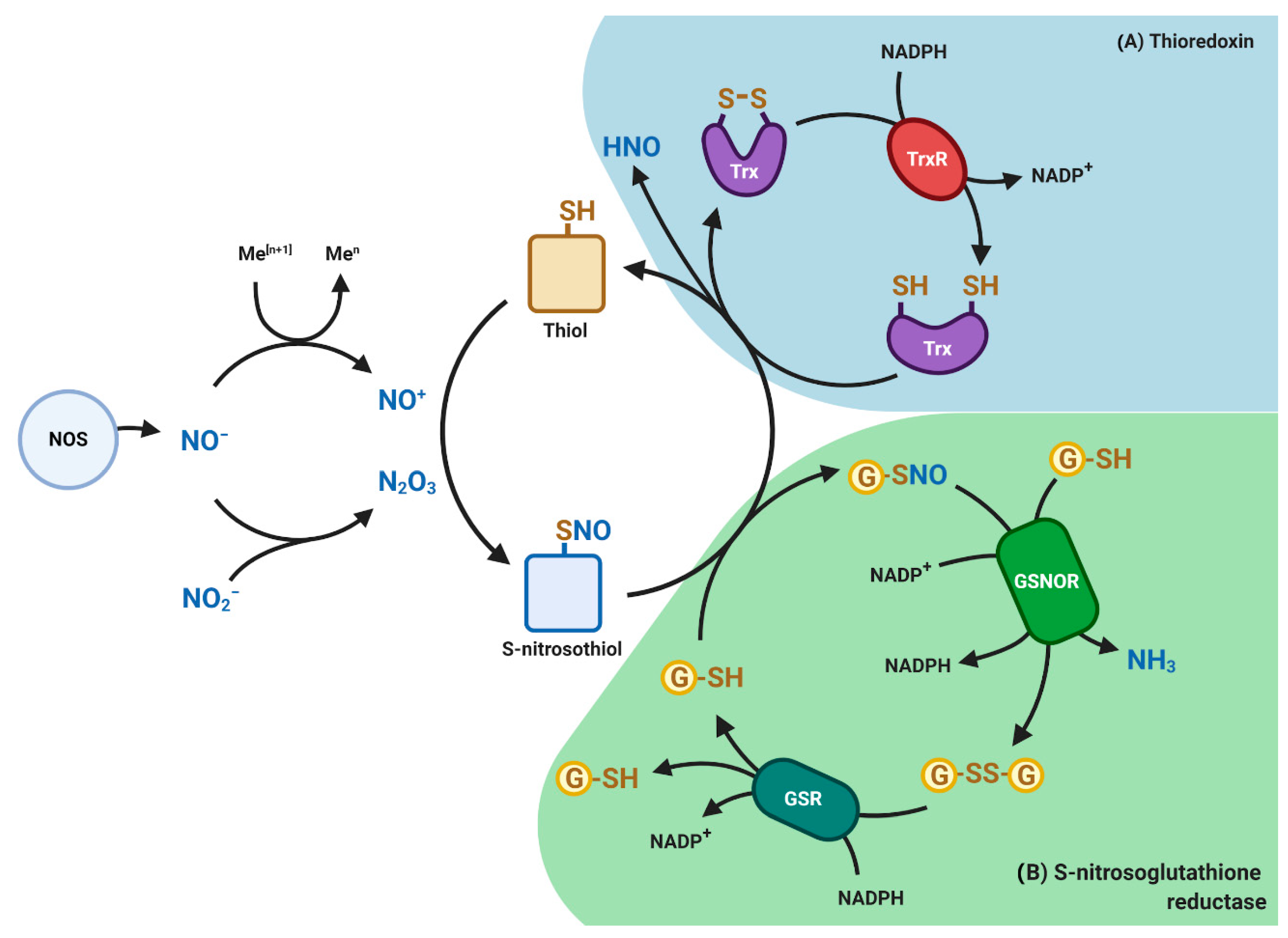
2.2. RNS Control Mechanisms
3. Glutathione, Redox and ROS in Healthy and Tumor Cells
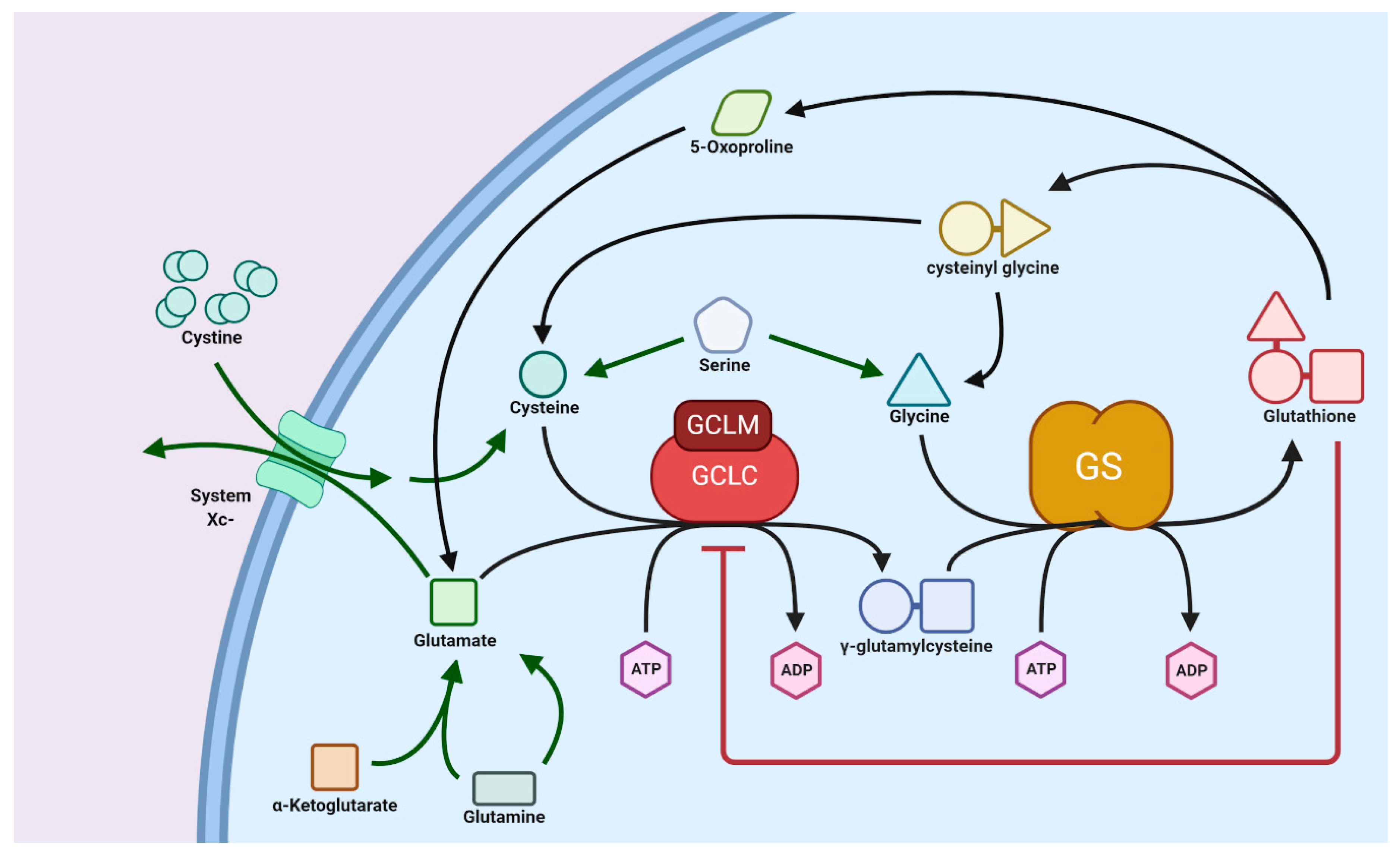
3.1. Glutathione Synthesis: Normal versus Tumor Cells
3.2. Transport of GSH
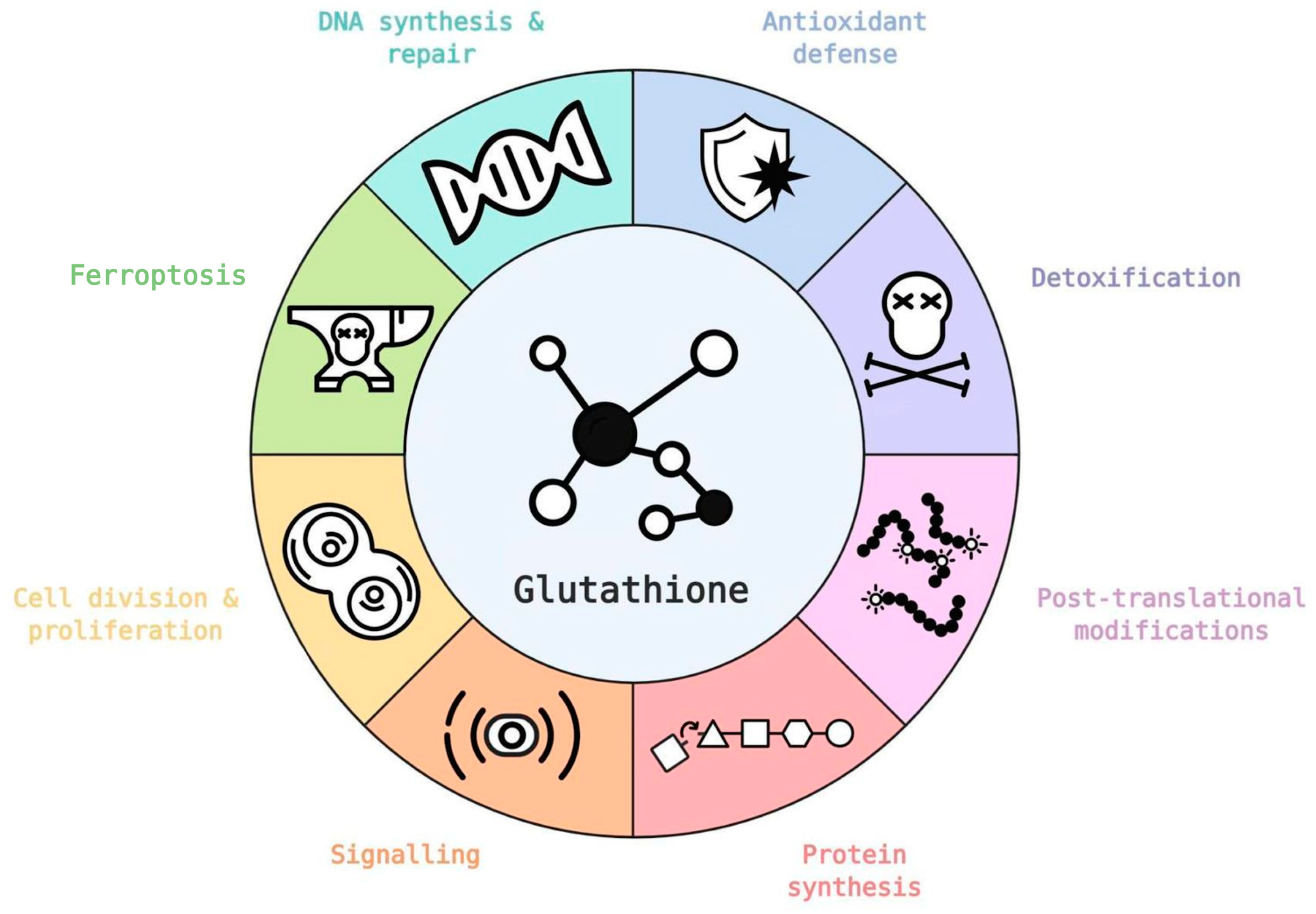
4. Glutathione as an Antioxidant
Glutathione S-Transferase
5. Glutathione, Inflammation, and Cancer
5.1. Role of Peroxiredoxin (PRDX)
5.2. Role of Glutathione Peroxidases (GPXs)
5.3. Role of S-Nitrosation
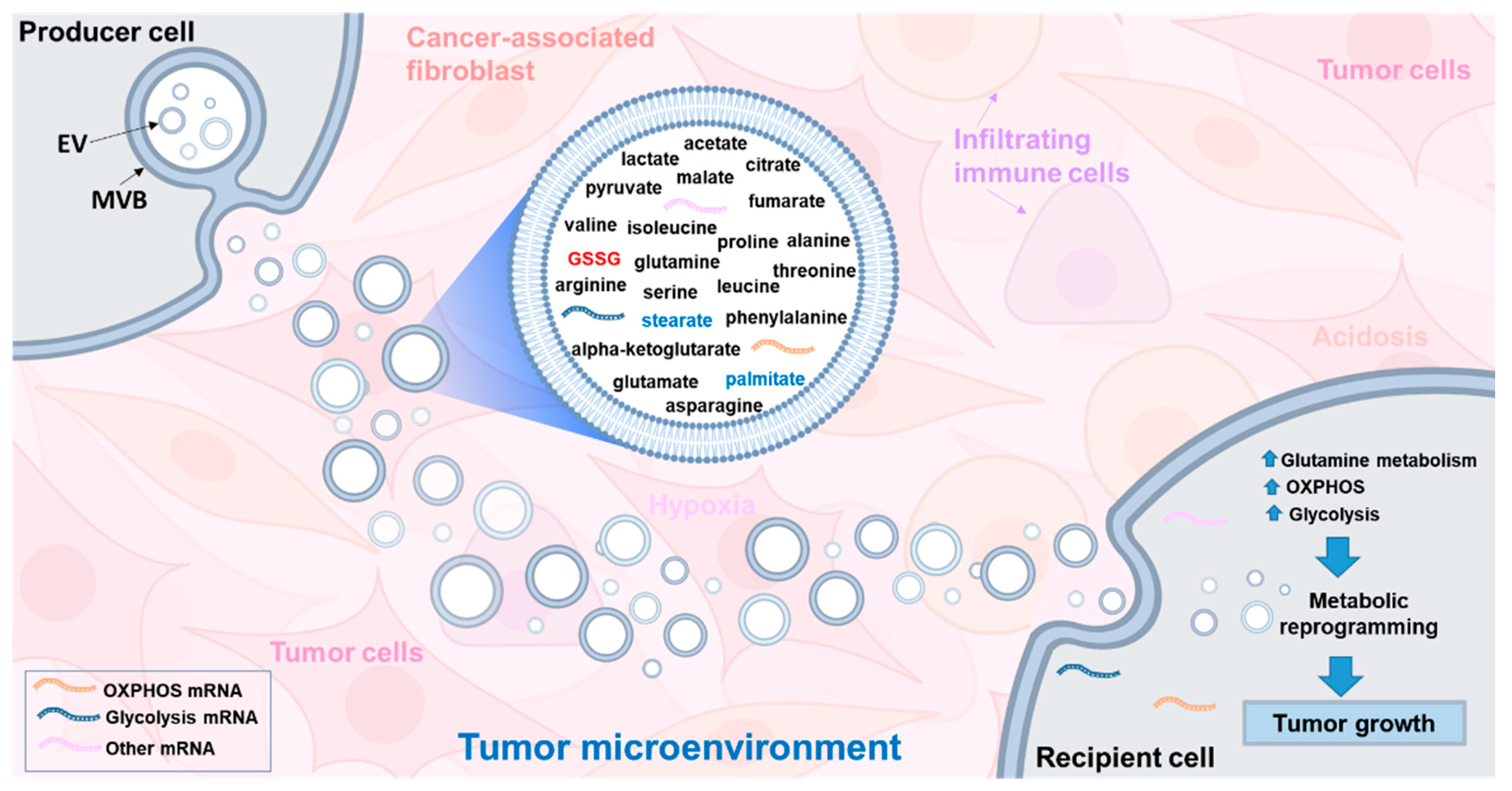
6. Glutathione and Tumor Microenvironment
7. Examples of Glutathione-Related Tumor Therapies
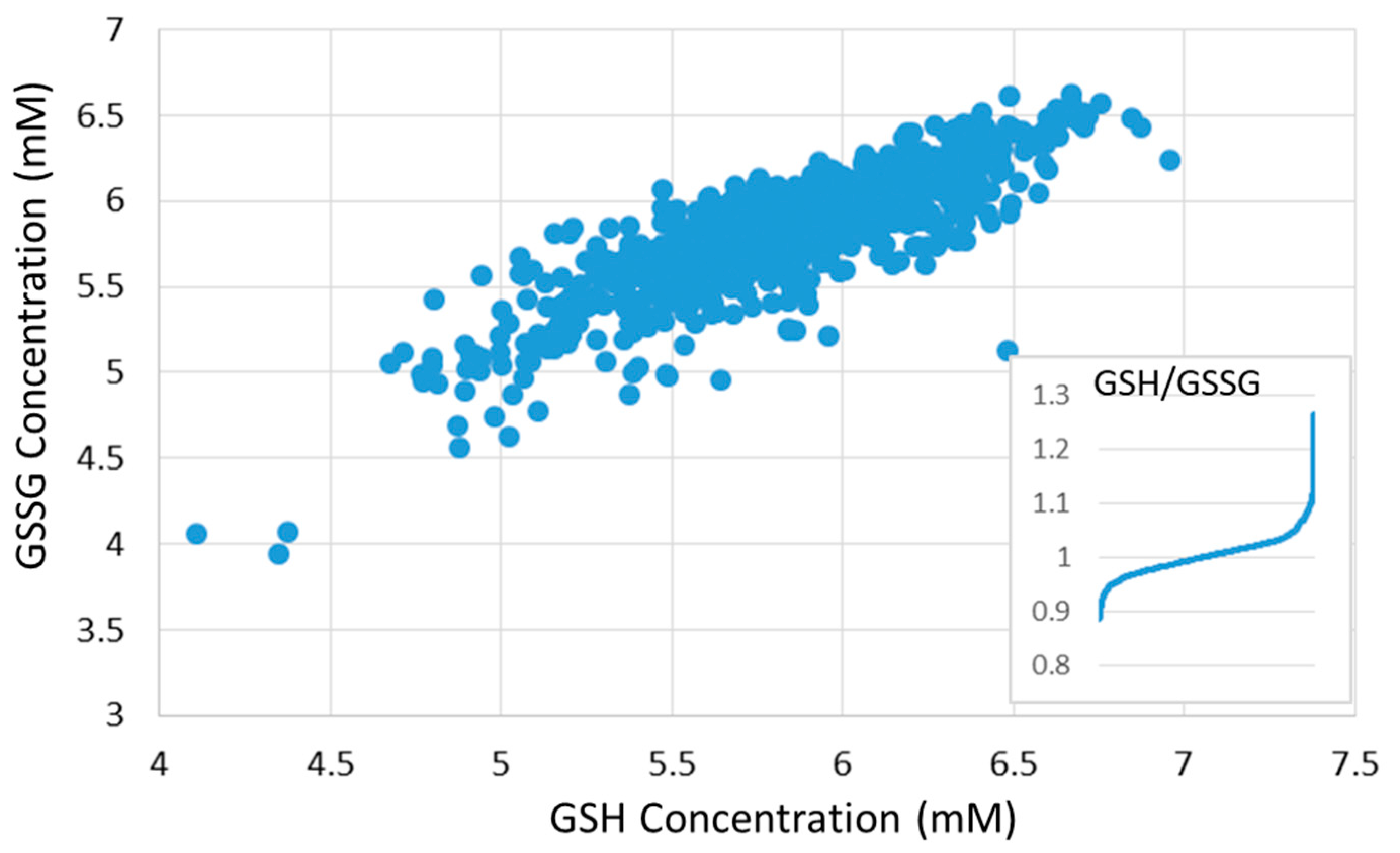
8. Computer Modeling of GSH Metabolism
9. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef]
- Vyssokikh, M.Y.; Holtze, S.; Averina, O.A.; Lyamzaev, K.G.; Panteleeva, A.A.; Marey, M.V.; Zinovkin, R.A.; Severin, F.F.; Skulachev, M.V.; Fasel, N.J.; et al. Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc. Natl. Acad. Sci. USA 2020, 117, 6491–6501. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M.-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Board, P.G.; Menon, D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim. Biophys. Acta 2013, 1830, 3267–3288. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Gill, R.; Mailloux, R.J. Protein S-glutathionylation: The linchpin for the transmission of regulatory information on redox buffering capacity in mitochondria. Chem. Biol. Interact. 2019, 299, 151–162. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Superoxide as an intracellular radical sink. Free Radic. Biol. Med. 1993, 14, 85–90. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Nóbrega-Pereira, S. NADPH: New oxygen for the ROS theory of aging. Oncotarget 2016, 7, 50814–50815. [Google Scholar] [CrossRef]
- Smith, C.D.; Schmidt, C.A.; Lin, C.-T.; Fisher-Wellman, K.H.; Neufer, P.D. Flux through mitochondrial redox circuits linked to nicotinamide nucleotide transhydrogenase generates counterbalance changes in energy expenditure. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Kitajima, T.; Hara, K. A model of the mechanism of cooperativity and associativity of long-term potentiation in the hippocampus: A fundamental mechanism of associative memory and learning. Biol. Cybern. 1991, 64, 365–371. [Google Scholar] [CrossRef]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef]
- Mailloux, R.J.; McBride, S.L.; Harper, M.-E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 2013, 38, 592–602. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Xuan, J.Y.; Beauchamp, B.; Jui, L.; Lou, M.; Harper, M.-E. Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J. Biol. Chem. 2013, 288, 8365–8379. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, A.; Mailloux, R.J.; Adjeitey, C.N.-K.; Harper, M.-E. Glutathionylation of UCP2 sensitizes drug resistant leukemia cells to chemotherapeutics. Biochim. Biophys. Acta 2013, 1833, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol. 2020, 32, 101472. [Google Scholar] [CrossRef] [PubMed]
- Scirè, A.; Cianfruglia, L.; Minnelli, C.; Bartolini, D.; Torquato, P.; Principato, G.; Galli, F.; Armeni, T. Glutathione compartmentalization and its role in glutathionylation and other regulatory processes of cellular pathways. BioFactors 2018, 45, 152–168. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.-W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.T.; Stamler, J.S. Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 2012, 287, 4411–4418. [Google Scholar] [CrossRef]
- Rizza, S.; Filomeni, G. Chronicles of a reductase: Biochemistry, genetics and physio-pathological role of GSNOR. Free Radic. Biol. Med. 2017, 110, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Furuta, S. Basal S-Nitrosylation Is the Guardian of Tissue Homeostasis. Trends Cancer Res. 2017, 3, 744–748. [Google Scholar] [CrossRef]
- Cichon, M.A.; Radisky, D.C. ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-kB-dependent activation of Snail. Oncotarget 2014, 5, 2827–2838. [Google Scholar] [CrossRef]
- Choi, J.-A.; Lee, J.-W.; Kim, H.; Kim, E.-Y.; Seo, J.-M.; Ko, J.; Kim, J.-H. Pro-survival of estrogen receptor-negative breast cancer cells is regulated by a BLT2–reactive oxygen species-linked signaling pathway. Carcinog 2009, 31, 543–551. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Talbott, S.J.; Rojanasakul, Y.; Nimmannit, U.; Pongrakhananon, V.; Wang, L.; Chanvorachote, P. Regulation of Lung Cancer Cell Migration and Invasion by Reactive Oxygen Species and Caveolin-1. J. Biol. Chem. 2010, 285, 38832–38840. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Chandel, N.S. Regulation of redox balance in cancer and T cells. J. Biol. Chem. 2018, 293, 7499–7507. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dange ROS liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet. 2011, 20, 4605–4616. [Google Scholar] [CrossRef]
- Iommarini, L.; Kurelac, I.; Capristo, M.; Calvaruso, M.A.; Giorgio, V.; Bergamini, C.; Ghelli, A.; Nanni, P.; De Giovanni, C.; Carelli, V.; et al. Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum. Mol. Genet. 2014, 23, 1453–1466. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharm. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci. Rep. 2019, 9, 17639. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef]
- Noun Project. Available online: https://thenounproject.com/ (accessed on 15 September 2020).
- Lien, E.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumor suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Vernier, M.; Dufour, C.R.; McGuirk, S.; Scholtes, C.; Li, X.; Bourmeau, G.; Kuasne, H.; Park, M.; St-Pierre, J.; Audet-Walsh, E.; et al. Estrogen-related receptors are targetable ROS sensors. Genes Dev. 2020, 34, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M.; Cormier, K.; Touaibia, M.; Reyjal, J.; Robichaud, S.; Belbraouet, M.; Turcotte, S. 1H NMR metabolomics analysis of renal cell carcinoma cells: Effect of VHL inactivation on metabolism. Int. J. Cancer 2016, 138, 2439–2449. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the γ-glutamyl cycle. Iubmb Life 2018, 70, 585–592. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Yuan, J.; Zhang, Y.; Jin, X. Altered glutamate cysteine ligase expression and activity in renal cell carcinoma. Biomed. Rep. 2014, 2, 831–834. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Ortmayr, K.; Dubuis, S.; Zampieri, M. Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun. 2019, 10, 1841. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M.; Ferguson, D.; Culf, A.; Morin, P., Jr.; Touaibia, M. 1H NMR metabolomics analysis of glioblastoma subtypes: Correlation between metabolomics and gene expression characteristics. J. Biol. Chem. 2012, 287, 20164–20175. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ning, S.; Ghandi, M.; Kryukov, G.V.; Gopal, S.; Deik, A.; Souza, A.; Pierce, K.; Keskula, P.; Hernandez, D.; et al. The landscape of cancer cell line metabolism. Nat. Med. 2019, 25, 850–860. [Google Scholar] [CrossRef]
- Park, J.O.; Rubin, S.A.; Xu, Y.-F.; Amador-Noguez, D.; Fan, J.; Shlomi, T.; Shlomi, T.; Rabinowitz, J.D. Metabolite concentrations, fluxes, and free energies imply efficient enzyme usage. Nat. Chem. Biol. 2016, 12, 482. [Google Scholar] [CrossRef]
- Jeong, E.M.; Yoon, J.-H.; Lim, J.; Shin, J.-W.; Cho, A.Y.; Heo, J.; Lee, K.B.; Lee, J.-H.; Lee, W.J.; Kim, H.-J.; et al. Real-Time Monitoring of Glutathione in Living Cells Reveals that High Glutathione Levels Are Required to Maintain Stem Cell Function. Stem Cell Rep. 2018, 10, 600–614. [Google Scholar] [CrossRef]
- Zaman, G.J.; Lankelma, J.; Van Tellingen, O.; Beijnen, J.; Dekker, H.; Paulusma, C.C.; Elferink, R.P.O.; Baas, F.; Borst, P. Role of glutathione in the export of compounds from cells by the multidrug-resistance-associated protein. Proc. Natl. Acad. Sci. USA 1995, 92, 7690–7694. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Leier, I.; Buchholz, U.; Center, M.; Keppler, D. ATP-dependent transport of glutathione S-conjugates by the multidrug resistance-associated protein. Cancer Res. 1994, 54, 4833–4836. [Google Scholar]
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Asp. Med. 2009, 30, 13. [Google Scholar] [CrossRef]
- Delic, M.; Mattanovich, D.; Gasser, B. Monitoring intracellular redox conditions in the endoplasmic reticulum of living yeasts. FEMS Microbiol. Lett. 2010, 306, 61–66. [Google Scholar] [CrossRef]
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef]
- Kojer, K.; Bien, M.; Gangel, H.; Morgan, B.; Dick, T.P.; Riemer, J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012, 31, 3169–3182. [Google Scholar] [CrossRef]
- Booty, L.M.; King, M.S.; Thangaratnarajah, C.; Majd, H.; James, A.M.; Kunji, E.R.; Murphy, M.P. The mitochondrial dicarboxylate and 2-oxoglutarate carriers do not transport glutathione. FEBS Lett. 2015, 589, 621–628. [Google Scholar] [CrossRef]
- Markovic, J.; Borrás, C.; Ortega, Á.; Sastre, J.; Viña, J.; Pallardó, F.V. Glutathione Is Recruited into the Nucleus in Early Phases of Cell Proliferation. J. Biol. Chem. 2007, 282, 20416–20424. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Markovic, J.; Dasí, F.; Queval, G.; Schnaubelt, D.; Foyer, C.H.; Pallardó, F.V. Nuclear glutathione. Biochim. Biophys. Acta 2013, 1830, 3304–3316. [Google Scholar] [CrossRef] [PubMed]
- Markovic, J.; García-Gimenez, J.L.; Gimeno, A.; Viña, J.; Pallardó, F.V. Role of glutathione in cell nucleus. Free Radic. Res. 2010, 44, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D.W.; McConkey, D.J.; McDonnell, T.J.; Brisbay, S.; Meyn, R.E. Bcl-2 expression causes redistribution of glutathione to the nucleus. Proc. Natl. Acad. Sci. USA 1998, 95, 2956–2960. [Google Scholar] [CrossRef]
- Ponsero, A.J.; Igbaria, A.; Darch, M.A.; Miled, S.; Outten, C.E.; Winther, J.R.; Palais, G.; D’Autréaux, B.; Delaunay-Moisan, A.; Toledano, M.B. Endoplasmic Reticulum Transport of Glutathione by Sec61 Is Regulated by Ero1 and Bip. Mol. Cell 2017, 67, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Tomonobu, N.; Komalasari, N.L.G.Y.; Sumardika, I.W.; Jiang, F.; Chen, Y.; Yamamoto, K.-I.; Kinoshita, R.; Murata, H.; Inoue, Y.; Sakaguchi, M. Xylitol acts as an anticancer monosaccharide to induce selective cancer death via regulation of the glutathione level. Chem. Biol. Interact. 2020, 324, 109085. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, G.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Rubino, F.M. Toxicity of Glutathione-Binding Metals: A Review of Targets and Mechanisms. Toxics 2015, 3, 20–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Sig. Transduct. Target Ther. 2020, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharm. 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Morel, F.; Aninat, C. The glutathione transferase kappa family. Drug Metab. Rev. 2011, 43, 281–291. [Google Scholar] [CrossRef]
- Dong, S.-C.; Sha, H.-H.; Xu, X.-Y.; Hu, T.-M.; Lou, R.; Li, H.; Wu, J.-Z.; Dan, C.; Feng, J. Glutathione S-transferase π: A potential role in antitumor therapy. Drug Des. Dev. Ther. 2018, 12, 3535–3547. [Google Scholar] [CrossRef]
- Pasello, M.; Michelacci, F.; Scionti, I.; Hattinger, C.M.; Zuntini, M.; Caccuri, A.M.; Scotlandi, K.; Picci, P.; Serra, M. Overcoming Glutathione S-Transferase P1-Related Cisplatin Resistance in Osteosarcoma. Cancer Res. 2008, 68, 6661–6668. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef]
- Ritchie, K.J.; Walsh, S.; Sansom, O.J.; Henderson, C.J.; Wolf, C.R. Markedly enhanced colon tumorigenesis in ApcMin mice lacking glutathione S-transferase Pi. Proc. Natl. Acad. Sci. USA 2009, 106, 20859–20864. [Google Scholar] [CrossRef] [PubMed]
- Zendehdel, K.; Bahmanyar, S.; McCarthy, S.; Nyren, O.; Andersson, B.; Ye, W. Genetic polymorphisms of glutathione S-transferase genes GSTP1, GSTM1, and GSTT1 and risk of esophageal and gastric cardia cancers. Cancer Causes Control. 2009, 20, 2031–2038. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Song, K.; Yi, J.; Shen, X.; Cai, Y. Genetic Polymorphisms of Glutathione S-Transferase Genes GSTM1, GSTT1 and Risk of Hepatocellular Carcinoma. PLoS ONE 2012, 7, e48924. [Google Scholar] [CrossRef] [PubMed]
- Seitz, G.; Bonin, M.; Fuchs, J.; Poths, S.; Ruck, P.; Warmann, S.W.; Armeanu-Ebinger, S. Inhibition of glutathione-S-transferase as a treatment strategy for multidrug resistance in childhood rhabdomyosarcoma. Int. J. Oncol. 2010, 36, 491–500. [Google Scholar] [CrossRef]
- Tew, K.D. Glutathione-Associated Enzymes In Anticancer Drug Resistance. Cancer Res. 2016, 76, 7–9. [Google Scholar] [CrossRef]
- Su, F.; Hu, X.; Jia, W.; Gong, C.; Song, E.; Hamar, P. Glutathion s transferase π indicates chemotherapy resistance in breast cancer. J. Surg. Res. 2003, 113, 102–108. [Google Scholar] [CrossRef]
- Esteller, M.; Corn, P.G.; Urena, J.M.; Gabrielson, E.; Baylin, S.B.; Herman, J.G. Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res. 1998, 58, 4515–4518. [Google Scholar]
- Horowitz, M.; Esakov, E.; Rose, P.; Reizes, O. Signaling within the epithelial ovarian cancer tumor microenvironment: The challenge of tumor heterogeneity. Ann. Transl. Med. 2020, 8, 905. [Google Scholar] [CrossRef]
- Song, Y.; Chen, J.; Liu, K.; Zhou, K.; Lu, Y.; Wang, X.; Yang, Y.; Liu, X. Glutathione S-Transferase Pi 1 (GSTP1) Gene 313 A/G (rs1695) polymorphism is associated with the risk of urinary bladder cancer: Evidence from a systematic review and meta-analysis based on 34 case-control studies. Gene 2019, 719, 144077. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Diotallevi, M.; Checconi, P.; Palamara, A.T.; Celestino, I.; Coppo, L.; Holmgren, A.; Abbas, K.; Peyrot, F.; Mengozzi, M.; Ghezzi, P. Glutathione Fine-Tunes the Innate Immune Response toward Antiviral Pathways in a Macrophage Cell Line Independently of Its Antioxidant Properties. Front. Immunol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Lu, C.-T.; Lee, T.-Y.; Chen, Y.-J. dbGSH: A database of S-glutathionylation. Bioinformatics 2014, 30, 2386–2388. [Google Scholar] [CrossRef] [PubMed]
- Lermant, A.; Murdoch, C.E. Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells. Antioxidants 2019, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Hao, L.; Liu, X.; Sun, G.; Xu, R.; Wang, H.; Liu, C. Comprehensive analysis of peroxiredoxins expression profiles and prognostic values in breast cancer. Biomark Res. 2019, 7, 16. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, C.; Xiao, G.; Shan, H.; Tang, L.; Yi, Y.; Yu, W.; Gu, Y. S-nitrosylation of the Peroxiredoxin-2 promotes S-nitrosoglutathione-mediated lung cancer cells apoptosis via AMPK-SIRT1 pathway. Cell Death Dis. 2019, 10, 329. [Google Scholar] [CrossRef]
- Cai, C.-Y.; Zhai, L.-L.; Wu, Y.; Tang, Z.-G. Expression and clinical value of peroxiredoxin-1 in patients with pancreatic cancer. Eur. J. Surg. Oncol. 2015, 41, 228–235. [Google Scholar] [CrossRef]
- Salzano, S.; Checconi, P.; Hanschmann, E.M.; Lillig, C.H.; Bowler, L.D.; Chan, P.; Vaudry, D.; Mengozzi, M.; Coppo, L.; Sacre, S.; et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. USA 2014, 111, 12157–12162. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, P.; Zhang, C.; Chiewchengchol, D.; Zhao, F.; Li, J.; Venkataraman, A.; Zhou, Z.; Zhou, W.; Yu, H.; et al. Positive Regulation of Interleukin-1 Beta Bioactivity by Physiological ROS-Mediated Cysteine S-Glutathionylation. SSRN Electron. J. 2018. [Google Scholar] [CrossRef]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Litmanovich, A.; Khazim, K.; Cohen, I. The Role of Interleukin-1 in the Pathogenesis of Cancer and its Potential as a Therapeutic Target in Clinical Practice. Oncol. Ther. 2018, 6, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Kipp, A. Glutathione peroxidases in different stages of carcinogenesis. Biochim. Biophys. Acta 2009, 1790, 1555–1568. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, Y.; Guo, S.; Wang, G. Glutathione peroxidases as oncotargets. Oncotarget 2017, 8, 80093–80102. [Google Scholar] [CrossRef]
- Kipp, A.P. Selenium-Dependent Glutathione Peroxidases During Tumor Development. Adv. Cancer Res. 2017, 136, 109–138. [Google Scholar]
- Esworthy, R.S.; Kim, B.-W.; Chow, J.; Shen, B.; Doroshow, J.H.; Chu, F.-F. Nox1 causes ileocolitis in mice deficient in glutathione peroxidase-1 and -2. Free Radic. Biol. Med. 2014, 68, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, S.C.; Gollowitzer, A.; Laoukili, J.; Kranenburg, O.; Werz, O.; Koeberle, A.; Kipp, A.P. Distinct and overlapping functions of glutathione peroxidases 1 and 2 in limiting NF-κB-driven inflammation through redox-active mechanisms. Redox Biol. 2020, 28, 101388. [Google Scholar] [CrossRef]
- Chang, C.; Worley, B.L.; Phaëton, R.; Hempel, N. Extracellular Glutathione Peroxidase GPx3 and Its Role in Cancer. Cancers 2020, 12, 2197. [Google Scholar] [CrossRef]
- Barrett, C.W.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T.; et al. Tumor Suppressor Function of the Plasma Glutathione Peroxidase Gpx3 in Colitis-Associated Carcinoma. Cancer Res. 2013, 73, 1245–1255. [Google Scholar] [CrossRef]
- Maiorino, M.; Scapin, M.; Ursini, F.; Biasolo, M.; Bosello, V.; Flohé, L. Distinct promoters determine alternative transcription of gpx-4 into phospholipid-hydroperoxide glutathione peroxidase variants. J. Biol. Chem. 2003, 278, 34286–34290. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Deng, X.; Xie, X.; Liu, Y.; Friedmann Angeli, J.P.; Lai, L. Activation of Glutathione Peroxidase 4 as a Novel Anti-inflammatory Strategy. Front. Pharmacol. 2018, 9, 1120. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumor progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Somasundaram, V.; Basudhar, D.; Bharadwaj, G.; No, J.H.; Ridnour, L.A.; Cheng, R.Y.; Fujita, M.; Thomas, U.D.; Anderson, S.K.; McVicar, D.W.; et al. Molecular Mechanisms of Nitric Oxide in Cancer Progression, Signal Transduction, and Metabolism. Antioxid. Redox Signal. 2019, 30, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.Y.; Basudhar, D.; Ridnour, L.A.; Heinecke, J.L.; Kesarwala, A.H.; Glynn, S.A.; Switzer, C.H.; Ambs, S.; Miranda, K.M.; Wink, D.A. Gene expression profiles of NO- and HNO-donor treated breast cancer cells: Insights into tumor response and resistance pathways. Nitric Oxide 2014, 43, 17–28. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.-O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Switzer, C.H.; Glynn, S.A.; Cheng, R.Y.S.; Ridnour, L.A.; Green, J.E.; Ambs, S.; Wink, D.A. S-nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol. Cancer Res. 2012, 10, 1203–1215. [Google Scholar] [CrossRef]
- Cañas, A.; López-Sánchez, L.M.; Peñarando, J.; Valverde, A.; Conde, F.M.; Hernández, V.; Fuentes, E.; López-Pedrera, C.; De La Haba-Rodríguez, J.R.; Aranda, E.; et al. Altered S-nitrosothiol homeostasis provides a survival advantage to breast cancer cells in HER2 tumors and reduces their sensitivity to trastuzumab. Biochim. Biophys. Acta 2016, 1862, 601–610. [Google Scholar] [CrossRef]
- Ehrenfeld, P.; Cordova, F.; Duran, W.N.; Sanchez, F.A. S-nitrosylation and its role in breast cancer angiogenesis and metastasis. Nitric Oxide 2019, 87, 52–59. [Google Scholar] [CrossRef]
- Bennett, J.M.; Foon, K.A. Immunologic Approaches to the Classification and Management of Lymphomas and Leukemias. Springer Sci. Bus. Media 2012, 68, 1–31. [Google Scholar]
- Vader, P.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Emerging targets for cancer therapy. Trends Mol. Med. 2014, 20, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Lotvall, J.; Valadi, H. Cell to cell signalling via exosomes through esRNA. Cell Adh. Migr. 2007, 1, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1–4. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Giannatale, A.D.; Ceder, S.; et al. Tumor exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [PubMed]
- Atay, S.; Godwin, A.K. Tumor-derived exosomes. Commun. Integr. Biol. 2014, 7, e28231. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.; Taylor, M.A. Functional Roles for Exosomal MicroRNAs in the Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2017, 15, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2019, 10, 3137. [Google Scholar] [CrossRef]
- Horimoto, Y.; Polanska, U.M.; Takahashi, Y.; Orimo, A. Emerging roles of the tumor-associated stroma in promoting tumor metastasis. Cell Adh. Migr. 2012, 6, 193–202. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of Exosomes in Crosstalk Between Cancer-Associated Fibroblasts and Cancer Cells. Front Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; Lucas, F.A.S.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, 10250. [Google Scholar] [CrossRef]
- Zeng, A.; Wei, Z.; Rabinovsky, R.; Jun, H.J.; El Fatimy, R.; Deforzh, E.; Arora, R.; Yao, Y.; Yao, S.; Yan, W.; et al. Glioblastoma-Derived Extracellular Vesicles Facilitate Transformation of Astrocytes via Reprogramming Oncogenic Metabolism. iScience 2020, 23, 101420. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumor microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH Is a Key Factor for Exosome Traffic in Tumor Cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; Spugnini, E.; Mizzoni, D.; Di Raimo, R.; Fais, S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Cancer Metastasis Rev. 2019, 38, 93–101. [Google Scholar] [CrossRef]
- Čuperlović-Culf, M.; Khieu, N.H.; Surendra, A.; Hewitt, M.; Charlebois, C.; Sandhu, J.K. Analysis and Simulation of Glioblastoma Cell Lines-Derived Extracellular Vesicles Metabolome. Metabolites 2020, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, V.V.; Gillies, R.J. Janus-faced tumor microenvironment and redox. Antioxid. Redox Signal 2014, 21, 723–729. [Google Scholar] [CrossRef]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1–dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system x- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, G.; Li, R.; Guan, M.; Wang, X.; Zou, T.; Zhang, Y.; Wang, C.; Shu, C.; Hong, H.; et al. Biodegradable, Hydrogen Peroxide, and Glutathione Dual Responsive Nanoparticles for Potential Programmable Paclitaxel Release. J. Am. Chem. Soc. 2018, 140, 7373–7376. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Tu, J.; Wang, J.; Shajii, A.; Kong, N.; Feng, C.; Zhang, Y.; Yu, M.; Xie, T.; Bharwani, Z.; et al. Glutathione-Responsive Prodrug Nanoparticles for Effective Drug Delivery and Cancer Therapy. ACS Nano 2018, 13, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.; Jung, E.; Noh, J.; Hyun, H.; Seon, S.; Hong, S.; Kim, D.; Lee, D. Glutathione-Depleting Pro-Oxidant as a Selective Anticancer Therapeutic Agent. ACS Omega 2019, 4, 10070–10077. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M. Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling. Metabolites 2018, 11, 4. [Google Scholar] [CrossRef]
- Reed, M.C.; Thomas, R.L.; Pavisic, J.; Jill James, S.; Ulrich, C.M.; Frederik Nijhout, H. A mathematical model of glutathione metabolism. Theor. Biol. Med. Model. 2008, 5, 8. [Google Scholar] [CrossRef]
- Kelly, R.A.; Leedale, J.A.; Calleja, D.; Enoch, S.J.; Harrell, A.; Chadwick, A.E.; Webb, S. Modelling changes in glutathione homeostasis as a function of quinone redox metabolism. Sci. Rep. 2019, 9, 6333. [Google Scholar] [CrossRef]
- Roy, M.; Finley, S.D. Computational Model Predicts the Effects of Targeting Cellular Metabolism in Pancreatic Cancer. Front. Physiol. 2017, 8, 217. [Google Scholar] [CrossRef]
- Schomburg, I. BRENDA, enzyme data and metabolic information. Nucleic Acids Res. 2002, 30, 47–49. [Google Scholar] [CrossRef]
- Alam, M.T.; Olin-Sandoval, V.; Stincone, A.; Keller, M.A.; Zelezniak, A.; Luisi, B.F.; Ralser, M. The self-inhibitory nature of metabolic networks and its alleviation through compartmentalization. Nat. Commun. 2017, 8, 16018. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.; Nielsen, J. Genome scale metabolic modeling of cancer. Metab. Eng. 2017, 43, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.P.; Bintener, T.; Ternes, D.; Kulms, D.; Haan, S.; Letellier, E.; Sauter, T. Identifying and targeting cancer-specific metabolism with network-based drug target prediction. Ebio Med. 2019, 43, 98–106. [Google Scholar] [CrossRef]
- Metzcar, J.; Wang, Y.; Heiland, R.; Macklin, P. A Review of Cell-Based Computational Modeling in Cancer Biology. J. Clin. Cancer Inf. 2019, 3, 1–13. [Google Scholar] [CrossRef]
- Roy, M.; Finley, S.D. Metabolic reprogramming dynamics in tumor spheroids: Insights from a multicellular, multiscale model. PLoS Comput. Biol. 2019, 15, e1007053. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. https://doi.org/10.3390/biom10101429
Kennedy L, Sandhu JK, Harper M-E, Cuperlovic-Culf M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules. 2020; 10(10):1429. https://doi.org/10.3390/biom10101429
Chicago/Turabian StyleKennedy, Luke, Jagdeep K. Sandhu, Mary-Ellen Harper, and Miroslava Cuperlovic-Culf. 2020. "Role of Glutathione in Cancer: From Mechanisms to Therapies" Biomolecules 10, no. 10: 1429. https://doi.org/10.3390/biom10101429
APA StyleKennedy, L., Sandhu, J. K., Harper, M.-E., & Cuperlovic-Culf, M. (2020). Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules, 10(10), 1429. https://doi.org/10.3390/biom10101429