Prion-Like Propagation Mechanisms in Tauopathies and Traumatic Brain Injury: Challenges and Prospects


Abstract
:1. Introduction
2. Tau Protein
2.1. The MAPT Gene Encodes Various Isoforms of Tau Protein
2.2. Tauopathies Are Diverse But Can Be Categorized in Various Useful Ways
2.3. Human Genetics of MAPT Implicates Tau in Various Dementias
2.4. Physiological Functions of Tau
2.5. Tau Domains and PTMs in Native and Aggregated Tau Structures
2.6. PTMs Correlate with Accumulation of Tau Aggregates: Cause and Effect
2.7. Other Biochemical Aspects That Impact Accumulation of Tau Aggregates
3. “Prion-Like” Properties of Tau
3.1. Prion-Like Diseases: Defining Characteristics
3.2. Evidence Supporting the Prion-Like Seeding and Spreading of Tau In Vitro and In Vivo
3.3. Prion-Like Strains of Tau May Exist and Be Fertile Ground for Innovation
3.4. Mechanisms and Factors Underlying Prion-Like Tau Transmissions Remain Mysterious
3.5. How Does Tau Spread Intercellularly?
3.6. How Does Tau Get Released into the Extracellular Space during Prion-Like Spreading?
3.7. Mechanisms Mediating the Cellular Uptake of Tau Aggregates
3.8. Factors Contributing to the Propagation of Tau
4. Prion-Like Properties of Tauopathy Following TBI
4.1. Overview of TBI
4.2. Prion-Like Proprieties of Tau in TBI and CTE
4.3. CTE May Be a Unique Prion-Like Strain of Tauopathy
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrer, I.; Lopez-Gonzalez, I.; Carmona, M.; Arregui, L.; Dalfo, E.; Torrejon-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef] [Green Version]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizynski, B.; Dzwolak, W.; Nieznanski, K. Amyloidogenesis of Tau protein. Protein Sci. 2017, 26, 2126–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Jakes, R.; Vanmechelen, E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 1995, 189, 167–169. [Google Scholar] [CrossRef]
- Kopke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [PubMed]
- Holmes, B.B.; Diamond, M.I. Prion-like properties of Tau protein: The importance of extracellular Tau as a therapeutic target. J. Biol. Chem. 2014, 289, 19855–19861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986, 387, 271–280. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Schuck, T.; Schmidt, M.L.; Lee, V.M. Distribution of tau proteins in the normal human central and peripheral nervous system. J. Histochem Cytochem 1989, 37, 209–215. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Kubo, A.; Misonou, H.; Matsuyama, M.; Nomori, A.; Wada-Kakuda, S.; Takashima, A.; Kawata, M.; Murayama, S.; Ihara, Y.; Miyasaka, T. Distribution of endogenous normal tau in the mouse brain. J. Comp. Neurol. 2019, 527, 985–998. [Google Scholar] [CrossRef]
- Kubo, A.; Ueda, S.; Yamane, A.; Wada-Kakuda, S.; Narita, M.; Matsuyama, M.; Nomori, A.; Takashima, A.; Kato, T.; Onodera, O.; et al. Ectopic Expression Induces Abnormal Somatodendritic Distribution of Tau in the Mouse Brain. J. Neurosci. 2019, 39, 6781–6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotti, C.G.; Banker, G.A.; Binder, L.I. The expression and distribution of the microtubule-associated proteins tau and microtubule-associated protein 2 in hippocampal neurons in the rat in situ and in cell culture. Neuroscience 1987, 23, 121–130. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempf, M.; Clement, A.; Faissner, A.; Lee, G.; Brandt, R. Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J. Neurosci. 1996, 16, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgrafe, K.; Mandelkow, E.M.; et al. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Magnoni, S.; Esparza, T.J.; Conte, V.; Carbonara, M.; Carrabba, G.; Holtzman, D.M.; Zipfel, G.J.; Stocchetti, N.; Brody, D.L. Tau elevations in the brain extracellular space correlate with reduced amyloid-beta levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain 2012, 135, 1268–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutajangout, A.; Boom, A.; Leroy, K.; Brion, J.P. Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer’s disease. FEBS Lett. 2004, 576, 183–189. [Google Scholar] [CrossRef]
- Trabzuni, D.; Wray, S.; Vandrovcova, J.; Ramasamy, A.; Walker, R.; Smith, C.; Luk, C.; Gibbs, J.R.; Dillman, A.; Hernandez, D.G.; et al. MAPT expression and splicing is differentially regulated by brain region: Relation to genotype and implication for tauopathies. Hum. Mol. Genet. 2012, 21, 4094–4103. [Google Scholar] [CrossRef] [Green Version]
- Boyne, L.J.; Tessler, A.; Murray, M.; Fischer, I. Distribution of Big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol. 1995, 358, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.; Baas, P.W. Resurrecting the Mysteries of Big Tau. Trends Neurosci. 2020, 10.1016/j.tins.2020.04.007. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Butner, K.A.; Kirschner, M.W. Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 1991, 115, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Kellogg, E.H.; Hejab, N.M.A.; Poepsel, S.; Downing, K.H.; DiMaio, F.; Nogales, E. Near-atomic model of microtubule-tau interactions. Science 2018, 360, 1242–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadavath, H.; Jaremko, M.; Jaremko, L.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Folding of the Tau Protein on Microtubules. Angew Chem. Int. Ed. Engl. 2015, 54, 10347–10351. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gotz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Song, X.; Nisbet, R.; Gotz, J. Co-immunoprecipitation with Tau Isoform-specific Antibodies Reveals Distinct Protein Interactions and Highlights a Putative Role for 2N Tau in Disease. J. Biol. Chem. 2016, 291, 8173–8188. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef]
- Luna-Muñoz, J.; Harrington, C.R.; Wischik, C.M.; Flores-Rodríguez, P.; Avila, J.; Zamudio, S.R.; De la Cruz, F.; Mena, R.; Meraz-Ríos, M.A.; Floran-Garduño, B. Phosphorylation of tau protein associated as a protective mechanism in the presence of toxic, C-terminally truncated tau in Alzheimer’s disease. Understanding Alzheimer’s Disease 2013, 89. [Google Scholar]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; von Bergen, M.; Biernat, J.; Fischer, D.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. The “jaws” of the tau-microtubule interaction. J. Biol. Chem. 2007, 282, 12230–12239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischik, C.M.; Novak, M.; Thogersen, H.C.; Edwards, P.C.; Runswick, M.J.; Jakes, R.; Walker, J.E.; Milstein, C.; Roth, M.; Klug, A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4506–4510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, F.X.; Luiken, J.A.; Bolhuis, P.G. Primary Fibril Nucleation of Aggregation Prone Tau Fragments PHF6 and PHF6. J. Phys. Chem. B 2017, 121, 3250–3261. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Murrell, J.R.; Farlow, M.R.; Ghetti, B. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 4113–4118. [Google Scholar] [CrossRef] [Green Version]
- Gotz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu. Rev. Pathol. 2019, 14, 239–261. [Google Scholar] [CrossRef]
- Forrest, S.L.; Halliday, G.M.; McCann, H.; McGeachie, A.B.; McGinley, C.V.; Hodges, J.R.; Piguet, O.; Kwok, J.B.; Spillantini, M.G.; Kril, J.J. Heritability in frontotemporal tauopathies. Alzheimers Dement. 2019, 11, 115–124. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta. Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Rohan, Z.; Matej, R. Current concepts in the classification and diagnosis of frontotemporal lobar degenerations: A practical approach. Arch. Pathol. Lab. Med. 2014, 138, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef]
- Forrest, S.L.; Kril, J.J.; Stevens, C.H.; Kwok, J.B.; Hallupp, M.; Kim, W.S.; Huang, Y.; McGinley, C.V.; Werka, H.; Kiernan, M.C.; et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 2018, 141, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Stein, T.D.; Kiernan, P.T.; Alvarez, V.E. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015, 25, 350–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Irwin, D.J.; Brettschneider, J.; McMillan, C.T.; Cooper, F.; Olm, C.; Arnold, S.E.; Van Deerlin, V.M.; Seeley, W.W.; Miller, B.L.; Lee, E.B.; et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann. Neurol. 2016, 79, 272–287. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef]
- Ksiezak-Reding, H.; Tracz, E.; Yang, L.S.; Dickson, D.W.; Simon, M.; Wall, J.S. Ultrastructural instability of paired helical filaments from corticobasal degeneration as examined by scanning transmission electron microscopy. Am. J. Pathol. 1996, 149, 639–651. [Google Scholar]
- Ling, H.; Kovacs, G.G.; Vonsattel, J.P.; Davey, K.; Mok, K.Y.; Hardy, J.; Morris, H.R.; Warner, T.T.; Holton, J.L.; Revesz, T. Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain 2016, 139, 3237–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervos-Navarro, J.; Schumacher, K. Neurofibrillary pathology in progressive supranuclear palsy (PSP). J. Neural Transm. Suppl. 1994, 42, 153–164. [Google Scholar] [CrossRef]
- Williams, D.R.; Holton, J.L.; Strand, C.; Pittman, A.; de Silva, R.; Lees, A.J.; Revesz, T. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain 2007, 130, 1566–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.; Doherty, K.M.; Silveira-Moriyama, L.; Bandopadhyay, R.; Lashley, T.; Mamais, A.; Hondhamuni, G.; Wray, S.; Newcombe, J.; O’Sullivan, S.S.; et al. Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: An emerging group of 4-repeat tauopathies. Acta Neuropathol. 2011, 122, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Ruberu, N.N.; Sawabe, M.; Arai, T.; Tanaka, N.; Kakuta, Y.; Yamanouchi, H.; Murayama, S. Staging of argyrophilic grains: An age-associated tauopathy. J. Neuropathol. Exp. Neurol. 2004, 63, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Arena, J.D.; Smith, D.H.; Lee, E.B.; Gibbons, G.S.; Irwin, D.J.; Robinson, J.L.; Lee, V.M.; Trojanowski, J.Q.; Stewart, W.; Johnson, V.E. Tau immunophenotypes in chronic traumatic encephalopathy recapitulate those of ageing and Alzheimer’s disease. Brain 2020, 143, 1572–1587. [Google Scholar] [CrossRef]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Chen, J.; Yu, J.T.; Wojta, K.; Wang, H.F.; Zetterberg, H.; Blennow, K.; Yokoyama, J.S.; Weiner, M.W.; Kramer, J.H.; Rosen, H.; et al. Genome-wide association study identifies MAPT locus influencing human plasma tau levels. Neurology 2017, 88, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Pittman, A.M.; Myers, A.J.; Abou-Sleiman, P.; Fung, H.C.; Kaleem, M.; Marlowe, L.; Duckworth, J.; Leung, D.; Williams, D.; Kilford, L.; et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J. Med. Genet. 2005, 42, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Melquist, S.; Cruts, M.; Theuns, J.; Del-Favero, J.; Poorkaj, P.; Baker, M.; Sleegers, K.; Crook, R.; De Pooter, T.; et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum. Mol. Genet. 2005, 14, 3281–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittman, A.M.; Fung, H.C.; de Silva, R. Untangling the tau gene association with neurodegenerative disorders. Hum. Mol. Genet. 2006, 15 (Suppl. 2), R188–R195. [Google Scholar] [CrossRef]
- Baker, M.; Litvan, I.; Houlden, H.; Adamson, J.; Dickson, D.; Perez-Tur, J.; Hardy, J.; Lynch, T.; Bigio, E.; Hutton, M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 1999, 8, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Helgason, A.; Thorleifsson, G.; Steinthorsdottir, V.; Masson, G.; Barnard, J.; Baker, A.; Jonasdottir, A.; Ingason, A.; Gudnadottir, V.G.; et al. A common inversion under selection in Europeans. Nat. Genet. 2005, 37, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.; Fung, H.C.; Steele, J.; Eerola, J.; Tienari, P.; Pittman, A.; Silva, R.; Myers, A.; Vrieze, F.W.; Singleton, A.; et al. The tau H2 haplotype is almost exclusively Caucasian in origin. Neurosci. Lett. 2004, 369, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Andreadis, A.; Trojanowski, J.Q.; Dickson, D.W.; Kang, D.; Chen, X.; Wiederholt, W.; Hansen, L.; Masliah, E.; Thal, L.J.; et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann. Neurol. 1997, 41, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Pittman, A.M.; Myers, A.J.; Duckworth, J.; Bryden, L.; Hanson, M.; Abou-Sleiman, P.; Wood, N.W.; Hardy, J.; Lees, A.; de Silva, R. The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum. Mol. Genet. 2004, 13, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houlden, H.; Baker, M.; Morris, H.R.; MacDonald, N.; Pickering-Brown, S.; Adamson, J.; Lees, A.J.; Rossor, M.N.; Quinn, N.P.; Kertesz, A.; et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 2001, 56, 1702–1706. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Haggiagi, A.; Liu, X.; Wasserscheid, J.; Nelson, P.T.; Dewar, K.; Clark, L.N.; Crary, J.F. The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol. 2012, 124, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Caffrey, T.M.; Joachim, C.; Wade-Martins, R. Haplotype-specific expression of the N-terminal exons 2 and 3 at the human MAPT locus. Neurobiol. Aging 2008, 29, 1923–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.C.; Bechy, A.L.; Denk, F.; Collins, E.; Gavriliouk, M.; Zaugg, J.B.; Ryan, B.J.; Wade-Martins, R.; Caffrey, T.M. Haplotype-specific MAPT exon 3 expression regulated by common intronic polymorphisms associated with Parkinsonian disorders. Mol. Neurodegener. 2017, 12, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffrey, T.M.; Joachim, C.; Paracchini, S.; Esiri, M.M.; Wade-Martins, R. Haplotype-specific expression of exon 10 at the human MAPT locus. Hum. Mol. Genet. 2006, 15, 3529–3537. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Invest. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Jiang, Z.; Cote, J.; Kwon, J.M.; Goate, A.M.; Wu, J.Y. Aberrant splicing of tau pre-mRNA caused by intronic mutations associated with the inherited dementia frontotemporal dementia with parkinsonism linked to chromosome 17. Mol. Cell Biol. 2000, 20, 4036–4048. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Liu, F. Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 2014, 30, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Crowther, R.A. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999, 450, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Strang, K.H.; Croft, C.L.; Sorrentino, Z.A.; Chakrabarty, P.; Golde, T.E.; Giasson, B.I. Distinct differences in prion-like seeding and aggregation between Tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem. 2018, 293, 2408–2421. [Google Scholar] [CrossRef] [Green Version]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, B.; Gamblin, T.C. FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry 2012, 51, 8597–8607. [Google Scholar] [CrossRef] [Green Version]
- Jordan, B.D.; Relkin, N.R.; Ravdin, L.D.; Jacobs, A.R.; Bennett, A.; Gandy, S. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA 1997, 278, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Mannix, R.; Meehan, W.P., III. Evaluating the Effects of APOE4 after Mild Traumatic Brain Injury in Experimental Models. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F.H., Ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Dardiotis, E.; Fountas, K.N.; Dardioti, M.; Xiromerisiou, G.; Kapsalaki, E.; Tasiou, A.; Hadjigeorgiou, G.M. Genetic association studies in patients with traumatic brain injury. Neurosurg. Focus 2010, 28, E9. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Mez, J.; Crary, J.F.; Tripodis, Y.; Alvarez, V.E.; Mahar, I.; Huber, B.R.; Alosco, M.L.; Nicks, R.; Abdolmohammadi, B.; et al. Variation in TMEM106B in chronic traumatic encephalopathy. Acta Neuropathol. Commun. 2018, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Bieniek, K.F.; Ross, O.A.; Cormier, K.A.; Walton, R.L.; Soto-Ortolaza, A.; Johnston, A.E.; DeSaro, P.; Boylan, K.B.; Graff-Radford, N.R.; Wszolek, Z.K.; et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta. Neuropathol. 2015, 130, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Abdolmohammadi, B.; Dupre, A.; Evers, L.; Mez, J. Genetics of Chronic Traumatic Encephalopathy. Semin Neurol. 2020, 40, 420–429. [Google Scholar] [CrossRef]
- Bennett, E.R.; Reuter-Rice, K.; Laskowitz, D.T. Genetic Influences in Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Hirokawa, N.; Shiomura, Y.; Okabe, S. Tau proteins: The molecular structure and mode of binding on microtubules. J. Cell Biol. 1988, 107, 1449–1459. [Google Scholar] [CrossRef]
- Prezel, E.; Elie, A.; Delaroche, J.; Stoppin-Mellet, V.; Bosc, C.; Serre, L.; Fourest-Lieuvin, A.; Andrieux, A.; Vantard, M.; Arnal, I. Tau can switch microtubule network organizations: From random networks to dynamic and stable bundles. Mol. Biol. Cell 2018, 29, 154–165. [Google Scholar] [CrossRef]
- Samsonov, A.; Yu, J.Z.; Rasenick, M.; Popov, S.V. Tau interaction with microtubules in vivo. J. Cell Sci. 2004, 117, 6129–6141. [Google Scholar] [CrossRef] [Green Version]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Rook, S.L. Expression of tau protein in non-neuronal cells: Microtubule binding and stabilization. J. Cell Sci. 1992, 102 Pt 2, 227–237. [Google Scholar]
- Qiang, L.; Sun, X.; Austin, T.O.; Muralidharan, H.; Jean, D.C.; Liu, M.; Yu, W.; Baas, P.W. Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr. Biol. 2018, 28, 2181–2189.e2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Chaudhary, A.R.; Berger, F.; Berger, C.L.; Hendricks, A.G. Tau directs intracellular trafficking by regulating the forces exerted by kinesin and dynein teams. Traffic 2018, 19, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Ramos, A.; Diaz-Hernandez, M.; Rubio, A.; Diaz-Hernandez, J.I.; Miras-Portugal, M.T.; Avila, J. Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 2009, 19, 708–717. [Google Scholar] [CrossRef]
- Regan, P.; Piers, T.; Yi, J.H.; Kim, D.H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef] [Green Version]
- Regan, P.; Whitcomb, D.J.; Cho, K. Physiological and Pathophysiological Implications of Synaptic Tau. Neuroscientist 2017, 23, 137–151. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef] [Green Version]
- Sohn, P.D.; Huang, C.T.; Yan, R.; Fan, L.; Tracy, T.E.; Camargo, C.M.; Montgomery, K.M.; Arhar, T.; Mok, S.A.; Freilich, R.; et al. Pathogenic Tau Impairs Axon Initial Segment Plasticity and Excitability Homeostasis. Neuron 2019, 104, 458–470.e455. [Google Scholar] [CrossRef]
- Gunawardana, C.G.; Mehrabian, M.; Wang, X.; Mueller, I.; Lubambo, I.B.; Jonkman, J.E.; Wang, H.; Schmitt-Ulms, G. The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol. Cell Proteom. 2015, 14, 3000–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.A.; Hamm, M.J.; Meier, S.E.; Weiss, B.E.; Nation, G.K.; Chishti, E.A.; Arango, J.P.; Chen, J.; Zhu, H.; Blalock, E.M.; et al. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 2019, 137, 571–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, M.; Magrin, C.; Pedrioli, G.; Pinton, S.; Salvade, A.; Papin, S.; Paganetti, P. Tau affects P53 function and cell fate during the DNA damage response. Commun. Biol. 2020, 3, 245. [Google Scholar] [CrossRef] [PubMed]
- Bukar Maina, M.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Baquero, J.; Varriano, S.; Ordonez, M.; Kuczaj, P.; Murphy, M.R.; Aruggoda, G.; Lundine, D.; Morozova, V.; Makki, A.E.; Alonso, A.D.C.; et al. Nuclear Tau, p53 and Pin1 Regulate PARN-Mediated Deadenylation and Gene Expression. Front. Mol. Neurosci. 2019, 12, 242. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Kramer, E.M.; Cardine, A.M.; Schraven, B.; Brandt, R.; Trotter, J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 2002, 22, 698–707. [Google Scholar] [CrossRef] [Green Version]
- Seiberlich, V.; Bauer, N.G.; Schwarz, L.; Ffrench-Constant, C.; Goldbaum, O.; Richter-Landsberg, C. Downregulation of the microtubule associated protein tau impairs process outgrowth and myelin basic protein mRNA transport in oligodendrocytes. Glia 2015, 63, 1621–1635. [Google Scholar] [CrossRef]
- Sayas, C.L.; Medina, M.; Cuadros, R.; Olla, I.; Garcia, E.; Perez, M.; Ferrer, I.; Hernandez, F.; Avila, J. Role of tau N-terminal motif in the secretion of human tau by End Binding proteins. PLoS ONE 2019, 14, e0210864. [Google Scholar] [CrossRef] [Green Version]
- Stefanoska, K.; Volkerling, A.; Bertz, J.; Poljak, A.; Ke, Y.D.; Ittner, L.M.; Ittner, A. An N-terminal motif unique to primate tau enables differential protein-protein interactions. J. Biol. Chem. 2018, 293, 3710–3719. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Ishiguro, K.; Hisanaga, S. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, B.L.; Denis, P.E.; Panda, D.; Radeke, M.J.; Miller, H.P.; Wilson, L.; Feinstein, S.C. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol. Biol. Cell. 1997, 8, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Gustke, N.; Trinczek, B.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein and interactions with microtubules. Biochemistry 1994, 33, 9511–9522. [Google Scholar] [CrossRef] [PubMed]
- McKibben, K.M.; Rhoades, E. Independent tubulin binding and polymerization by the proline-rich region of Tau is regulated by Tau’s N-terminal domain. J. Biol. Chem. 2019, 294, 19381–19394. [Google Scholar] [CrossRef]
- Arnold, C.S.; Johnson, G.V.; Cole, R.N.; Dong, D.L.; Lee, M.; Hart, G.W. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J. Biol. Chem. 1996, 271, 28741–28744. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, T.; Uryu, K.; Giasson, B.I.; Ischiropoulos, H.; LightFoot, R.; Bellmann, C.; Richter-Landsberg, C.; Lee, V.M.; Trojanowski, J.Q. Nitration of tau protein is linked to neurodegeneration in tauopathies. Am. J. Pathol. 2003, 163, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Kondo, J.; Ihara, Y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 1987, 235, 1641–1644. [Google Scholar] [CrossRef]
- Nakamura, K.; Greenwood, A.; Binder, L.; Bigio, E.H.; Denial, S.; Nicholson, L.; Zhou, X.Z.; Lu, K.P. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 2012, 149, 232–244. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Lee, J.H.; Jeon, J.H.; Lee, M.J. Degradation or aggregation: The ramifications of post-translational modifications on tau. BMB Rep. 2018, 51, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Gan, L. Acetylated tau in Alzheimer’s disease: An instigator of synaptic dysfunction underlying memory loss: Increased levels of acetylated tau blocks the postsynaptic signaling required for plasticity and promotes memory deficits associated with tauopathy. Bioessays 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Hanger, D.P.; Byers, H.L.; Wray, S.; Leung, K.Y.; Saxton, M.J.; Seereeram, A.; Reynolds, C.H.; Ward, M.A.; Anderton, B.H. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 2007, 282, 23645–23654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mair, W.; Muntel, J.; Tepper, K.; Tang, S.; Biernat, J.; Seeley, W.W.; Kosik, K.S.; Mandelkow, E.; Steen, H.; Steen, J.A. FLEXITau: Quantifying Post-translational Modifications of Tau Protein in Vitro and in Human Disease. Anal. Chem. 2016, 88, 3704–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Seereeram, A.; Byers, H.L.; Leung, K.Y.; Ward, M.A.; Anderton, B.H.; Hanger, D.P. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008, 22, 3186–3195. [Google Scholar] [CrossRef]
- LaPointe, N.E.; Morfini, G.; Pigino, G.; Gaisina, I.N.; Kozikowski, A.P.; Binder, L.I.; Brady, S.T. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J. Neurosci. Res. 2009, 87, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Biernat, J.; von Bergen, M.; Mandelkow, E.; Mandelkow, E.M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Simic, G.; Babic Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buee, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, S.; Saxton, M.; Anderton, B.H.; Hanger, D.P. Direct analysis of tau from PSP brain identifies new phosphorylation sites and a major fragment of N-terminally cleaved tau containing four microtubule-binding repeats. J. Neurochem. 2008, 105, 2343–2352. [Google Scholar] [CrossRef] [PubMed]
- Mercken, M.; Vandermeeren, M.; Lubke, U.; Six, J.; Boons, J.; Van de Voorde, A.; Martin, J.J.; Gheuens, J. Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol. 1992, 84, 265–272. [Google Scholar] [CrossRef]
- Noble, W.; Hanger, D.P.; Miller, C.C.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef] [Green Version]
- Lasagna-Reeves, C.A.; de Haro, M.; Hao, S.; Park, J.; Rousseaux, M.W.; Al-Ramahi, I.; Jafar-Nejad, P.; Vilanova-Velez, L.; See, L.; De Maio, A.; et al. Reduction of Nuak1 Decreases Tau and Reverses Phenotypes in a Tauopathy Mouse Model. Neuron 2016, 92, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Sundermann, F.; Fernandez, M.P.; Morgan, R.O. An evolutionary roadmap to the microtubule-associated protein MAP Tau. BMC Genom. 2016, 17, 264. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef]
- Karagoz, G.E.; Duarte, A.M.; Akoury, E.; Ippel, H.; Biernat, J.; Moran Luengo, T.; Radli, M.; Didenko, T.; Nordhues, B.A.; Veprintsev, D.B.; et al. Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell 2014, 156, 963–974. [Google Scholar] [CrossRef] [Green Version]
- Arosio, P.; Knowles, T.P.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J. Biol. Chem. 2001, 276, 48165–48174. [Google Scholar] [CrossRef] [Green Version]
- Mirbaha, H.; Holmes, B.B.; Sanders, D.W.; Bieschke, J.; Diamond, M.I. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem. 2015, 290, 14893–14903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 2011, 286, 23063–23076. [Google Scholar] [CrossRef] [Green Version]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 2010, 49, 10039–10041. [Google Scholar] [CrossRef]
- Sahara, N.; Maeda, S.; Yoshiike, Y.; Mizoroki, T.; Yamashita, S.; Murayama, M.; Park, J.M.; Saito, Y.; Murayama, S.; Takashima, A. Molecular chaperone-mediated tau protein metabolism counteracts the formation of granular tau oligomers in human brain. J. Neurosci. Res. 2007, 85, 3098–3108. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular tau oligomers as intermediates of tau filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef] [Green Version]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Gerson, J.E.; Sengupta, U.; Guerrero-Munoz, M.J.; Lasagna-Reeves, C.A.; Kayed, R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J. Alzheimers Dis. 2014, 40 (Suppl. 1), S97–S111. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.; Jimenez, J.S.; Sayas, C.L.; Bolos, M.; Zabala, J.C.; Rivas, G.; Hernandez, F. Tau Structures. Front. Aging Neurosci. 2016, 8, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegmann, S.; Medalsy, I.D.; Mandelkow, E.; Muller, D.J. The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectrolyte brush. Proc. Natl. Acad. Sci. USA 2013, 110, E313–E321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartner, U.; Janke, C.; Holzer, M.; Vanmechelen, E.; Arendt, T. Postmortem changes in the phosphorylation state of tau-protein in the rat brain. Neurobiol. Aging 1998, 19, 535–543. [Google Scholar] [CrossRef]
- Oka, T.; Tagawa, K.; Ito, H.; Okazawa, H. Dynamic changes of the phosphoproteome in postmortem mouse brains. PLoS ONE 2011, 6, e21405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Combs, C.K.; Pilcher, W.H.; Song, L.Y.; Utal, A.K.; Coleman, P.D. Low initial tau phosphorylation in human brain biopsy samples. Neurobiol. Aging 1997, 18, 475–481. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Hu, W.; Xie, S.; Gong, C.X.; Iqbal, K.; Liu, F. Rapid alteration of protein phosphorylation during postmortem: Implication in the study of protein phosphorylation. Sci. Rep. 2015, 5, 15709. [Google Scholar] [CrossRef] [Green Version]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef]
- Barthelemy, N.R.; Li, Y.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.S.; Buckles, V.; Fagan, A.M.; Perrin, R.J.; Goate, A.M.; et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med. 2020, 26, 398–407. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid Med. Cell Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef] [Green Version]
- Necula, M.; Kuret, J. Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J. Biol. Chem. 2004, 279, 49694–49703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiris, E.; Ventimiglia, D.; Sargin, M.E.; Gaylord, M.R.; Altinok, A.; Rose, K.; Manjunath, B.S.; Jordan, M.A.; Wilson, L.; Feinstein, S.C. Combinatorial Tau pseudophosphorylation: Markedly different regulatory effects on microtubule assembly and dynamic instability than the sum of the individual parts. J. Biol. Chem. 2011, 286, 14257–14270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Gamblin, T.C. Pseudohyperphosphorylation causing AD-like changes in tau has significant effects on its polymerization. Biochemistry 2009, 48, 6002–6011. [Google Scholar] [CrossRef] [Green Version]
- Rankin, C.A.; Sun, Q.; Gamblin, T.C. Tau phosphorylation by GSK-3beta promotes tangle-like filament morphology. Mol. Neurodegener. 2007, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, E.S.; Shin, R.W.; Billingsley, M.L.; Van deVoorde, A.; O’Connor, M.; Trojanowski, J.Q.; Lee, V.M. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar] [CrossRef]
- Taniguchi-Watanabe, S.; Arai, T.; Kametani, F.; Nonaka, T.; Masuda-Suzukake, M.; Tarutani, A.; Murayama, S.; Saito, Y.; Arima, K.; Yoshida, M.; et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 2016, 131, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. Elife 2019, 8. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hasegawa, M.; Ono, T.; Tashima, K.; Ihara, Y.; Miyamoto, E. Dephosphorylation of fetal-tau and paired helical filaments-tau by protein phosphatases 1 and 2A and calcineurin. J. Biochem. 1995, 118, 1224–1231. [Google Scholar] [CrossRef]
- Miyamoto, T.; Stein, L.; Thomas, R.; Djukic, B.; Taneja, P.; Knox, J.; Vossel, K.; Mucke, L. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 2017, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.T.; Wang, X.; Wang, C.; Sohn, P.D.; Theofilas, P.; Sidhu, M.; Arevalo, J.B.; Heinsen, H.; Huang, E.J.; Rosen, H.; et al. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathol. 2013, 125, 581–593. [Google Scholar] [CrossRef] [Green Version]
- Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Madden, V.; Tripathy, A.; Yuan, C.X.; Cohen, T.J. A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Sci. Rep. 2017, 7, 44102. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yahya, M.; Lashuel, H.A. Protein Semisynthesis Provides Access to Tau Disease-Associated Post-translational Modifications (PTMs) and Paves the Way to Deciphering the Tau PTM Code in Health and Diseased States. J. Am. Chem. Soc. 2018, 140, 6611–6621. [Google Scholar] [CrossRef] [Green Version]
- Tracy, T.; Claiborn, K.C.; Gan, L. Regulation of Tau Homeostasis and Toxicity by Acetylation. Adv. Exp. Med. Biol. 2019, 1184, 47–55. [Google Scholar] [CrossRef]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Yuzwa, S.A.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Cheung, A.H.; Okon, M.; McIntosh, L.P.; Vocadlo, D.J. O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J. Mol. Biol. 2014, 426, 1736–1752. [Google Scholar] [CrossRef] [PubMed]
- Losev, Y.; Paul, A.; Frenkel-Pinter, M.; Abu-Hussein, M.; Khalaila, I.; Gazit, E.; Segal, D. Novel model of secreted human tau protein reveals the impact of the abnormal N-glycosylation of tau on its aggregation propensity. Sci. Rep. 2019, 9, 2254. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.; Mukrasch, M.D.; von Bergen, M.; Klos-Witkowska, A.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural and microtubule binding properties of tau mutants of frontotemporal dementias. Biochemistry 2007, 46, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Drombosky, K.W.; Hou, Z.; Sari, L.; Kashmer, O.M.; Ryder, B.D.; Perez, V.A.; Woodard, D.R.; Lin, M.M.; Diamond, M.I.; et al. Tau local structure shields an amyloid-forming motif and controls aggregation propensity. Nat. Commun. 2019, 10, 2493. [Google Scholar] [CrossRef] [Green Version]
- Mok, S.A.; Condello, C.; Freilich, R.; Gillies, A.; Arhar, T.; Oroz, J.; Kadavath, H.; Julien, O.; Assimon, V.A.; Rauch, J.N.; et al. Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat. Struct. Mol. Biol. 2018, 25, 384–393. [Google Scholar] [CrossRef]
- Tatebayashi, Y.; Miyasaka, T.; Chui, D.H.; Akagi, T.; Mishima, K.; Iwasaki, K.; Fujiwara, M.; Tanemura, K.; Murayama, M.; Ishiguro, K.; et al. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. USA 2002, 99, 13896–13901. [Google Scholar] [CrossRef] [Green Version]
- Tanemura, K.; Akagi, T.; Murayama, M.; Kikuchi, N.; Murayama, O.; Hashikawa, T.; Yoshiike, Y.; Park, J.M.; Matsuda, K.; Nakao, S.; et al. Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol. Dis. 2001, 8, 1036–1045. [Google Scholar] [CrossRef] [Green Version]
- Barghorn, S.; Zheng-Fischhofer, Q.; Ackmann, M.; Biernat, J.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 2000, 39, 11714–11721. [Google Scholar] [CrossRef] [PubMed]
- Fichou, Y.; Lin, Y.; Rauch, J.N.; Vigers, M.; Zeng, Z.; Srivastava, M.; Keller, T.J.; Freed, J.H.; Kosik, K.S.; Han, S. Cofactors are essential constituents of stable and seeding-active tau fibrils. Proc. Natl. Acad. Sci. USA 2018, 115, 13234–13239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Wilson, D.M.; Binder, L.I. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 2181–2195. [Google Scholar]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef]
- Cremers, C.M.; Knoefler, D.; Gates, S.; Martin, N.; Dahl, J.U.; Lempart, J.; Xie, L.; Chapman, M.R.; Galvan, V.; Southworth, D.R.; et al. Polyphosphate: A Conserved Modifier of Amyloidogenic Processes. Mol. Cell 2016, 63, 768–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.J.; DeTure, M.A.; McBride, M.; Dickson, D.W.; Petrucelli, L. Three repeat isoforms of tau inhibit assembly of four repeat tau filaments. PLoS ONE 2010, 5, e10810. [Google Scholar] [CrossRef] [PubMed]
- Schoch, K.M.; DeVos, S.L.; Miller, R.L.; Chun, S.J.; Norrbom, M.; Wozniak, D.F.; Dawson, H.N.; Bennett, C.F.; Rigo, F.; Miller, T.M. Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model. Neuron 2016, 90, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Harrison, J.; Deaton, C.A.; Johnson, G.V.W. Tau Clearance Mechanisms. Adv. Exp. Med. Biol. 2019, 1184, 57–68. [Google Scholar] [CrossRef]
- Patel, T.K.; Habimana-Griffin, L.; Gao, X.; Xu, B.; Achilefu, S.; Alitalo, K.; McKee, C.A.; Sheehan, P.W.; Musiek, E.S.; Xiong, C.; et al. Dural lymphatics regulate clearance of extracellular tau from the CNS. Mol. Neurodegener. 2019, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, Z.T.; Mok, S.A.; Gestwicki, J.E. Therapeutic Strategies for Restoring Tau Homeostasis. Cold Spring Harb Perspect Med. 2018, 8. [Google Scholar] [CrossRef]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Kovacech, B.; Novak, M. Tau truncation is a productive posttranslational modification of neurofibrillary degeneration in Alzheimer’s disease. Curr. Alzheimer Res. 2010, 7, 708–716. [Google Scholar] [CrossRef]
- Ravalin, M.; Theofilas, P.; Basu, K.; Opoku-Nsiah, K.A.; Assimon, V.A.; Medina-Cleghorn, D.; Chen, Y.F.; Bohn, M.F.; Arkin, M.; Grinberg, L.T.; et al. Specificity for latent C termini links the E3 ubiquitin ligase CHIP to caspases. Nat. Chem. Biol. 2019, 15, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [Green Version]
- Bokenberger, K.; Sjolander, A.; Dahl Aslan, A.K.; Karlsson, I.K.; Akerstedt, T.; Pedersen, N.L. Shift work and risk of incident dementia: A study of two population-based cohorts. Eur. J. Epidemiol. 2018, 33, 977–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhan, G.; Fenik, P.; Brandes, M.; Bell, P.; Francois, N.; Shulman, K.; Veasey, S. Chronic Sleep Disruption Advances the Temporal Progression of Tauopathy in P301S Mutant Mice. J. Neurosci. 2018, 38, 10255–10270. [Google Scholar] [CrossRef] [Green Version]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiei, S.S.; Guerrero-Munoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Wegmann, S.; Kopeikina, K.J.; Hawkes, J.; Rudinskiy, N.; Andermann, M.L.; Spires-Jones, T.L.; Bacskai, B.J.; Hyman, B.T. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 510–514. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res. 2006, 54, 197–201. [Google Scholar] [CrossRef]
- Gerson, J.; Castillo-Carranza, D.L.; Sengupta, U.; Bodani, R.; Prough, D.S.; DeWitt, D.S.; Hawkins, B.E.; Kayed, R. Tau Oligomers Derived from Traumatic Brain Injury Cause Cognitive Impairment and Accelerate Onset of Pathology in Htau Mice. J. Neurotrauma 2016, 33, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flach, K.; Hilbrich, I.; Schiffmann, A.; Gartner, U.; Kruger, M.; Leonhardt, M.; Waschipky, H.; Wick, L.; Arendt, T.; Holzer, M. Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 2012, 287, 43223–43233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; Davidowitz, E.; Lopez, P.; Emadi, S.; Moe, J.; Sierks, M. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell. Biol. 2013, 2013, 260787. [Google Scholar] [CrossRef]
- Swanson, E.; Breckenridge, L.; McMahon, L.; Som, S.; McConnell, I.; Bloom, G.S. Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction. J. Alzheimers. Dis. 2017, 58, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.T.; Donzelli, S.; Chiki, A.; Syed, M.M.K.; Lashuel, H.A. A simple, versatile and robust centrifugation-based filtration protocol for the isolation and quantification of alpha-synuclein monomers, oligomers and fibrils: Towards improving experimental reproducibility in alpha-synuclein research. J. Neurochem. 2020, 153, 103–119. [Google Scholar] [CrossRef]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [Green Version]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Bolton, D.C.; Groth, D.F.; Bowman, K.A.; Cochran, S.P.; McKinley, M.P. Further purification and characterization of scrapie prions. Biochemistry 1982, 21, 6942–6950. [Google Scholar] [CrossRef]
- Wille, H.; Requena, J.R. The Structure of PrP(Sc) Prions. Pathogens 2018, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Brandel, J.P.; Knight, R. Variant Creutzfeldt-Jakob disease. Handb Clin. Neurol. 2018, 153, 191–205. [Google Scholar] [CrossRef]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cescatti, M.; Saverioni, D.; Capellari, S.; Tagliavini, F.; Kitamoto, T.; Ironside, J.; Giese, A.; Parchi, P. Analysis of Conformational Stability of Abnormal Prion Protein Aggregates across the Spectrum of Creutzfeldt-Jakob Disease Prions. J. Virol. 2016, 90, 6244–6254. [Google Scholar] [CrossRef] [Green Version]
- Parchi, P.; Strammiello, R.; Notari, S.; Giese, A.; Langeveld, J.P.; Ladogana, A.; Zerr, I.; Roncaroli, F.; Cras, P.; Ghetti, B.; et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: An updated classification. Acta Neuropathol. 2009, 118, 659–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, G.; Seeger, H.; Bogousslavsky, J.; Tolnay, M.; Janzer, R.C.; Aguzzi, A.; Glatzel, M. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med. 2006, 3, e14. [Google Scholar] [CrossRef] [PubMed]
- Capellari, S.; Strammiello, R.; Saverioni, D.; Kretzschmar, H.; Parchi, P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: Insights into phenotypic variability and disease pathogenesis. Acta Neuropathol. 2011, 121, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Sidle, K.C.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Caroni, P. Selective neuronal vulnerability in neurodegenerative diseases: From stressor thresholds to degeneration. Neuron 2011, 71, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erana, H. The Prion 2018 round tables (II): Abeta, tau, alpha-synuclein... are they prions, prion-like proteins, or what? Prion 2019, 13, 41–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell. Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, A.J.; Yu, P.; Miller, B.B.; Shcherbinin, S.; Dickson, J.; Navitsky, M.; Joshi, A.D.; Devous, M.D., Sr.; Mintun, M.S. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016, 139, 1539–1550. [Google Scholar] [CrossRef] [Green Version]
- Lowe, V.J.; Wiste, H.J.; Senjem, M.L.; Weigand, S.D.; Therneau, T.M.; Boeve, B.F.; Josephs, K.A.; Fang, P.; Pandey, M.K.; Murray, M.E.; et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 2018, 141, 271–287. [Google Scholar] [CrossRef]
- Cope, T.E.; Rittman, T.; Borchert, R.J.; Jones, P.S.; Vatansever, D.; Allinson, K.; Passamonti, L.; Vazquez Rodriguez, P.; Bevan-Jones, W.R.; O’Brien, J.T.; et al. Tau burden and the functional connectome in Alzheimer’s disease and progressive supranuclear palsy. Brain 2018, 141, 550–567. [Google Scholar] [CrossRef]
- Alonso, A.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [Green Version]
- Clavaguera, F.; Hench, J.; Goedert, M.; Tolnay, M. Invited review: Prion-like transmission and spreading of tau pathology. Neuropathol. Appl. Neurobiol. 2015, 41, 47–58. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. Propagation of Tau aggregates. Mol. Brain 2017, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.L.; Narasimhan, S.; Changolkar, L.; He, Z.; Stieber, A.; Zhang, B.; Gathagan, R.J.; Iba, M.; McBride, J.D.; Trojanowski, J.Q.; et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 2016, 213, 2635–2654. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Hench, J.; Lavenir, I.; Schweighauser, G.; Frank, S.; Goedert, M.; Tolnay, M. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014, 127, 299–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morozova, O.A.; March, Z.M.; Robinson, A.S.; Colby, D.W. Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein. Biochemistry 2013, 52, 6960–6967. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Cavallini, A.; Angers, R.; Glover, S.; Murray, T.K.; Barnham, L.; Jackson, S.; O’Neill, M.J.; Isaacs, A.M.; Hutton, M.L.; et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J. Biol. Chem. 2015, 290, 1049–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Narasimhan, S.; Changolkar, L.; Riddle, D.M.; Kats, A.; Stieber, A.; Weitzman, S.A.; Zhang, B.; Li, Z.; Roberson, E.D.; Trojanowski, J.Q.; et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Sharma, A.M.; Thomas, T.L.; Woodard, D.R.; Kashmer, O.M.; Diamond, M.I. Tau monomer encodes strains. Elife 2018, 7. [Google Scholar] [CrossRef]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, e1006323. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.J. Tauopathies as clinicopathological entities. Parkinsonism Relat. Disord. 2016, 22 (Suppl. 1), S29–S33. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Leugers, C.J. Tau and tauopathies. Prog. Mol. Biol. Transl. Sci. 2012, 107, 263–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.E.; Ghoshal, N.; Wall, J.S.; Binder, L.I.; Ksiezak-Reding, H. Structural analysis of Pick’s disease-derived and in vitro-assembled tau filaments. Am. J. Pathol. 2001, 158, 1481–1490. [Google Scholar] [CrossRef]
- Komori, T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. 1999, 9, 663–679. [Google Scholar] [CrossRef]
- Jellinger, K.; Danielczyk, W.; Fischer, P.; Gabriel, E. Clinicopathological analysis of dementia disorders in the elderly. J. Neurol. Sci. 1990, 95, 239–258. [Google Scholar] [CrossRef]
- Sergeant, N.; Wattez, A.; Delacourte, A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: Tau pathologies with exclusively “exon 10” isoforms. J. Neurochem. 1999, 72, 1243–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Abdolmohammadi, B.; Stein, T.D. The neuropathology of chronic traumatic encephalopathy. Handb Clin. Neurol. 2018, 158, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G. Tauopathies. Handb Clin. Neurol. 2017, 145, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Audouard, E.; Houben, S.; Masaracchia, C.; Yilmaz, Z.; Suain, V.; Authelet, M.; De Decker, R.; Buee, L.; Boom, A.; Leroy, K.; et al. High-Molecular-Weight Paired Helical Filaments from Alzheimer Brain Induces Seeding of Wild-Type Mouse Tau into an Argyrophilic 4R Tau Pathology in Vivo. Am. J. Pathol. 2016, 186, 2709–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; McBride, J.D.; Xu, H.; Changolkar, L.; Kim, S.J.; Zhang, B.; Narasimhan, S.; Gibbons, G.S.; Guo, J.L.; Kozak, M.; et al. Transmission of tauopathy strains is independent of their isoform composition. Nat. Commun. 2020, 11, 7. [Google Scholar] [CrossRef]
- Ferrer, I.; Zelaya, M.V.; Aguilo Garcia, M.; Carmona, M.; Lopez-Gonzalez, I.; Andres-Benito, P.; Lidon, L.; Gavin, R.; Garcia-Esparcia, P.; Del Rio, J.A. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol. 2020, 30, 298–318. [Google Scholar] [CrossRef]
- Woerman, A.L.; Aoyagi, A.; Patel, S.; Kazmi, S.A.; Lobach, I.; Grinberg, L.T.; McKee, A.C.; Seeley, W.W.; Olson, S.H.; Prusiner, S.B. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc. Natl. Acad. Sci. USA 2016, 113, E8187–E8196. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaniyappan, S.; Tepper, K.; Biernat, J.; Chandupatla, R.R.; Hubschmann, S.; Irsen, S.; Bicher, S.; Klatt, C.; Mandelkow, E.M.; Mandelkow, E. FRET-based Tau seeding assay does not represent prion-like templated assembly of Tau filaments. Mol. Neurodegener. 2020, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Buist, A.; Soares, A.; Callaerts, K.; Calafate, S.; Stevenaert, F.; Daniels, J.P.; Zoll, B.E.; Crowe, A.; Brunden, K.R.; et al. The Dynamics and Turnover of Tau Aggregates in Cultured Cells: Insights into therapies for tauopathies. J. Biol. Chem. 2016, 291, 13175–13193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644.e612. [Google Scholar] [CrossRef] [PubMed]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Linehan, J.M.; et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardivel, M.; Begard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buee, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.P.; Buee, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef] [Green Version]
- Demaegd, K.; Schymkowitz, J.; Rousseau, F. Transcellular Spreading of Tau in Tauopathies. Chembiochem 2018, 19, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M. Neuroscience. Garbage truck of the brain. Science 2013, 340, 1529–1530. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [Green Version]
- Hadjihambi, A.; Harrison, I.F.; Costas-Rodriguez, M.; Vanhaecke, F.; Arias, N.; Gallego-Duran, R.; Mastitskaya, S.; Hosford, P.S.; Olde Damink, S.W.M.; Davies, N.; et al. Impaired brain glymphatic flow in experimental hepatic encephalopathy. J. Hepatol. 2019, 70, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, X.; Dage, J.L.; Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 2012, 48, 356–366. [Google Scholar] [CrossRef]
- Karch, C.M.; Jeng, A.T.; Goate, A.M. Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J. Biol. Chem. 2012, 287, 42751–42762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Yamada, K.; Cirrito, J.R.; Stewart, F.R.; Jiang, H.; Finn, M.B.; Holmes, B.B.; Binder, L.I.; Mandelkow, E.M.; Diamond, M.I.; Lee, V.M.; et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011, 31, 13110–13117. [Google Scholar] [CrossRef]
- Buch, K.; Riemenschneider, M.; Bartenstein, P.; Willoch, F.; Muller, U.; Schmolke, M.; Nolde, T.; Steinmann, C.; Guder, W.G.; Kurz, A. [Tau protein. A potential biological indicator for early detection of Alzheimer disease]. Nervenarzt 1998, 69, 379–385. [Google Scholar] [CrossRef]
- Rubenstein, R.; Chang, B.; Davies, P.; Wagner, A.K.; Robertson, C.S.; Wang, K.K. A novel, ultrasensitive assay for tau: Potential for assessing traumatic brain injury in tissues and biofluids. J. Neurotrauma 2015, 32, 342–352. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Kovac, A.; Majerova, P.; Bullock, K.M.; Shi, M.; Zhang, J. Tau Proteins Cross the Blood-Brain Barrier. J. Alzheimers Dis. 2017, 55, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Wang, C.; Tang, Y.; Mok, S.A.; Tsai, R.M.; Rojas, J.C.; Karydas, A.; Miller, B.L.; Boxer, A.L.; et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol. Neurodegener. 2020, 15, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viotti, C. ER to Golgi-Dependent Protein Secretion: The Conventional Pathway. Methods Mol. Biol. 2016, 1459, 3–29. [Google Scholar] [CrossRef]
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Merezhko, M.; Brunello, C.A.; Yan, X.; Vihinen, H.; Jokitalo, E.; Uronen, R.L.; Huttunen, H.J. Secretion of Tau via an Unconventional Non-vesicular Mechanism. Cell Rep. 2018, 25, 2027–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Muller, H.M.; Nachman, E.; Steringer, J.P.; Ruiz de Almodovar, C.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Mohamed, N.V.; Desjardins, A.; Lippe, R.; Fon, E.A.; Leclerc, N. Rab7A regulates tau secretion. J. Neurochem. 2017, 141, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Avila, J.; Hernandez, F. Propagation of Tau via Extracellular Vesicles. Front. Neurosci 2019, 13, 698. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Gotz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.S.; Kim, D.K.; Kim, Y.K.; Gho, Y.S. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013, 13, 1554–1571. [Google Scholar] [CrossRef]
- Fevrier, B.; Raposo, G. Exosomes: Endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 2004, 16, 415–421. [Google Scholar] [CrossRef]
- Korkut, C.; Li, Y.; Koles, K.; Brewer, C.; Ashley, J.; Yoshihara, M.; Budnik, V. Regulation of postsynaptic retrograde signaling by presynaptic exosome release. Neuron 2013, 77, 1039–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.; Garcia-Garcia, E.; Gomez-Ramos, A.; Falcon-Perez, J.M.; Diaz-Hernandez, M.; Hernandez, F.; Avila, J. Tau overexpression results in its secretion via membrane vesicles. Neurodegener. Dis. 2012, 10, 73–75. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Kruger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Dujardin, S.; Begard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.C.; Bousset, L.; Melki, R.; et al. Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef] [PubMed]
- Amyere, M.; Mettlen, M.; Van Der Smissen, P.; Platek, A.; Payrastre, B.; Veithen, A.; Courtoy, P.J. Origin, originality, functions, subversions and molecular signalling of macropinocytosis. Int. J. Med. Microbiol. 2002, 291, 487–494. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Varghese, M.; Ksiezak-Reding, H.; Dzhun, A.; Wang, J.; Pasinetti, G.M. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J. Biol. Chem. 2012, 287, 20522–20533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Miyata, H.; Kametani, F.; Nonaka, T.; Akiyama, H.; Hisanaga, S.; Hasegawa, M. Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 2015, 129, 895–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Morozova, V.; Cohen, L.S.; Makki, A.E.; Shur, A.; Pilar, G.; El Idrissi, A.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef] [Green Version]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horonchik, L.; Tzaban, S.; Ben-Zaken, O.; Yedidia, Y.; Rouvinski, A.; Papy-Garcia, D.; Barritault, D.; Vlodavsky, I.; Taraboulos, A. Heparan sulfate is a cellular receptor for purified infectious prions. J. Biol. Chem. 2005, 280, 17062–17067. [Google Scholar] [CrossRef] [Green Version]
- Rauch, J.N.; Chen, J.J.; Sorum, A.W.; Miller, G.M.; Sharf, T.; See, S.K.; Hsieh-Wilson, L.C.; Kampmann, M.; Kosik, K.S. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 2018, 8, 6382. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.D.; Wassmer, T.; Fraser, G.; Smith, J.; Perkinton, M.; Billinton, A.; Livesey, F.J. Extracellular Monomeric and Aggregated Tau Efficiently Enter Human Neurons through Overlapping but Distinct Pathways. Cell Rep. 2018, 22, 3612–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Jadhav, H.R.; Bhatt, T. Dynamin Functions and Ligands: Classical Mechanisms Behind. Mol. Pharmacol. 2017, 91, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calafate, S.; Flavin, W.; Verstreken, P.; Moechars, D. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 2016, 17, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Parton, R.G.; Dotti, C.G. Cell biology of neuronal endocytosis. J. Neurosci. Res. 1993, 36, 1–9. [Google Scholar] [CrossRef]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; de Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Wauters, M.; Wattiez, R.; Ris, L. Internalization of the Extracellular Full-Length Tau Inside Neuro2A and Cortical Cells Is Enhanced by Phosphorylation. Biomolecules 2016, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- de Calignon, A.; Polydoro, M.; Suarez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Dujardin, S.; Lecolle, K.; Caillierez, R.; Begard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Auregan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.; Cooper, J.; Murray, T.K.; Garn, K.; McNaughton, E.; Clarke, H.; Parhizkar, S.; Ward, M.A.; Cavallini, A.; Jackson, S.; et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014, 127, 667–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fein, J.A.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Yang, F.; Cole, G.M.; Gylys, K.H. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am. J. Pathol. 2008, 172, 1683–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolow, S.; Henkins, K.M.; Bilousova, T.; Gonzalez, B.; Vinters, H.V.; Miller, C.A.; Cornwell, L.; Poon, W.W.; Gylys, K.H. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J. Neurochem. 2015, 133, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Pickett, E.K.; Henstridge, C.M.; Allison, E.; Pitstick, R.; Pooler, A.; Wegmann, S.; Carlson, G.; Hyman, B.T.; Spires-Jones, T.L. Spread of tau down neural circuits precedes synapse and neuronal loss in the rTgTauEC mouse model of early Alzheimer’s disease. Synapse 2017, 71. [Google Scholar] [CrossRef]
- Ahmadian, N.; Hejazi, S.; Mahmoudi, J.; Talebi, M. Tau Pathology of Alzheimer Disease: Possible Role of Sleep Deprivation. Basic Clin. Neurosci. 2018, 9, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Dunnett, S.; Ho, Y.S.; Chang, R.C. The role of sleep deprivation and circadian rhythm disruption as risk factors of Alzheimer’s disease. Front. Neuroendocrinol. 2019, 54, 100764. [Google Scholar] [CrossRef]
- Özcan, G.G.; Lim, S.; Leighton, P.L.A.; Allison, W.T.; Rihel, J. Bi-directional modification of sleep and wake by amyloid beta oligomers. bioRxiv 2019, 10.1101/610014, 610014. [Google Scholar] [CrossRef] [Green Version]
- Luther, M.; Poppert Cordts, K.M.; Williams, C.N. Sleep disturbances after pediatric traumatic brain injury: A systematic review of prevalence, risk factors, and association with recovery. Sleep 2020. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, M.C.; Beaulieu-Bonneau, S.; Morin, C.M. Sleep-wake disturbances after traumatic brain injury. Lancet Neurol. 2015, 14, 746–757. [Google Scholar] [CrossRef]
- Wei, L.; Wen, Y.T.; Thompson, H.J.; Liu, C.Y.; Su, Y.K.; Chen, P.Y.; Chen, C.Y.; Chuang, Y.H.; Lin, Y.J.; Chen, C.T.; et al. Sleep Disturbances Following Traumatic Brain Injury in Older Adults: A Comparison Study. J. Head Trauma Rehabil. 2020. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, J.D.; Wolfe, M.; Mysiw, W.J.; Jackson, R.D.; Bogner, J.A. Early identification of mild traumatic brain injury in female victims of domestic violence. Am. J. Obstet Gynecol. 2003, 188, S71–S76. [Google Scholar] [CrossRef]
- Jordan, B.D. The clinical spectrum of sport-related traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.S.; Jing, R.; McFaull, S.R.; Cusimano, M.D. Health & Economic Burden of Traumatic Brain Injury in the Emergency Department. Can. J. Neurol. Sci. 2016, 43, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Cernak, I.; Noble-Haeusslein, L.J. Traumatic brain injury: An overview of pathobiology with emphasis on military populations. J. Cereb Blood Flow Metab. 2010, 30, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forde, C.T.; Karri, S.K.; Young, A.M.; Ogilvy, C.S. Predictive markers in traumatic brain injury: Opportunities for a serum biosignature. Br. J. Neurosurg. 2014, 28, 8–15. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blennow, K.; Hardy, J.; Zetterberg, H. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Haces, M.; Tang, J.; Acosta, G.; Fernandez, J.; Shi, R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeKosky, S.T.; Blennow, K.; Ikonomovic, M.D.; Gandy, S. Acute and chronic traumatic encephalopathies: Pathogenesis and biomarkers. Nat. Rev. Neurol. 2013, 9, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Masel, B.E.; DeWitt, D.S. Traumatic brain injury: A disease process, not an event. J. Neurotrauma 2010, 27, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Plassman, B.L.; Havlik, R.J.; Steffens, D.C.; Helms, M.J.; Newman, T.N.; Drosdick, D.; Phillips, C.; Gau, B.A.; Welsh-Bohmer, K.A.; Burke, J.R.; et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 2000, 55, 1158–1166. [Google Scholar] [CrossRef]
- Guo, Z.; Cupples, L.A.; Kurz, A.; Auerbach, S.H.; Volicer, L.; Chui, H.; Green, R.C.; Sadovnick, A.D.; Duara, R.; DeCarli, C.; et al. Head injury and the risk of AD in the MIRAGE study. Neurology 2000, 54, 1316–1323. [Google Scholar] [CrossRef]
- Schofield, P.W.; Tang, M.; Marder, K.; Bell, K.; Dooneief, G.; Chun, M.; Sano, M.; Stern, Y.; Mayeux, R. Alzheimer’s disease after remote head injury: An incidence study. J. Neurol. Neurosurg. Psychiatry 1997, 62, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Shively, S.; Scher, A.I.; Perl, D.P.; Diaz-Arrastia, R. Dementia resulting from traumatic brain injury: What is the pathology? Arch. Neurol. 2012, 69, 1245–1251. [Google Scholar] [CrossRef]
- Dhandapani, S.; Manju, D.; Sharma, B.; Mahapatra, A. Prognostic significance of age in traumatic brain injury. J. Neurosci. Rural Pract. 2012, 3, 131–135. [Google Scholar] [CrossRef]
- Graves, A.B.; White, E.; Koepsell, T.D.; Reifler, B.V.; van Belle, G.; Larson, E.B.; Raskind, M. The association between head trauma and Alzheimer’s disease. Am. J. Epidemiol. 1990, 131, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Washington, P.M.; Villapol, S.; Burns, M.P. Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp. Neurol. 2016, 275 Pt 3, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Millspaugh, J. Dementia pugilistica. US Naval Med. Bull. 1937, 35, e303. [Google Scholar]
- Martland, H.S. Punch drunk. J. Am. Med. Assoc. 1928, 91, 1103–1107. [Google Scholar] [CrossRef]
- Bieniek, K.F.; Blessing, M.M.; Heckman, M.G.; Diehl, N.N.; Serie, A.M.; Paolini, M.A., 2nd; Boeve, B.F.; Savica, R.; Reichard, R.R.; Dickson, D.W. Association between contact sports participation and chronic traumatic encephalopathy: A retrospective cohort study. Brain Pathol. 2020, 30, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, N.; Culver, C.; Dams-O’Connor, K.; Hammond, F.; Diaz-Arrastia, R. Clinical phenotype of dementia after traumatic brain injury. J. Neurotrauma 2013, 30, 1117–1122. [Google Scholar] [CrossRef] [Green Version]
- Stern, R.A.; Daneshvar, D.H.; Baugh, C.M.; Seichepine, D.R.; Montenigro, P.H.; Riley, D.O.; Fritts, N.G.; Stamm, J.M.; Robbins, C.A.; McHale, L.; et al. Clinical presentation of chronic traumatic encephalopathy. Neurology 2013, 81, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.C.; Hall, R.C.; Chapman, M.J. Definition, diagnosis, and forensic implications of postconcussional syndrome. Psychosomatics 2005, 46, 195–202. [Google Scholar] [CrossRef] [Green Version]
- McKee, A.C.; Gavett, B.E.; Stern, R.A.; Nowinski, C.J.; Cantu, R.C.; Kowall, N.W.; Perl, D.P.; Hedley-Whyte, E.T.; Price, B.; Sullivan, C.; et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 918–929. [Google Scholar] [CrossRef]
- Critchley, M. Medical aspects of boxing, particularly from a neurological standpoint. Br. Med. J. 1957, 1, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Grahmann, H.; Ule, G. [Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica & traumatic encephalopathy of boxers)]. Psychiatr. Neurol. 1957, 134, 261–283. [Google Scholar]
- Schwab, N.; Hazrati, L.N. Assessing the Limitations and Biases in the Current Understanding of Chronic Traumatic Encephalopathy. J. Alzheimers Dis. 2018, 64, 1067–1076. [Google Scholar] [CrossRef]
- Maas, A.I.; Menon, D.K.; Steyerberg, E.W.; Citerio, G.; Lecky, F.; Manley, G.T.; Hill, S.; Legrand, V.; Sorgner, A.; Participants, C.-T.; et al. Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI): A prospective longitudinal observational study. Neurosurgery 2015, 76, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgoraptis, N.; Li, L.M.; Whittington, A.; Zimmerman, K.A.; Maclean, L.M.; McLeod, C.; Ross, E.; Heslegrave, A.; Zetterberg, H.; Passchier, J.; et al. In vivo detection of cerebral tau pathology in long-term survivors of traumatic brain injury. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meaney, D.F.; Smith, D.H. Biomechanics of concussion. Clin Sports Med. 2011, 30, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giza, C.C.; Hovda, D.A. The new neurometabolic cascade of concussion. Neurosurgery 2014, 75 (Suppl. 4), S24–S33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi, A.; Zhang, J.H.; Obenaus, A. Response of the cerebral vasculature following traumatic brain injury. J. Cereb Blood Flow Metab. 2017, 37, 2320–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.S.; Craft, R.; Yu, G.Q.; Ho, K.; Wang, X.; Mohan, G.; Mangnitsky, S.; Ponnusamy, R.; Mucke, L. Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS ONE 2014, 9, e115765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, B.R.; Meabon, J.S.; Martin, T.J.; Mourad, P.D.; Bennett, R.; Kraemer, B.C.; Cernak, I.; Petrie, E.C.; Emery, M.J.; Swenson, E.R.; et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J. Alzheimers. Dis. 2013, 37, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.T.; Sanchez, L.; Esparza, T.J.; Brody, D.L. Distinct temporal and anatomical distributions of amyloid-beta and tau abnormalities following controlled cortical impact in transgenic mice. PLoS ONE 2011, 6, e25475. [Google Scholar] [CrossRef] [Green Version]
- Corsellis, J.A.; Bruton, C.J.; Freeman-Browne, D. The aftermath of boxing. Psychol. Med. 1973, 3, 270–303. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cairns, N.J.; Dickson, D.W.; Folkerth, R.D.; Keene, C.D.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.P.; Stewart, W.; et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016, 131, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, A.C.; Stern, R.A.; Nowinski, C.J.; Stein, T.D.; Alvarez, V.E.; Daneshvar, D.H.; Lee, H.S.; Wojtowicz, S.M.; Hall, G.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Ost, M.; Nylen, K.; Csajbok, L.; Ohrfelt, A.O.; Tullberg, M.; Wikkelso, C.; Nellgard, P.; Rosengren, L.; Blennow, K.; Nellgard, B. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology 2006, 67, 1600–1604. [Google Scholar] [CrossRef] [PubMed]
- Franz, G.; Beer, R.; Kampfl, A.; Engelhardt, K.; Schmutzhard, E.; Ulmer, H.; Deisenhammer, F. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology 2003, 60, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Liliang, P.C.; Liang, C.L.; Lu, K.; Wang, K.W.; Weng, H.C.; Hsieh, C.H.; Tsai, Y.D.; Chen, H.J. Relationship between injury severity and serum tau protein levels in traumatic brain injured rats. Resuscitation 2010, 81, 1205–1208. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Uryu, K.; Higuchi, M.; Longhi, L.; Hoover, R.; Fujimoto, S.; McIntosh, T.; Lee, V.M.; Trojanowski, J.Q. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J. Neurotrauma 2005, 22, 1134–1141. [Google Scholar] [CrossRef] [Green Version]
- Zanier, E.R.; Bertani, I.; Sammali, E.; Pischiutta, F.; Chiaravalloti, M.A.; Vegliante, G.; Masone, A.; Corbelli, A.; Smith, D.H.; Menon, D.K.; et al. Induction of a transmissible tau pathology by traumatic brain injury. Brain 2018, 141, 2685–2699. [Google Scholar] [CrossRef]
- Bittar, A.; Bhatt, N.; Hasan, T.F.; Montalbano, M.; Puangmalai, N.; McAllen, S.; Ellsworth, A.; Carretero Murillo, M.; Taglialatela, G.; Lucke-Wold, B.; et al. Neurotoxic tau oligomers after single versus repetitive mild traumatic brain injury. Brain Commun. 2019, 1, fcz004. [Google Scholar] [CrossRef]
- Ojo, J.O.; Mouzon, B.; Greenberg, M.B.; Bachmeier, C.; Mullan, M.; Crawford, F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J. Neuropathol. Exp. Neurol. 2013, 72, 137–151. [Google Scholar] [CrossRef] [Green Version]
- Geddes, J.F.; Vowles, G.H.; Nicoll, J.A.; Revesz, T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999, 98, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G., 3rd; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Sullan, M.J.; Asken, B.M.; Jaffee, M.S.; DeKosky, S.T.; Bauer, R.M. Glymphatic system disruption as a mediator of brain trauma and chronic traumatic encephalopathy. Neurosci. Biobehav. Rev. 2018, 84, 316–324. [Google Scholar] [CrossRef]
- Piantino, J.; Lim, M.M.; Newgard, C.D.; Iliff, J. Linking Traumatic Brain Injury, Sleep Disruption and Post-Traumatic Headache: A Potential Role for Glymphatic Pathway Dysfunction. Curr. Pain Headache Rep. 2019, 23, 62. [Google Scholar] [CrossRef]
- Mathias, J.L.; Alvaro, P.K. Prevalence of sleep disturbances, disorders, and problems following traumatic brain injury: A meta-analysis. Sleep Med. 2012, 13, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.K.; Leonessa, F.; Ling, G.S. Blast TBI Models, Neuropathology, and Implications for Seizure Risk. Front. Neurol. 2014, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinsky, M.; Storzbach, D.; Goy, E.; Evrard, C. Traumatic brain injury and psychogenic seizures in veterans. J. Head Trauma Rehabil. 2015, 30, E65–E70. [Google Scholar] [CrossRef]
- Asikainen, I.; Kaste, M.; Sarna, S. Early and late posttraumatic seizures in traumatic brain injury rehabilitation patients: Brain injury factors causing late seizures and influence of seizures on long-term outcome. Epilepsia 1999, 40, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Temkin, N.R. Risk factors for posttraumatic seizures in adults. Epilepsia 2003, 44, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Parko, K.L. Clinical approach to posttraumatic epilepsy. Semin Neurol. 2015, 35, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Schierhout, G.; Roberts, I. Prophylactic antiepileptic agents after head injury: A systematic review. J. Neurol. Neurosurg. Psychiatry 1998, 64, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, C.R.; McGuire, J.L.; DePasquale, E.A.; Gardner, A.E.; Floyd, C.L.; McCullumsmith, R.E. Glutamate Neurotransmission in Rodent Models of Traumatic Brain Injury. J. Neurotrauma 2017, 34, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ji, B.; Cheng, B.; Chen, J.; Bai, B. Neuroprotection of microRNA in neurological disorders (Review). Biomed. Rep. 2014, 2, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.; Shultz, S.R.; Hovens, C.M.; Velakoulis, D.; Jones, N.C.; O’Brien, T.J. Hyperphosphorylated tau is implicated in acquired epilepsy and neuropsychiatric comorbidities. Mol. Neurobiol. 2014, 49, 1532–1539. [Google Scholar] [CrossRef]
- Crespo-Biel, N.; Canudas, A.M.; Camins, A.; Pallas, M. Kainate induces AKT, ERK and cdk5/GSK3beta pathway deregulation, phosphorylates tau protein in mouse hippocampus. Neurochem. Int. 2007, 50, 435–442. [Google Scholar] [CrossRef]
- Liang, Z.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Dysregulation of tau phosphorylation in mouse brain during excitotoxic damage. J. Alzheimers Dis. 2009, 17, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.F.; Zeng, C.; Ma, Y.F.; Guo, T.H.; Chen, J.M.; Chen, Y.; Cai, X.F.; Li, F.R.; Wang, X.H.; Huang, W.J.; et al. Potential roles of Cdk5/p35 and tau protein in hippocampal mossy fiber sprouting in the PTZ kindling model. Clin. Lab. 2010, 56, 127–136. [Google Scholar]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. Revisiting the Impact of Neurodegenerative Proteins in Epilepsy: Focus on Alpha-Synuclein, Beta-Amyloid, and Tau. Biology 2020, 9, 122. [Google Scholar] [CrossRef]
- Bugay, V.; Bozdemir, E.; Vigil, F.A.; Holstein, D.M.; Chun, S.H.; Elliot, W.; Sprague, C.; Cavazos, J.E.; Zamora, D.O.; Rule, G.; et al. A mouse model of repetitive blast traumatic brain injury reveals post-trauma seizures and increased neuronal excitability. J. Neurotrauma 2019. [Google Scholar] [CrossRef]
- Akman, O.; Demiralp, T.; Ates, N.; Onat, F.Y. Electroencephalographic differences between WAG/Rij and GAERS rat models of absence epilepsy. Epilepsy Res. 2010, 89, 185–193. [Google Scholar] [CrossRef]
- Kandratavicius, L.; Balista, P.A.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, E.H.; Garcia-Cairasco, N.; Bueno-Junior, L.S.; Leite, J.P. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, N.; Harada, R.; Ishiki, A.; Kikuchi, A.; Nakamura, T.; Kudo, Y. The development and validation of tau PET tracers: Current status and future directions. Clin. Transl. Imaging 2018, 6, 305–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, E.R.; Leuzy, A.; Gauthier, S.; Rosa-Neto, P. Developments in Tau PET Imaging. Can. J. Neurol. Sci. 2014, 41, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alyenbaawi, H.; Kanyo, R.; Locskai, L.F.; Kamali-Jamil, R.; DuVal, M.G.; Bai, Q.; Wille, H.; Burton, E.A.; Allison, W.T. Seizures are a druggable mechanistic link between TBI and subsequent tauopathy. eLife 2020. In Revision. [Google Scholar]
- Castellani, R.J.; Perry, G. Dementia Pugilistica Revisited. J. Alzheimers. Dis. 2017, 60, 1209–1221. [Google Scholar] [CrossRef] [Green Version]
- Hof, P.R.; Bouras, C.; Buee, L.; Delacourte, A.; Perl, D.P.; Morrison, J.H. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol. 1992, 85, 23–30. [Google Scholar] [CrossRef]
- Katsumoto, A.; Takeuchi, H.; Tanaka, F. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front. Neurol. 2019, 10, 980. [Google Scholar] [CrossRef]
- Nam, W.H.; Choi, Y.P. In vitro generation of tau aggregates conformationally distinct from parent tau seeds of Alzheimer’s brain. Prion 2019, 13, 1–12. [Google Scholar] [CrossRef]
- Bessen, R.A.; Marsh, R.F. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 1992, 66, 2096–2101. [Google Scholar] [CrossRef] [Green Version]


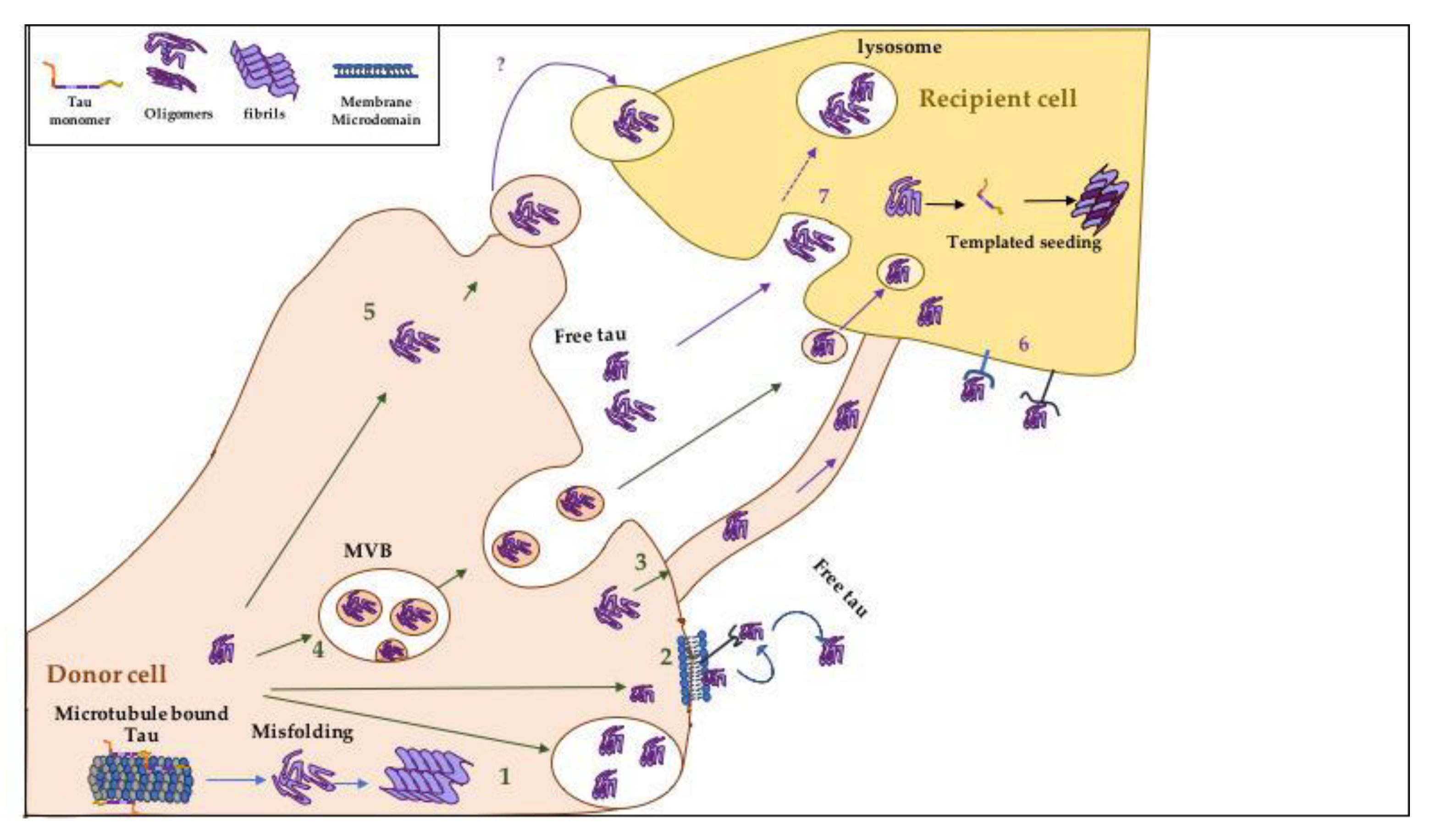
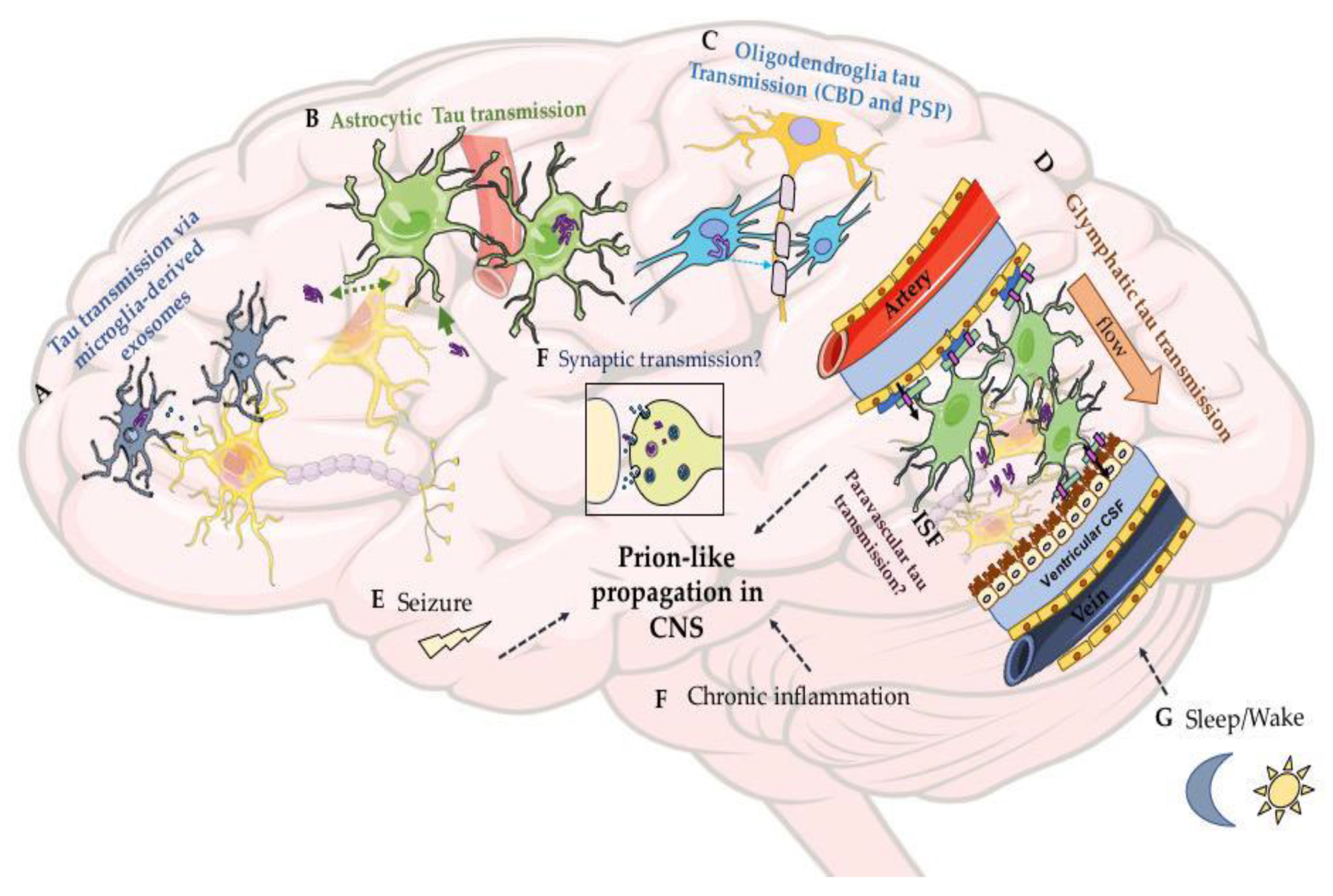

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Tauopathies | The Form of Tau Aggregates | Tau Filaments Composition and Ultrastructure | Supportive Pathological Features Not Related to Phosphorylated Tau (P-Tau) | Cells Affected by Tau Pathology and Disease Progression |
|---|---|---|---|---|
| Pick’s Disease (PiD) (primary tauopathy) | Round cytoplasmic tau aggregates (known as Pick bodies), ramified astrocytic filaments, and neuropil threads. | Filaments consist of 3R tau isoforms. Most of tau filaments (93%) are narrow filaments (with a slight helical twist), previously mentioned as straight filaments (SF), and the remaining small percent are classified as wide filaments [47]. | Neuronal loss, gliosis, and ballooned cells known as Pick cells | Tau pathology is observed in both neuronal and glial cells. Tauopathy is found restricted to neocortical and limbic areas in the frontotemporal regions, then spreads to subcortical structure and brainstem, followed by the motor cortex. In severe cases, it is found in the visual cortex [48]. |
| Corticobasal Degeneration (CBD) (primary tauopathy) | Filaments take the shape of comma or coil-like (coiled bodies) pre-tangles, astrocytic plaques, and neuritic threads. | Tau filaments composed of 4R isoforms. The filaments in CBD are heterogeneous, containing narrow single-stranded filaments and wide double-stranded filaments, with the first being three times more abundant [49,50]. | Neuronal loss and degeneration of the substantia nigra in mild and severe cases. | Tau pathology affecting both neurons and glia. Based on a recent neuropathological analysis of CBD cases, tau pathology is limited to the anterior of the frontal cortex, amygdala, and basal ganglia in earlier stages, then progresses to affect more prominently the frontal and parietal cortices, amygdala, caudate, subthalamic nucleus, and pontine tegmentum in late stages [51]. |
| Progressive Supranuclear Palsy (PSP) (primary tauopathy) | Neurofibrillary tangles, globose tangles, coiled bodies, and star-like or tufted astrocytes. | Filaments consists of 4R tau isoforms. Tau filaments formed are SF with rare twisted filaments [52]. | Neuronal loss and gliosis. | Tauopathy affects both neurons and glia. Pathology is restricted in an earlier stage to the pallido-luyso-nigral regions then proceeds to involve the basal ganglia. In later stages, the pathology progresses to involve areas in the frontal and parietal lobes such as the subthalamic nucleus and the substantia nigra [53]. |
| Globular Glial Tauopathy (GGT) (primary tauopathy) | Large and globular tau inclusions in oligodendrocyte and astrocytes. | Filaments composed mainly of 4R isoforms. Information regarding the ultrastructure of tau filaments is still unknown. | Atrophy, neuronal loss, and gliosis in the primary cortex and or in the corticospinal tract (some cases). | Tau filaments found in both neurons and glial cells. Rare 4R tauopathy and cases present with different patterns of neuropathology depending on the affected area. In a subtype of GGT that causes motor neuron disease, tau pathology is observed throughout the primary motor cortex. In a second subtype, which is associated with frontotemporal dementia, tau pathology is detected throughout the frontotemporal cortex [54]. Further information regarding the progression of tauopathy is still needed in GGT. |
| Argyrophilic Grain Disease (AGD) (primary tauopathy) | Argyrophilic grains, oligodendritic coiled bodies, neuronal pretangles. | Tau filaments composed of 4R isoforms. | Spongy ambient gyrus degeneration. | Tau filaments affects both neurons and glia. Tauopathy affects the ambient gyrus initially and then progresses through the medial temporal lobe. Tau pathology in advanced cases seen in septum and insular cortex [55]. |
| Chronic Traumatic Encephalopathy (CTE) (primary tauopathy) | Neurofibrillary tangles, dot-like or grain like neurites, and astrocytic tangles and thorn- shaped astrocytes | While neuronal tau filaments consist of 3R and 4R isoforms, astroglia tau filaments are composed mainly of 4R isoforms [56]. 90 percent of the filaments are helical filaments called type 1 which are distinct from PHFs and SF found in AD. The remaining are similar to PHFs in AD [57]. | TDP-43 Neuronal and glial Inclusions in severe cases. Axonal injury and degeneration Diffuse Aβ plaques (present in ~50% of cases) [46]. | Tau aggregates affect both neurons and glia. The pathology begins in the frontal cortex around small vessels then spreads throughout the cortex. The pathology affects the outer layer of the cerebral cortex, unlike AD (more details mentioned in Section 3.4 and 3.5) |
| Alzheimer’s Disease (AD) (secondary tauopathy) | Tau filaments in the form of neurofibrillary tangles, neuropil threads. | Tau filaments composed of both 3R and 4R isoforms and found as paired helical filaments (PHFs) and SFs [58]. | Progressive accumulation of Aβ plaques in distinct patterns (one of the main hallmarks of AD) [59]. | Tau pathology seen predominantly in neurons. It starts at the entorhinal and transentorhinal regions, then progresses to the limbic regions including the hippocampus and, at later stages, tauopathy is observed throughout the neocortex [41]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alyenbaawi, H.; Allison, W.T.; Mok, S.-A. Prion-Like Propagation Mechanisms in Tauopathies and Traumatic Brain Injury: Challenges and Prospects. Biomolecules 2020, 10, 1487. https://doi.org/10.3390/biom10111487
Alyenbaawi H, Allison WT, Mok S-A. Prion-Like Propagation Mechanisms in Tauopathies and Traumatic Brain Injury: Challenges and Prospects. Biomolecules. 2020; 10(11):1487. https://doi.org/10.3390/biom10111487
Chicago/Turabian StyleAlyenbaawi, Hadeel, W. Ted Allison, and Sue-Ann Mok. 2020. "Prion-Like Propagation Mechanisms in Tauopathies and Traumatic Brain Injury: Challenges and Prospects" Biomolecules 10, no. 11: 1487. https://doi.org/10.3390/biom10111487