More than Meets the ISG15: Emerging Roles in the DNA Damage Response and Beyond



Abstract
:
1. Ubiquitin and Ubiquitin-Like Proteins (UBLs)—An Overview
2. ISG15 and ISGylation


3. ISG15: More Than an Antiviral Protein
4. The DNA Damage Response (DDR)
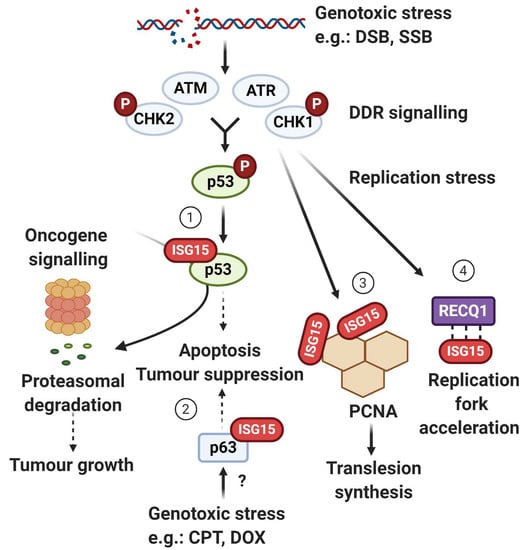
5. ISG15 System and p53—A Complex Relationship
5.1. ISG15 as a p53 Degradation Signal
5.2. p53-Mediated Induction of ISG15 System
5.3. ISG15 Enhances p53 Transactivity
6. ISGylation of ∆Np63 and Tumourigenesis
7. Translesion DNA Synthesis (TLS)—A New Terminator Model
8. ISG15 in Replication Fork Progression
9. Further Roles in Genome Stability—Bright Prospects for ISG15
10. Conclusions
Funding
Conflicts of Interest
References
- Cappadocia, L.; Lima, C.D. Ubiquitin-like protein conjugation: Structures, chemistry, and mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef]
- Hartmann-Petersen, R.; Gordon, C. Integral UBL domain proteins: A family of proteasome interacting proteins. Semin. Cell Dev. Biol. 2004, 15, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Madsen, L.; Schulze, A.; Seeger, M.; Hartmann-Petersen, R. Ubiquitin domain proteins in disease. BMC Biochem. 2007, 8, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeller, D.; Hecker, C.-M.; Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 2006, 6, 776–788. [Google Scholar] [CrossRef]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of Proteins by Ubiquitin and Ubiquitin-Like Proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.; Lerman, L.O.; Lerman, A. Ubiquitin and Ubiquitin-Like Proteins in Protein Regulation. Circ. Res. 2007, 100, 1276–1291. [Google Scholar] [CrossRef] [Green Version]
- Osborne, H.C.; Irving, E.; Schmidt, C.K. The Ubiquitin/UBL Drug Target Repertoire. Trends Mol. Med. 2020, 9–10. [Google Scholar] [CrossRef]
- Jackson, S.P.; Durocher, D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.S.; Jackson, S.P. Ubiquitylation, neddylation and the DNA damage response. Open Biol. 2015, 5, 150018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa, I.C.; Schmidt, C.K. Ubiquitin-like proteins in the DNA damage response: The next generation. Essays Biochem. 2020, 64, 737–752. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Lou, Z. Ubiquitin and ubiquitin-like molecules in DNA double strand break repair. Cell Biosci. 2020, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, A.L.; Ahrens, P.; Bright, P.M.; Ankel, H. Interferon induced a 15-kilodalton protein exhibiting marked homology to ubiquitin. J. Biol. Chem. 1987, 262, 11315–11323. [Google Scholar] [PubMed]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, P.J.; Broeze, R.J.; Lengyel, P. Accumulation of an mRNA and protein in interferon-treated Ehrlich ascites tumour cells. Nature 1979, 279, 523–525. [Google Scholar] [CrossRef]
- Loeb, K.R.; Haas, A.L. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J. Biol. Chem. 1992, 267, 7806–7813. [Google Scholar]
- D’Cunha, J.; Knight, E.; Haas, A.L.; Truitt, R.L.; Borden, E.C. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc. Natl. Acad. Sci. USA 1996, 93, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, J.; Wang, M.; Fu, Z.; Klein, J.M.; Haas, A.L.; Kim, J.-J.P. Crystal Structure of the Interferon-Induced Ubiquitin-Like Protein ISG15. J. Biol. Chem. 2005, 280, 27356–27365. [Google Scholar] [CrossRef] [Green Version]
- Dao, C.T.; Zhang, D.-E. ISG15: A ubiquitin-like enigma. Front. Biosci. 2005, 10, 2701–2722. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.G.; Yan, X.Z.; Xie, Y.Y.; Gao, X.C.; Song, A.X.; Zhang, D.E.; Hu, H.Y. Different roles for two ubiquitin-like domains of ISG15 in protein modification. J. Biol. Chem. 2008, 283, 13370–13377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso, M.C.; Djoric, N.; Walser, F.; Hess, S.; Schmid, F.M.; Burger, S.; Knobeloch, K.-P.; Penengo, L. Interferon-stimulated gene 15 accelerates replication fork progression inducing chromosomal breakage. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, D.E. Interferon-stimulated gene 15 and the protein ISGylation system. J. Interf. Cytokine Res. 2011, 31, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magor, K.E.; Navarro, D.M.; Barber, M.R.W.; Petkau, K.; Fleming-Canepa, X.; Blyth, G.A.D.; Blaine, A.H. Defense genes missing from the flight division. Dev. Comp. Immunol. 2013, 41, 377–388. [Google Scholar] [CrossRef]
- Daczkowski, C.M.; Dzimianski, J.V.; Clasman, J.R.; Goodwin, O.; Mesecar, A.D.; Pegan, S.D. Structural Insights into the Interaction of Coronavirus Papain-Like Proteases and Interferon-Stimulated Gene Product 15 from Different Species. J. Mol. Biol. 2017, 429, 1661–1683. [Google Scholar] [CrossRef]
- Langley, C.; Goodwin, O.; Dzimianski, J.V.; Daczkowski, C.M.; Pegan, S.D. Structure of interferon-stimulated gene product 15 (ISG15) from the bat species Myotis davidii and the impact of interdomain ISG15 interactions on viral protein engagement. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 21–31. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, X. Structural insights into the species preference of the influenza B virus NS1 protein in ISG15 binding. Protein Cell 2019, 10, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Zuin, A.; Isasa, M.; Crosas, B. Ubiquitin Signaling: Extreme Conservation as a Source of Diversity. Cells 2014, 3, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Knigth, E.; Fahey, D.; Cordova, B.; Hillman, M.; Kutny, R.; Reich, N.; Blomstrom, D. A 15-kDa interferon-induced protein is derived by COOH-terminal processing of a 17-kDa precursor. J. Biol. Chem. 1988, 263, 4520–4522. [Google Scholar]
- Potter, J.L.; Narasimhan, J.; Mende-Mueller, L.; Haas, A.L. Precursor processing of pro-ISG15/UCRP, an interferon-β-induced ubiquitin-like protein. J. Biol. Chem. 1999, 274, 25061–25068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durfee, L.A.; Huibregtse, J.M. The ISG15 Conjugation System. Methods Mol. Biol. 2012, 832, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, K., II; Giannakopoulos, N.V.; Virgin, H.W.; Zhang, D.-E. Interferon-Inducible Ubiquitin E2, Ubc8, Is a Conjugating Enzyme for Protein ISGylation. Mol. Cell. Biol. 2004, 24, 9592–9600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastur, A. Herc5, an Interferon-Induced HECT E3 Enzyme, Is Required for Conjugation of ISG15 in Human Cells. J. Biol. Chem. 2006, 281, 4334–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Zhang, D.-E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef] [Green Version]
- Okumura, F.; Zou, W.; Zhang, D.E. ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes Dev. 2007, 21, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Kroismayr, R.; Baranyi, U.; Stehlik, C.; Dorfleutner, A.; Binder, B.R.; Lipp, J. HERC5, a HECT E3 ubiquitin ligase tightly regulated in LPS activated endothelial cells. J. Cell Sci. 2004, 117, 4749–4756. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.J.Y.; Pung, Y.F.; Sze, N.S.-K.; Chin, K.-C. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. USA 2006, 103, 10735–10740. [Google Scholar] [CrossRef] [Green Version]
- Duda, D.M.; Olszewski, J.L.; Schuermann, J.P.; Kurinov, I.; Miller, D.J.; Nourse, A.; Alpi, A.F.; Schulman, B.A. Structure of HHARI, a RING-IBR-RING ubiquitin ligase: Autoinhibition of an Ariadne-family E3 and insights into ligation mechanism. Structure 2013, 21, 1030–1041. [Google Scholar] [CrossRef] [Green Version]
- Martín-Vicente, M.; Medrano, L.M.; Resino, S.; García-Sastre, A.; Martínez, I. TRIM25 in the regulation of the antiviral innate immunity. Front. Immunol. 2017, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Beaudenon, S.L.; Kelley, M.L.; Waddell, M.B.; Yuan, W.; Schulman, B.A.; Huibregtse, J.M.; Krug, R.M. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-/-induced ubiquitin-like protein. Proc. Natl. Acad. Sci. USA 2004, 101, 7578–7582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durfee, L.A.; Kelley, M.L.; Huibregtse, J.M. The basis for selective E1-E2 interactions in the ISG15 conjugation system. J. Biol. Chem. 2008, 283, 23895–23902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malakhov, M.P.; Malakhova, O.A.; Kim, K., II; Ritchie, K.J.; Zhang, D.E. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J. Biol. Chem. 2002, 277, 9976–9981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basters, A.; Geurink, P.P.; El Oualid, F.; Ketscher, L.; Casutt, M.S.; Krause, E.; Ovaa, H.; Knobeloch, K.P.; Fritz, G. Molecular characterization of ubiquitin-specific protease 18 reveals substrate specificity for interferon-stimulated gene 15. FEBS J. 2014, 281, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.J.; Hahn, C.S.; Kim, K., II; Yan, M.; Rosario, D.; Li, L.; de la Torre, J.C.; Zhang, D.-E. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat. Med. 2004, 10, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Ketscher, L.; Hannß, R.; Morales, D.J.; Basters, A.; Guerra, S.; Goldmann, T.; Hausmann, A.; Prinz, M.; Naumann, R.; Pekosz, A.; et al. Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc. Natl. Acad. Sci. USA 2015, 112, 1577–1582. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.-Y. DNA damage induces nuclear translocation of parkin. J. Biomed. Sci. 2009, 16, 67. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.-Y. Regulation of DNA repair by parkin. Biochem. Biophys. Res. Commun. 2009, 382, 321–325. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Nam, H.-J.; Gao, B.; Yin, P.; Qin, B.; Yi, S.-Y.; Ham, H.; Evans, D.; Kim, S.-H.; et al. Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1. Mol. Cell 2015, 60, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Ma, X.; Tu, Y.; Huang, M.; Liu, H.; Wang, F.; Gong, J.; Wang, J.; Li, X.; Chen, Q.; et al. Parkin regulates translesion DNA synthesis in response to UV radiation. Oncotarget 2017, 8, 36423–36437. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-W.; Wang, X.-F.; Ni, S.-J.; Qin, W.; Zhao, L.-Q.; Hua, R.-X.; Lu, Y.-W.; Li, J.; Dimri, G.P.; Guo, W.-J. UBTD1 induces cellular senescence through an UBTD1-Mdm2/p53 positive feedback loop. J. Pathol. 2015, 235, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Chabot, B. The RNA Splicing Response to DNA Damage. Biomolecules 2015, 5, 2935–2977. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.Y. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 2003, 17, 1630–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, M.; Pieroni, S.; Brunacci, C.; Piobbico, D.; Bartoli, D.; Bellet, M.M.; Colombo, E.; Pelicci, P.G.; Della Fazia, M.A.; Servillo, G. Hepatocyte odd protein shuttling (HOPS) is a bridging protein in the nucleophosmin-p19Arf network. Oncogene 2013, 32, 3350–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Zhang, Y.; Chen, J.; Zhou, B.; Chen, G.; Chen, P. Tmub1 Suppresses Hepatocellular Carcinoma by Promoting the Ubiquitination of ΔNp63 Isoforms. Mol. Ther. Oncolytics 2020, 18, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; Piobbico, D.; Chiacchiaretta, M.; Brunacci, C.; Pieroni, S.; Bartoli, D.; Gargaro, M.; Fallarino, F.; Puccetti, P.; Soddu, S.; et al. HOPS/TMUB1 retains p53 in the cytoplasm and sustains p53-dependent mitochondrial apoptosis. EMBO Rep. 2020, 21, 1–18. [Google Scholar] [CrossRef]
- Mistry, H.; Tamblyn, L.; Butt, H.; Sisgoreo, D.; Gracias, A.; Larin, M.; Gopalakrishnan, K.; Hande, M.; McPherson, J. UHRF1 is a genome caretaker that facilitates the DNA damage response to γ-irradiation. Genome Integr. 2010, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ma, H.; Inuzuka, H.; Diao, J.; Lan, F.; Shi, Y.G.; Wei, W.; Shi, Y. DNA Damage Regulates UHRF1 Stability via the SCF β-TrCP E3 Ligase. Mol. Cell. Biol. 2013, 33, 1139–1148. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, H.; Chen, Y.; Yang, X.; Wang, P.; Liu, T.; Deng, M.; Qin, B.; Correia, C.; Lee, S.; et al. A cell cycle-dependent BRCA1–UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat. Commun. 2016, 7, 10201. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Uckelmann, M.; Densham, R.M.; Baas, R.; Winterwerp, H.H.K.; Fish, A.; Sixma, T.K.; Morris, J.R. USP48 restrains resection by site-specific cleavage of the BRCA1 ubiquitin mark from H2A. Nat. Commun. 2018, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velimezi, G.; Robinson-Garcia, L.; Muñoz-Martínez, F.; Wiegant, W.W.; da Silva, J.F.; Owusu, M.; Moder, M.; Wiedner, M.; Rosenthal, S.B.; Fisch, K.M.; et al. Map of synthetic rescue interactions for the Fanconi anemia DNA repair pathway identifies USP48. Nat. Commun. 2018, 9, 2280. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Chan, H.-H.; Chen, S.-H.; Sarvagalla, S.; Chen, P.-S.; Coumar, M.S.; Cheng, S.M.; Chang, Y.-C.; Lin, C.-H.; Leung, E.; et al. BIRC5/Survivin is a novel ATG12–ATG5 conjugate interactor and an autophagy-induced DNA damage suppressor in human cancer and mouse embryonic fibroblast cells. Autophagy 2020, 16, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenciute, G.; Liu, S.; Yucer, N.; Shi, Y.; Ortiz, P.; Liu, Q.; Kim, B.-J.; Odejimi, A.O.; Leng, M.; Qin, J.; et al. Nuclear BAG6-UBL4A-GET4 Complex Mediates DNA Damage Signaling and Cell Death. J. Biol. Chem. 2013, 288, 20547–20557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Dai, J.; Ma, M.; Xu, T.; Kong, Y.; Cui, C.; Chi, Z.; Si, L.; Tang, H.; Yang, L.; et al. RBCK1 promotes p53 degradation via ubiquitination in renal cell carcinoma. Cell Death Dis. 2019, 10, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-H.; O’Connor, D.; Brimmell, M.; Packham, G. The BAG-1 cochaperone is a negative regulator of p73-dependent transcription. Br. J. Cancer 2009, 100, 1347–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-Y.; Pfeiffer, H.K.; Mellert, H.S.; Stanek, T.J.; Sussman, R.T.; Kumari, A.; Yu, D.; Rigoutsos, I.; Thomas-Tikhonenko, A.; Seidel, H.E.; et al. Inhibition of the Single Downstream Target BAG1 Activates the Latent Apoptotic Potential of MYC. Mol. Cell. Biol. 2011, 31, 5037–5045. [Google Scholar] [CrossRef] [Green Version]
- Paredes, F.; Parra, V.; Torrealba, N.; Navarro-Marquez, M.; Gatica, D.; Bravo-Sagua, R.; Troncoso, R.; Pennanen, C.; Quiroga, C.; Chiong, M.; et al. HERPUD1 protects against oxidative stress-induced apoptosis through downregulation of the inositol 1,4,5-trisphosphate receptor. Free Radic. Biol. Med. 2016, 90, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Jachimowicz, R.D.; Beleggia, F.; Isensee, J.; Velpula, B.B.; Goergens, J.; Bustos, M.A.; Doll, M.A.; Shenoy, A.; Checa-Rodriguez, C.; Wiederstein, J.L.; et al. UBQLN4 Represses Homologous Recombination and Is Overexpressed in Aggressive Tumors. Cell 2019, 176, 505–519.e22. [Google Scholar] [CrossRef] [Green Version]
- Garvin, A.J.; Morris, J.R. SUMO, a small, but powerful, regulator of double-strand break repair. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160281. [Google Scholar] [CrossRef] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, M.; Saied, F. Multigrid solution of the Poisson-Boltzmann equation. J. Comput. Chem. 1993, 14, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Holst, M.J.; Saied, F. Numerical solution of the nonlinear Poisson-Boltzmann equation: Developing more robust and efficient methods. J. Comput. Chem. 1995, 16, 337–364. [Google Scholar] [CrossRef] [Green Version]
- Holst, M. Adaptive numerical treatment of elliptic systems on manifolds. Adv. Comput. Math. 2001, 15, 139–191. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bank, R.E.; Holst, M. A New Paradigm for Parallel Adaptive Meshing Algorithms. SIAM Rev. 2003, 45, 291–323. [Google Scholar] [CrossRef] [Green Version]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Malakhova, O.; Malakhov, M.; Hetherington, C.; Zhang, D.E. Lipopolysaccharide activates the expression of ISG15-specific protease UBP43 via interferon regulatory factor 3. J. Biol. Chem. 2002, 277, 14703–14711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitha-Rowe, I.; Hasse, B.A.; Dmitrovsky, E. Involvement of UBE1L in ISG15 Conjugation during Retinoid-induced Differentiation of Acute Promyelocytic Leukemia. J. Biol. Chem. 2004, 279, 18178–18187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, M. Cell cycle arrest and apoptosis provoked by UV radiation-induced DNA damage are transcriptionally highly divergent responses. Nucleic Acids Res. 2003, 31, 4779–4790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Hummer, B.T.; Li, X.; Hassel, B.A. Camptothecin Induces the Ubiquitin-like Protein, ISG15, and Enhances ISG15 Conjugation in Response to Interferon. J. Interf. Cytokine Res. 2004, 24, 647–654. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Jo, M.G.; Yoo, H.M.; Hong, S.H.; Park, J.M.; Ka, S.H.; Oh, K.H.; Seol, J.H.; Jung, Y.K.; Chung, C.H. Chemosensitivity is controlled by p63 modification with ubiquitin-like protein ISG15. J. Clin. Invest. 2012, 122, 2622–2636. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Yang, S.W.; Yu, K.R.; Ka, S.H.; Lee, S.W.; Seol, J.H.; Jeon, Y.J.; Chung, C.H. Modification of PCNA by ISG15 Plays a Crucial Role in Termination of Error-Prone Translesion DNA Synthesis. Mol. Cell 2014, 54, 626–638. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.J.; Park, J.H.; Chung, C.H. Interferon-Stimulated Gene 15 in the Control of Cellular Responses to Genotoxic Stress. Mol. Cells 2017, 40, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Yang, S.W.; Park, J.M.; Ka, S.H.; Kim, J.-H.; Kong, Y.-Y.; Jeon, Y.J.; Seol, J.H.; Chung, C.H. Positive feedback regulation of p53 transactivity by DNA damage-induced ISG15 modification. Nat. Commun. 2016, 7, 12513. [Google Scholar] [CrossRef] [Green Version]
- Radoshevich, L.; Impens, F.; Ribet, D.; Quereda, J.J.; Tham, T.N.; Nahori, M.A.; Bierne, H.; Dussurget, O.; Pizarro-Cerdá, J.; Knobeloch, K.P.; et al. ISG15 counteracts Listeria monocytogenes infection. Elife 2015, 4, 1–23. [Google Scholar] [CrossRef]
- Lertsooksawat, W.; Wongnoppavich, A.; Chairatvit, K. Up-regulation of interferon-stimulated gene 15 and its conjugation machinery, UbE1L and UbcH8 expression by tumor necrosis factor-α through p38 MAPK and JNK signaling pathways in human lung carcinoma. Mol. Cell. Biochem. 2019, 462, 51–59. [Google Scholar] [CrossRef]
- Swaim, C.D.; Scott, A.F.; Canadeo, L.A.; Huibregtse, J.M. Extracellular ISG15 Signals Cytokine Secretion through the LFA-1 Integrin Receptor. Mol. Cell 2017, 68, 581–590.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, P.F.; Mansur, D.S. Beyond ISGlylation: Functions of Free Intracellular and Extracellular ISG15. J. Interf. Cytokine Res. 2017, 37, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Kim, K., II; Luo, J.K.; Zou, W.; Kumar, K.G.S.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.-L.; Hassel, B.A. Proteasomes Modulate Conjugation to the Ubiquitin-like Protein, ISG15. J. Biol. Chem. 2003, 278, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.D.; Haas, A.L.; Wood, L.M.; Tsai, Y.C.; Pestka, S.; Rubin, E.H.; Saleem, A.; Nur-E-Kamal, A.; Liu, L.F. Elevated expression of ISG15 in tumor cells interferes with the ubiquitin/26S proteasome pathway. Cancer Res. 2006, 66, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.X.; Yang, K.; Liu, X.; Liu, X.Y.; Wei, B.; Shan, Y.F.; Zhu, L.H.; Wang, C. Positive Regulation of Interferon Regulatory Factor 3 Activation by Herc5 via ISG15 Modification. Mol. Cell. Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, L.M.; Sankar, S.; Reed, R.E.; Haas, A.L.; Liu, L.F.; McKinnon, P.; Desai, S.D. A Novel Role for ATM in Regulating Proteasome-Mediated Protein Degradation through Suppression of the ISG15 Conjugation Pathway. PLoS ONE 2011, 6, e16422. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, M.; Poluektova, L.Y.; Tuma, D.J.; Kharbanda, K.K.; Osna, N.A. Acetaldehyde Disrupts Interferon Alpha Signaling in Hepatitis C Virus-Infected Liver Cells by Up-Regulating USP18. Alcohol. Clin. Exp. Res. 2016, 40, 2329–2338. [Google Scholar] [CrossRef]
- Arimoto, K.-I.; Konishi, H.; Shimotohno, K. UbcH8 regulates ubiquitin and ISG15 conjugation to RIG-I. Mol. Immunol. 2008, 45, 1078–1084. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Zhang, H.; Zhu, M.; Chen, F.; Hu, Y.; Liu, H.; Zhu, H. Interferon-stimulated Gene 15 (ISG15) is a trigger for tumorigenesis and metastasis of hepatocellular carcinoma. Oncotarget 2014, 5, 8429–8441. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.-B.; Arimoto, K.; Motamedchaboki, K.; Yan, M.; Wolf, D.A.; Zhang, D.-E. Identification and characterization of a novel ISG15-ubiquitin mixed chain and its role in regulating protein homeostasis. Sci. Rep. 2015, 5, 12704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.J.; Blumen, S.; Pitha-Rowe, I.; Kitareewan, S.; Freemantle, S.J.; Feng, Q.; Dmitrovsky, E. UBE1L represses PML/RARα by targeting the PML domain for ISG15ylation. Mol. Cancer Ther. 2008, 7, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Sekula, D.; Guo, Y.; Liu, X.; Black, C.C.; Galimberti, F.; Shah, S.J.; Sempere, L.F.; Memoli, V.; Andersen, J.B.; et al. UBE1L causes lung cancer growth suppression by targeting cyclin D1. Mol. Cancer Ther. 2008, 7, 3780–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-F.; Wee, S.; Gunaratne, J.; Lane, D.P.; Bulavin, D.V. Isg15 controls p53 stability and functions. Cell Cycle 2014, 13, 2199–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.S.; Kwon, Y.J.; Chun, Y.J. CYP1B1 activates Wnt/β-catenin signaling through suppression of Herc5-mediated ISGylation for protein degradation on β-catenin in HeLa cells. Toxicol. Res. 2017, 33, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Li, Y.; Wang, H.; Zhao, J.; Zhao, Y.; Liu, Z.; Ma, H. FOXO3a is stabilized by USP18-mediated de-ISGylation and inhibits TGF-β1-induced fibronectin expression. J. Investig. Med. 2020, 68, 786–791. [Google Scholar] [CrossRef]
- Wan, X.; Chen, H.; Khan, M.A.; Xu, A.; Yang, F.; Zhang, Y.; Zhang, D. ISG15 inhibits IFN-α-resistant liver cancer cell growth. Biomed Res. Int. 2013, 2013, 570909. [Google Scholar] [CrossRef] [Green Version]
- Im, E.; Yoo, L.; Hyun, M.; Shin, W.H.; Chung, K.C. Covalent ISG15 conjugation positively regulates the ubiquitin E3 ligase activity of parkin. Open Biol. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yoo, L.; Yoon, A.R.; Yun, C.O.; Chung, K.C. Covalent ISG15 conjugation to CHIP promotes its ubiquitin E3 ligase activity and inhibits lung cancer cell growth in response to type i interferon article. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Nguyen, T.; Goins, W.F.; Chiocca, E.A. Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. J. Biol. Chem. 2015, 290, 1485–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, S.D.; Reed, R.E.; Babu, S.; Lorio, E.A. ISG15 deregulates autophagy in genotoxin-treated ataxia telangiectasia cells. J. Biol. Chem. 2013, 288, 2388–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.D.; Reed, R.E.; Juncker, M.A.; Fang, Z.; Desai, S.D. Evidence for the Deregulation of Protein Turnover Pathways in Atm-Deficient Mouse Cerebellum: An Organotypic Study. J. Neuropathol. Exp. Neurol. 2017, 76, 578–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falvey, C.M.; O’Donovan, T.R.; El-Mashed, S.; Nyhan, M.J.; O’Reilly, S.; McKenna, S.L. UBE2L6/UBCH8 and ISG15 attenuate autophagy in esophageal cancer cells. Oncotarget 2017, 8, 23479–23491. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Zheng, H.; Dong, W.; Lv, H.; Lin, J.; Guo, K.; Zhang, Y. Antiviral activity of ISG15 against classical swine fever virus replication in porcine alveolar macrophages via inhibition of autophagy by ISGylating BECN1. Vet. Res. 2020, 51, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Zhang, T.; Xiao, J.; Zhu, K.; Wei, R.; Wu, Z.; Meng, H.; Li, Y.; Yuan, J. Modification of BECN1 by ISG15 plays a crucial role in autophagy regulation by type I IFN/ interferon. Autophagy 2015, 11, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burks, J.; Reed, R.E.; Desai, S.D. ISGylation governs the oncogenic function of Ki-Ras in breast cancer. Oncogene 2014, 33, 794–803. [Google Scholar] [CrossRef]
- Wang, J.-M.; Liu, B.-Q.; Zhang, Q.; Hao, L.; Li, C.; Yan, J.; Zhao, F.-Y.; Qiao, H.-Y.; Jiang, J.-Y.; Wang, H.-Q. ISG15 suppresses translation of ABCC2 via ISGylation of hnRNPA2B1 and enhances drug sensitivity in cisplatin resistant ovarian cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118647. [Google Scholar] [CrossRef]
- Okumura, F.; Okumura, A.J.; Uematsu, K.; Hatakeyama, S.; Zhang, D.E.; Kamura, T. Activation of double-stranded rna-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 2013, 288, 2839–2847. [Google Scholar] [CrossRef] [Green Version]
- Holthaus, D.; Vasou, A.; Bamford, C.G.G.; Andrejeva, J.; Paulus, C.; Randall, R.E.; McLauchlan, J.; Hughes, D.J. Direct Antiviral Activity of IFN-Stimulated Genes Is Responsible for Resistance to Paramyxoviruses in ISG15-Deficient Cells. J. Immunol. 2020, 205, 261–271. [Google Scholar] [CrossRef]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Held, T.; Basler, M.; Knobeloch, K.; Groettrup, M. Evidence for an involvement of the ubiquitin-like modifier ISG15 in MHC class I antigen presentation. Eur. J. Immunol. 2020, eji.202048646. [Google Scholar] [CrossRef] [PubMed]
- Villarroya-Beltri, C.; Baixauli, F.; Mittelbrunn, M.; Fernández-Delgado, I.; Torralba, D.; Moreno-Gonzalo, O.; Baldanta, S.; Enrich, C.; Guerra, S.; Sánchez-Madrid, F. ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat. Commun. 2016, 7, 13588. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.H.; Yang, Y.C.; Hsieh, M.Y.; Yeh, Y.C.; Li, T.K. A negative feedback of the HIF-1α pathway via interferon-stimulated gene 15 and ISGylation. Clin. Cancer Res. 2013, 19, 5927–5939. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.-B.; Miyauchi-Ishida, S.; Arimoto, K.; Liu, D.; Yan, M.; Liu, C.-W.; Győrffy, B.; Zhang, D.-E. Type I IFN induces protein ISGylation to enhance cytokine expression and augments colonic inflammation. Proc. Natl. Acad. Sci. USA 2015, 112, 14313–14318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, P.F.; Van Weyenbergh, J.; Delgobo, M.; de Patricio, D.O.; Ferguson, B.J.; Guabiraba, R.; Dierckx, T.; Menezes, S.M.; Báfica, A.; Mansur, D.S. ISG15-Induced IL-10 Is a Novel Anti-Inflammatory Myeloid Axis Disrupted during Active Tuberculosis. J. Immunol. 2018, 200, 1434–1442. [Google Scholar] [CrossRef] [Green Version]
- Østvik, A.E.; Svendsen, T.D.; van Beelen Granlund, A.; Doseth, B.; Skovdahl, H.K.; Bakke, I.; Thorsvik, S.; Afroz, W.; Walaas, G.A.; Mollnes, T.E.; et al. Intestinal Epithelial Cells Express Immunomodulatory ISG15 during Active Ulcerative Colitis and Crohn’s Disease. J. Crohn’s Colitis 2020, 14, 920–934. [Google Scholar] [CrossRef]
- Swaim, C.D.; Canadeo, L.A.; Monte, K.J.; Khanna, S.; Lenschow, D.J.; Huibregtse, J.M. Modulation of Extracellular ISG15 Signaling by Pathogens and Viral Effector Proteins. Cell Rep. 2020, 31, 107772. [Google Scholar] [CrossRef]
- Desai, S.D.; Reed, R.E.; Burks, J.; Wood, L.M.; Pullikuth, A.K.; Haas, A.L.; Liu, L.F.; Breslin, J.W.; Meiners, S.; Sankar, S. ISG15 disrupts cytoskeletal architecture and promotes motility in human breast cancer cells. Exp. Biol. Med. 2012, 237, 38–49. [Google Scholar] [CrossRef]
- Cerikan, B.; Shaheen, R.; Colo, G.P.; Gläßer, C.; Hata, S.; Knobeloch, K.P.; Alkuraya, F.S.; Fässler, R.; Schiebel, E. Cell-Intrinsic Adaptation Arising from Chronic Ablation of a Key Rho GTPase Regulator. Dev. Cell 2016, 39, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Hermann, M.; Bogunovic, D. ISG15: In Sickness and in Health. Trends Immunol. 2017, 38, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Dzimianski, J.V.; Scholte, F.E.M.; Bergeron, É.; Pegan, S.D. ISG15: It’s Complicated. J. Mol. Biol. 2019, 431, 4203–4216. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Gao, Y.; Mutter-Rottmayer, E.; Zlatanou, A.; Vaziri, C.; Yang, Y. Mechanisms of Post-Replication DNA Repair. Genes 2017, 8, 64. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA Synthesis and Mutagenesis in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Sharma, S.; Brosh, R.M. Human RECQ1 Is a DNA Damage Responsive Protein Required for Genotoxic Stress Resistance and Suppression of Sister Chromatid Exchanges. PLoS ONE 2007, 2, e1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berti, M.; Chaudhuri, A.R.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, H.C.; Yaffe, M.B. Phospho-Ser/Thr-binding domains: Navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 2013, 14, 563–580. [Google Scholar] [CrossRef]
- Huang, Y.F.; Bulavin, D.V. Oncogene-mediated regulation of p53 ISGylation and functions. Oncotarget 2014, 5, 5808–5818. [Google Scholar] [CrossRef] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Mercer, W.E.; Wiman, K.G.; Kohn, K.W.; Pietenpol, J.A.; Kastan, M.B.; Kinzler, K.W.; Vogelstein, B. WAF1/CIP1 Is Induced in p53-mediated G1 Arrest and Apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Hummer, B.T.; Li, X.-L.; Hassel, B.A. Role for p53 in Gene Induction by Double-Stranded RNA. J. Virol. 2001, 75, 7774–7777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Zong, Z.; Wang, Q.; Zhang, Z.; Deng, H. ISG15 silencing increases cisplatin resistance via activating p53-mediated cell DNA repair. Oncotarget 2017, 8, 107452–107461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuffour, E.O.; König, R.; Häussinger, D.; Schulz, W.A.; Münk, C. ISG15 Deficiency Enhances HIV-1 Infection by Accumulating Misfolded p53. MBio 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forys, J.T.; Kuzmicki, C.E.; Saporita, A.J.; Winkeler, C.L.; Maggi, L.B.; Weber, J.D. ARF and p53 Coordinate Tumor Suppression of an Oncogenic IFN-β-STAT1-ISG15 Signaling Axis. Cell Rep. 2014, 7, 514–526. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.R.; Simmen, R.C.M.; Raj, V.R.; Van, T.T.; MacLeod, S.L.; Simmen, F.A. Krüppel-like factor 9 (KLF9) prevents colorectal cancer through inhibition of interferon-related signaling. Carcinogenesis 2015, 36, 946–955. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Hua, P.; Wen, J.; Hu, Y.; Yang, H.; Xie, X. Prognostic value of ISG15 mRNA level in drinkers with esophageal squamous cell cancers. Int. J. Clin. Exp. Pathol. 2015, 8, 10975–10984. [Google Scholar]
- Qiu, X.; Hong, Y.; Yang, D.; Xia, M.; Zhu, H.; Li, Q.; Xie, H.; Wu, Q.; Liu, C.; Zuo, C. ISG15 as a novel prognostic biomarker for hepatitis B virus-related hepatocellular carcinoma. Int. J. Clin. Exp. Med. 2015, 8, 17140–17150. [Google Scholar]
- Hollingsworth, J.; Lau, A.; Tone, A.; Kollara, A.; Allen, L.; Colgan, T.J.; Dube, V.; Rosen, B.; Murphy, K.J.; Greenblatt, E.M.; et al. BRCA1 Mutation Status and Follicular Fluid Exposure Alters NFκB Signaling and ISGylation in Human Fallopian Tube Epithelial Cells. Neoplasia 2018, 20, 697–709. [Google Scholar] [CrossRef]
- Andersen, J.B.; Aaboe, M.; Borden, E.C.; Goloubeva, O.G.; Hassel, B.A.; Ørntoft, T.F. Stage-associated overexpression of the ubiquitin-like protein, ISG15, in bladder cancer. Br. J. Cancer 2006, 94, 1465–1471. [Google Scholar] [CrossRef]
- Bektas, N.; Noetzel, E.; Veeck, J.; Press, M.F.; Kristiansen, G.; Naami, A.; Hartmann, A.; Dimmler, A.; Beckmann, M.W.; Knüchel, R.; et al. The ubiquitin-like molecule interferon-stimulated gene 15 (ISG15) is a potential prognostic marker in human breast cancer. Breast Cancer Res. 2008, 10, R58. [Google Scholar] [CrossRef] [Green Version]
- Ina, S.; Hirono, S.; Noda, T.; Yamaue, H. Identifying molecular markers for chemosensitivity to gemcitabine in pancreatic cancer: Increased expression of interferon-stimulated gene 15 kd is associated with intrinsic chemoresistance. Pancreas 2010, 39, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Jinawath, N.; Furukawa, Y.; Hasegawa, S.; Li, M.; Tsunoda, T.; Satoh, S.; Yamaguchi, T.; Imamura, H.; Inoue, M.; Shiozaki, H.; et al. Comparison of gene-expression profiles between diffuse- and intestinal-type gastric cancers using a genome-wide cDNA microarray. Oncogene 2004, 23, 6830–6844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laljee, R.P.; Muddaiah, S.; Salagundi, B.; Cariappa, P.M.; Indra, A.S.; Sanjay, V.; Ramanathan, A. Interferon Stimulated Gene—ISG15 Is a Potential Diagnostic Biomarker in Oral Squamous Cell Carcinomas. Asian Pacific J. Cancer Prev. 2013, 14, 1147–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padovan, E.; Terracciano, L.; Certa, U.; Jacobs, B.; Reschner, A.; Bolli, M.; Spagnoli, G.C.; Borden, E.C.; Heberer, M. Interferon stimulated gene 15 constitutively produced by melanoma cells induces e-cadherin expression on human dendritic cells. Cancer Res. 2002, 62, 3453–3458. [Google Scholar]
- Talvinen, K.; Tuikkala, J.; Grönroos, J.; Huhtinen, H.; Kronqvist, P.; Aittokallio, T.; Nevalainen, O.; Hiekkanen, H.; Nevalainen, T.; Sundström, J. Biochemical and clinical approaches in evaluating the prognosis of colon cancer. Anticancer Res. 2006, 26, 4745–4751. [Google Scholar]
- Wei, J.; Zaika, E.; Zaika, A. p53 Family: Role of Protein Isoforms in Human Cancer. J. Nucleic Acids 2012, 2012, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Candi, E.; Dinsdale, D.; Rufini, A.; Salomoni, P.; Knight, R.A.; Mueller, M.; Krammer, P.H.; Melino, G. TAp63 and ΔNp63 in Cancer and Epidermal Development. Cell Cycle 2007, 6, 274–284. [Google Scholar] [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA–protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Quinet, A.; Lemaçon, D.; Vindigni, A. Replication Fork Reversal: Players and Guardians. Mol. Cell 2017, 68, 830–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chen, H.; Zhang, J.; Wang, Y.; Simoneau, A.; Yang, H.; Levine, A.S.; Zou, L.; Chen, Z.; Lan, L. cGAS suppresses genomic instability as a decelerator of replication forks. Sci. Adv. 2020, 6, eabb8941. [Google Scholar] [CrossRef] [PubMed]
- Mosbech, A.; Gibbs-Seymour, I.; Kagias, K.; Thorslund, T.; Beli, P.; Povlsen, L.; Nielsen, S.V.; Smedegaard, S.; Sedgwick, G.; Lukas, C.; et al. DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 2012, 19, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Meroni, A.; Vindigni, A. ISG15 fast-tracks DNA replication. J. Cell Biol. 2020, 219, 8–9. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Desai, S.D. ISG15: A double edged sword in cancer. Oncoimmunology 2015, 4, 14–15. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.H.; Kim, H.K.; Kim, E.; Kim, I.H.; Kim, J.H.; Chang, H.J.; Choi, I.J.; Lim, H.S.; Kim, I.J.; Kang, H.C.; et al. Increased expression of metallothionein is associated with irinotecan resistance in gastric cancer. Cancer Res. 2004, 64, 4703–4706. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.D.; Wood, L.M.; Tsai, Y.C.; Hsieh, T.S.; Marks, J.R.; Scott, G.L.; Giovanella, B.C.; Liu, L.F. ISG15 as a novel tumor biomarker for drug sensitivity. Mol. Cancer Ther. 2008, 7, 1430–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessema, M.; Yingling, C.M.; Thomas, C.L.; Klinge, D.M.; Bernauer, A.M.; Liu, Y.; Dacic, S.; Siegfried, J.M.; Dahlberg, S.E.; Schiller, J.H.; et al. SULF2 methylation is prognostic for lung cancer survival and increases sensitivity to topoisomerase-I inhibitors via induction of ISG15. Oncogene 2012, 31, 4107–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Wei, J.; Wang, H.; Yue, G.; Yu, L.; Yang, Y.; Xie, L.; Zou, Z.; Qian, X.; Ding, Y.; et al. A three-gene signature as potential predictive biomarker for irinotecan sensitivity in gastric cancer. J. Transl. Med. 2013, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luszczek, W.; Cheriyath, V.; Mekhail, T.M.; Borden, E.C. Combinations of DNA methyltransferase and histone deacetylase inhibitors induce DNA damage in small cell lung cancer cells: Correlation of resistance with IFN-stimulated gene expression. Mol. Cancer Ther. 2010, 9, 2309–2321. [Google Scholar] [CrossRef] [Green Version]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.A.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, A.C.; Patin, E.C.; Harrington, K.J.; Melcher, A.A. The immunological consequences of radiation-induced DNA damage. J. Pathol. 2019, 247, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [Green Version]
- Jacquelot, N.; Yamazaki, T.; Roberti, M.P.; Duong, C.P.M.; Andrews, M.C.; Verlingue, L.; Ferrere, G.; Becharef, S.; Vétizou, M.; Daillère, R.; et al. Sustained type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019, 29, 846–861. [Google Scholar] [CrossRef]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere position effect: Regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014, 28, 2464–2476. [Google Scholar] [CrossRef] [Green Version]
- Lou, Z.; Wei, J.; Riethman, H.; Baur, J.A.; Voglauer, R.; Shay, J.W.; Wright, W.E. Telomere length regulates ISG15 expression in human cells. Aging 2009, 1, 608–621. [Google Scholar] [CrossRef] [Green Version]
- Hirashima, K.; Migita, T.; Sato, S.; Muramatsu, Y.; Ishikawa, Y.; Seimiya, H. Telomere Length Influences Cancer Cell Differentiation In Vivo. Mol. Cell. Biol. 2013, 33, 2988–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirashima, K.; Seimiya, H. Telomeric repeat-containing RNA/G-quadruplex-forming sequences cause genome-wide alteration of gene expression in human cancer cells in vivo. Nucleic Acids Res. 2015, 43, 2022–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.K.; Sharma, S.; Sengupta, S.; Saha, D.; Kumar, P.; Hussain, T.; Srivastava, V.; Roy, S.D.; Shay, J.W.; Chowdhury, S. Telomere length-dependent transcription and epigenetic modifications in promoters remote from telomere ends. PLoS Genet. 2018, 14, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.; Yan, M.; Yin, X.; Zhang, D. Hematopoietic cells from Ube1L-deficient mice exhibit an impaired proliferation defect under the stress of bone marrow transplantation. Blood Cells Mol. Dis. 2010, 45, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuillier, F.; Li, Z.; Commere, P.H.; Dynesen, L.T.; Pellegrini, S. USP18 and ISG15 coordinately impact on SKP2 and cell cycle progression. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Liu, T.; Jiang, X.; Guo, C.; Liu, A.; Xiao, X. Downregulation of USP18 inhibits growth and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma cells by suppressing BCL2L1. Exp. Cell Res. 2017, 358, 315–322. [Google Scholar] [CrossRef]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar] [CrossRef] [Green Version]
- Giannakopoulos, N.V.; Luo, J.-K.; Papov, V.; Zou, W.; Lenschow, D.J.; Jacobs, B.S.; Borden, E.C.; Li, J.; Virgin, H.W.; Zhang, D.-E. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem. Biophys. Res. Commun. 2005, 336, 496–506. [Google Scholar] [CrossRef]
- Takeuchi, T.; Inoue, S.; Yokosawa, H. Identification and Herc5-mediated ISGylation of novel target proteins. Biochem. Biophys. Res. Commun. 2006, 348, 473–477. [Google Scholar] [CrossRef]
- Sameer, A.S.; Nissar, S. XPD-the lynchpin of NER: Molecule, gene, polymorphisms, and role in colorectal carcinogenesis. Front. Mol. Biosci. 2018, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Dolinko, A.V.; Chinyengetere, F.; Stanton, B.; Bomberger, J.M.; Demidenko, E.; Zhou, D.C.; Gallagher, R.; Ma, T.; Galimberti, F.; et al. Blockade of the ubiquitin protease UBP43 destabilizes transcription factor PML/RARa and inhibits the growth of acute promyelocytic leukemia. Cancer Res. 2010, 70, 9875–9885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellaire, G.; Ching, R.W.; Ahmed, K.; Jalali, F.; Tse, K.C.K.; Bristow, R.G.; Bazett-Jones, D.P. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J. Cell Biol. 2006, 175, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-X.; Timanova-Atanasova, A.; Zhao, R.-X.; Chang, K.-S. PML Colocalizes with and Stabilizes the DNA Damage Response Protein TopBP1. Mol. Cell. Biol. 2003, 23, 4247–4256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Allinson, S.L.; Dianov, G.L. CHIP-Mediated Degradation and DNA Damage-Dependent Stabilization Regulate Base Excision Repair Proteins. Mol. Cell 2008, 29, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Sarkar, S.; Du, K.; Brautigan, D.L.; Abbas, T.; Larner, J.M. The E3 Ligase CHIP Mediates p21 Degradation to Maintain Radioresistance. Mol. Cancer Res. 2017, 15, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirawatnotai, S.; Sittithumcharee, G. Paradoxical roles of cyclin D1 in DNA stability. DNA Repair 2016, 42, 56–62. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2014, 28, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Boehm, E.; Gildenberg, M.; Washington, M. The many roles of PCNA in eukaryotic DNA replication. Enzymes 2016, 39, 231–254. [Google Scholar] [CrossRef]
- Di Masi, A.; Cilli, D.; Berardinelli, F.; Talarico, A.; Pallavicini, I.; Pennisi, R.; Leone, S.; Antoccia, A.; Noguera, N.I.; Lo-Coco, F.; et al. PML nuclear body disruption impairs DNA double-strand break sensing and repair in APL. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef]
- Mustachio, L.M.; Kawakami, M.; Lu, Y.; Rodriguez-Canales, J.; Mino, B.; Behrens, C.; Wistuba, I.; Bota-Rabassedas, N.; Yu, J.; Lee, J.J.; et al. The ISG15-specific protease USP18 regulates stability of PTEN. Oncotarget 2017, 8, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Ming, M.; He, Y.-Y. PTEN in DNA damage repair. Cancer Lett. 2012, 319, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, T.; Sommers, J.A.; Huang, J.; Seidman, M.M.; Brosh, R.M. Catalytic strand separation by RECQ1 is required for RPA-mediated response to replication stress. Curr. Biol. 2015, 25, 2830–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, C.D.; Spyracopoulos, L.; Glover, J.N.M. Ubc13: The Lys63 ubiquitin chain building machine. Oncotarget 2016, 7, 64471–64504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, K.; Schell, M.; Hoppe, T.; Kashkar, H. Regulation of the DNA damage response by ubiquitin conjugation. Front. Genet. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrecilla, I.; Oehler, J.; Ramadan, K. The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuper, J.; Braun, C.; Elias, A.; Michels, G.; Sauer, F.; Schmitt, D.R.; Poterszman, A.; Egly, J.M.; Kisker, C. In TFIIH, XPD Helicase Is Exclusively Devoted to DNA Repair. PLoS Biol. 2014, 12, e1001954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiff, T.; Shtivelman, E.; Jeggo, P.; Kysela, B. AHNAK interacts with the DNA ligase IV-XRCC4 complex and stimulates DNA ligase IV-mediated double-stranded ligation. DNA Repair 2004, 3, 245–256. [Google Scholar] [CrossRef]
- Leung, J.W.C.; Makharashvili, N.; Agarwal, P.; Chiu, L.-Y.; Pourpre, R.; Cammarata, M.B.; Cannon, J.R.; Sherker, A.; Durocher, D.; Brodbelt, J.S.; et al. ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev. 2017, 31, 260–274. [Google Scholar] [CrossRef] [Green Version]
- Massey, T.H.; Jones, L. The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech. 2018, 11, dmm031930. [Google Scholar] [CrossRef] [Green Version]
- Bártová, E.; Malyšková, B.; Komůrková, D.; Legartová, S.; Suchánková, J.; Krejčí, J.; Kozubek, S. Function of heterochromatin protein 1 during DNA repair. Protoplasma 2017, 254, 1233–1240. [Google Scholar] [CrossRef]
- Ismail, I.H.; Gagné, J.P.; Caron, M.C.; McDonald, D.; Xu, Z.; Masson, J.Y.; Poirier, G.G.; Hendzel, M.J. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 2012, 40, 5497–5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kari, V.; Mansour, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of CHD 1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2018, 19, 1609–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugasawa, K. Mechanism and Regulation of DNA Damage Recognition in Mammalian Nucleotide Excision Repair, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 45, ISBN 9780128173961. [Google Scholar]
- Teng, Y.; Lang, L.; Jauregui, C.E. The Complexity of DEK Signaling in Cancer Progression. Curr. Cancer Drug Targets 2018, 18, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.R.; Ananthapadmanabhan, V.; Swanson, S.; Saini, S.; Sesay, F.; Yakovlev, V.; Florens, L.; DeCaprio, J.A.; Washburn, M.P.; Dozmorov, M.; et al. DYRK1A regulates the recruitment of 53BP1 to the sites of DNA damage in part through interaction with RNF169. Cell Cycle 2019, 18, 531–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.M.; Jackson, S.P. Histone marks: Repairing DNA breaks within the context of chromatin. Biochem. Soc. Trans. 2012, 40, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715. [Google Scholar] [CrossRef] [Green Version]
- Peuscher, M.H.; Jacobs, J.J.L. DNA-damage response and repair activities at uncapped telomeres depend on RNF8. Nat. Cell Biol. 2011, 13, 1139–1145. [Google Scholar] [CrossRef]
- Moumen, A.; Masterson, P.; O’Connor, M.J.; Jackson, S.P. hnRNP K: An HDM2 Target and Transcriptional Coactivator of p53 in Response to DNA Damage. Cell 2005, 123, 1065–1078. [Google Scholar] [CrossRef] [Green Version]
- Moumen, A.; Magill, C.; Dry, K.; Jackson, S.P. ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle 2013, 12, 698–704. [Google Scholar] [CrossRef] [Green Version]
- Blasius, M.; Bartek, J. ATM targets hnRNPK to control p53. Cell Cycle 2013, 12, 1162. [Google Scholar] [CrossRef] [Green Version]
- Wiesmann, N.; Strozynski, J.; Beck, C.; Zimmermann, N.; Mendler, S.; Gieringer, R.; Schmidtmann, I.; Brieger, J. Knockdown of hnRNPK leads to increased DNA damage after irradiation and reduces survival of tumor cells. Carcinogenesis 2017, 38, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J.; et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 2012, 45, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo, S. DNA Damage and Lamins. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2014; Volume 773, pp. 377–399. ISBN 978-1-4899-8031-1. [Google Scholar]
- Li, W.; Bai, X.; Li, J.; Zhao, Y.; Liu, J.; Zhao, H.; Liu, L.; Ding, M. The nucleoskeleton protein IFFO1 immobilizes broken DNA and suppresses chromosome translocation during tumorigenesis. Nature Cell Biol. 2019, 21, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 Protects Telomeres from Oxidative Damage and Preserves Telomeric DNA for Extension by Telomerase. Cell Rep. 2016, 17, 3107–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cekan, P.; Hasegawa, K.; Pan, Y.; Tubman, E.; Odde, D.; Chen, J.-Q.; Herrmann, M.A.; Kumar, S.; Kalab, P. RCC1-dependent activation of Ran accelerates cell cycle and DNA repair, inhibiting DNA damage–induced cell senescence. Mol. Biol. Cell 2016, 27, 1346–1357. [Google Scholar] [CrossRef] [PubMed]
- Dworak, N.; Makosa, D.; Chatterjee, M.; Jividen, K.; Yang, C.S.; Snow, C.; Simke, W.C.; Johnson, I.G.; Kelley, J.B.; Paschal, B.M. A nuclear lamina-chromatin-Ran GTPase axis modulates nuclear import and DNA damage signaling. Aging Cell 2019, 18, e12851. [Google Scholar] [CrossRef] [Green Version]
- Kitange, G.J.; Mladek, A.C.; Schroeder, M.A.; Pokorny, J.C.; Carlson, B.L.; Zhang, Y.; Nair, A.A.; Lee, J.H.; Yan, H.; Decker, P.A.; et al. Retinoblastoma Binding Protein 4 Modulates Temozolomide Sensitivity in Glioblastoma by Regulating DNA Repair Proteins. Cell Rep. 2016, 14, 2587–2598. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gan, S.; Ren, L.; Yuan, L.; Liu, J.; Wang, W.; Wang, X.; Zhang, Y.; Jiang, J.; Zhang, F.; et al. Multifaceted regulation and functions of replication factor C family in human cancers. Am. J. Cancer Res. 2018, 8, 1343–1355. [Google Scholar]
- Dou, H.; Huang, C.; Singh, M.; Carpenter, P.B.; Yeh, E.T.H. Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein a complex. Mol. Cell 2010, 39, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Salas-Armenteros, I.; Pérez-Calero, C.; Bayona-Feliu, A.; Tumini, E.; Luna, R.; Aguilera, A. Human THO–Sin3A interaction reveals new mechanisms to prevent R-loops that cause genome instability. EMBO J. 2017, 36, 3532–3547. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro-Silva, C.; Vermeulen, W.; Lans, H. SWI/SNF: Complex complexes in genome stability and cancer. DNA Repair 2019, 77, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Enomoto, A.; Miyagawa, K. Serine-Threonine Kinase 38 regulates CDC25A stability and the DNA damage-induced G2/M checkpoint. Cell. Signal. 2015, 27, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Yu, J.; Nowsheen, S.; Zhao, F.; Wang, L.; Lou, Z. STK38 promotes ATM activation by acting as a reader of histone H4 ufmylation. Sci. Adv. 2020, 6, eaax8214. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Sun, Y.; Ji, P.; Kopetz, S.; Zhang, W. Topoisomerase IIα in chromosome instability and personalized cancer therapy. Oncogene 2015, 34, 4019–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, S.; Alam, S.K.; Roy, K.S.; Datta, A.; Nath, S.; Roychoudhury, S. E2 Ubiquitin-Conjugating Enzyme, UBE2C Gene, Is Reciprocally Regulated by Wild-type and Gain-of-Function Mutant p53. J. Biol. Chem. 2016, 291, 14231–14247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teloni, F.; Michelena, J.; Lezaja, A.; Kilic, S.; Ambrosi, C.; Menon, S.; Dobrovolna, J.; Imhof, R.; Janscak, P.; Baubec, T.; et al. Efficient Pre-mRNA Cleavage Prevents Replication-Stress-Associated Genome Instability. Mol. Cell 2019, 73, 670–683.e12. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Nicolai, S.; Mahen, R.; Raschellà, G.; Marini, A.; Pieraccioli, M.; Malewicz, M.; Venkitaraman, A.R.; Melino, G. ZNF281 is recruited on DNA breaks to facilitate DNA repair by non-homologous end joining. Oncogene 2020, 39, 754–766. [Google Scholar] [CrossRef]
- Malakhova, O.A.; Zhang, D.E. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J. Biol. Chem. 2008, 283, 8783–8787. [Google Scholar] [CrossRef] [Green Version]
- Minakawa, M.; Sone, T.; Takeuchi, T.; Yokosawa, H. Regulation of the nuclear factor (NF)-κB pathway by ISGylation. Biol. Pharm. Bull. 2008, 31, 2223–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria-Bretones, I.; Cepeda-García, C.; Checa-Rodriguez, C.; Heyer, V.; Reina-San-Martin, B.; Soutoglou, E.; Huertas, P. DNA end resection requires constitutive sumoylation of CtIP by CBX4. Nat. Commun. 2017, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Yokosawa, H. ISG15 modification of Ubc13 suppresses its ubiquitin-conjugating activity. Biochem. Biophys. Res. Commun. 2005, 336, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Bade, V.N.; Nickels, J.; Keusekotten, K.; Praefcke, G.J.K. Covalent protein modification with ISG15 via a conserved cysteine in the hinge region. PLoS ONE 2012, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakos, G.; Yu, L.; Gak, I.A.; Roumeliotis, T.I.; Liakopoulos, D.; Choudhary, J.S.; Mansfeld, J. An E2-ubiquitin thioester-driven approach to identify substrates modified with ubiquitin and ubiquitin-like molecules. Nat. Commun. 2018, 9, 4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef] [Green Version]
- Brüninghoff, K.; Aust, A.; Taupitz, K.F.; Wulff, S.; Dörner, W.; Mootz, H.D. Identification of SUMO binding proteins enriched after covalent photo-crosslinking. ACS Chem. Biol. 2020, 15, 2406–2414. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STI NG pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Neves-Costa, A.; Moita, L.F. Modulation of inflammation and disease tolerance by DNA damage response pathways. FEBS J. 2017, 284, 680–698. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.J.; Carrero, J.A.; Hung, P.J.; Tubbs, A.T.; Andrews, J.M.; Edelson, B.T.; Calderon, B.; Innes, C.L.; Paules, R.S.; Payton, J.E.; et al. A type I IFN-dependent DNA damage response regulates the genetic program and inflammasome activation in macrophages. Elife 2017, 6, e24655. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Duan, T.; Feng, Y.; Liu, Q.; Lin, M.; Cui, J.; Wang, R. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2018, 37, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bogunovic, D.; Payelle-Brogard, B.; Francois-Newton, V.; Speer, S.D.; Yuan, C.; Volpi, S.; Li, Z.; Sanal, O.; Mansouri, D.; et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 2015, 517, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thery, F.; Wu, N.C.; Luhmann, E.K.; Dussurget, O.; Foecke, M.; Bredow, C.; Jiménez-Fernández, D.; Leandro, K.; Beling, A.; et al. The in vivo ISGylome links ISG15 to metabolic pathways and autophagy upon Listeria monocytogenes infection. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimentally Validated Targets/Interactors | |
|---|---|
| Targets/Interactors (Targeted Residues) | Roles in DDR/Associated Pathways |
| CHIP (aka STUB1) (K143) [110] | Regulates proteins involved in BER [204] and cell cycle arrest [205] |
| Cyclin D1 [103] | Gatekeeping cyclin for DNA replication/ roles in HR [206] |
| p53 (K291, K292 among others) [88,104] | Master regulator of DNA damage response [207] |
| p63: ΔNp63α (K139, K324) [85]; TAp63α (K194, K397) [85] | Genome stability/instability, particularly in epithelial cancer cells under genotoxic stress, and depending on isotype [85] |
| Parkin (K349, K369) [108] | Promotes NER [47] and has important roles in mitosis [49] |
| PCNA (K164, K168) [86] | Facilitates TLS and has roles in mismatch repair [208] |
| PML-RARα [102] | Disrupts of PML nuclear body formation important for HR [209] |
| PTEN [210] | Roles in DSB repair and NER [211] |
| RECQ1 [23] | DNA helicase in replication stress response [212] |
| UBE2N (K92) [199] | Roles in DSB repair, NER and TLS [213] |
| Ubiquitin (K29) [101] | Roles in various DNA repair and signalling pathways [3,10,214] |
| VCP [198] | DSB repair through extraction of ubiquitylated substrates [215] |
| XPD (aka ERCC2) [199] | Helicase for NER [216] |
| Candidate Targets/Interactors | |
| Targets/Interactors | Roles in DDR/Associated Pathways |
| AHNAK [197] | Interacts with NHEJ proteins and may facilitate strand ligation [217] |
| ARID5B [197] | Involved in chromatin organisation and recruited to DNA damage sites [218] |
| ATXN2 [197] | Suggested protection against oxidative stress/potentially harmful R-loops [219] |
| CBX1 (aka HP1γ) [23] | Likely promotes recruitment of repair factors in various pathways |
| CBX3 (aka HP1β) [23] | Likely promotes recruitment of repair factors in various pathways [220] |
| CBX4 [197] | Mediates SUMO conjugation at DNA lesions and facilitates DSB repair [221] |
| CHD1 [197] | Opens chromatin around DSBs to allow for recruitment of HR proteins [222] |
| DDB1 [23] | Part of UV damage recognition complex in NER [223] |
| DDB2 [197] | Part of UV damage recognition complex in NER [223] |
| DEK [23] | Structural modulator of chromatin [224] |
| PRKDC (aka DNA-PKcs) [23] | Canonical factor in DSB repair by NHEJ [137] |
| DYRK1A [197] | Regulates recruitment of 53BP1 to DNA damage sites, inhibiting NHEJ [225] |
| H2A1B [23] | Contributes to higher order chromatin structure [226,227,228] |
| HNRNPK [197] | Contributes to DNA damage signalling [229,230,231,232] |
| HNRNPU (aka SAF-A) [197] | Regulator of DNA-end resection [233] |
| LMNA [197] | Important for DSB repair and telomere maintenance [234,235] |
| PRDX1 [38,197] | Protects telomeres from oxidative damage [236] |
| RAN [197] | Regulates nuclear import of ATM [237,238] |
| RBBP4 [197] | As part of chromatin remodelling complexes regulates DNA repair [239] |
| RFC2 [23] | DNA replication factor involved in PCNA-related repair mechanisms [240] |
| SENP1 [197] | SUMO-deconjugating enzyme that regulates p53 activity [241] |
| SIN3A [197] | Restricts formation of potentially harmful R-loop structures in DNA [242] |
| SMAD4 [197] | Promotes expression of DSB and NER repair proteins [243] |
| SMARCE1 (aka SMCE1) [23] | Chromatin remodelling via SWI/SNF complex [244] |
| STK38 [199] | Facilitates cell cycle arrest [245] and promotes activation of ATM [246] |
| TOP2A [197] | Checkpoint for chromosome decatenation during mitosis [247] |
| UBE2C [88] | Regulator of cell cycle progression and arrest [248] |
| WDR33 [197] | Prevents genome instability caused by unreleased nascent transcripts [249] |
| XRCC5 (aka Ku80) [197] | Essential factor in DSB repair by NHEJ [250] |
| XRCC6 (aka Ku70) [88,197] | Essential factor in DSB repair by NHEJ [250] |
| ZNF281 [197] | Helps recruit XRCC4 to DNA breaks for DSB repair by NHEJ [251] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandy, Z.; da Costa, I.C.; Schmidt, C.K. More than Meets the ISG15: Emerging Roles in the DNA Damage Response and Beyond. Biomolecules 2020, 10, 1557. https://doi.org/10.3390/biom10111557
Sandy Z, da Costa IC, Schmidt CK. More than Meets the ISG15: Emerging Roles in the DNA Damage Response and Beyond. Biomolecules. 2020; 10(11):1557. https://doi.org/10.3390/biom10111557
Chicago/Turabian StyleSandy, Zac, Isabelle Cristine da Costa, and Christine K. Schmidt. 2020. "More than Meets the ISG15: Emerging Roles in the DNA Damage Response and Beyond" Biomolecules 10, no. 11: 1557. https://doi.org/10.3390/biom10111557