The Role of APP O-Glycosylation in Alzheimer’s Disease

Abstract
:1. Introduction
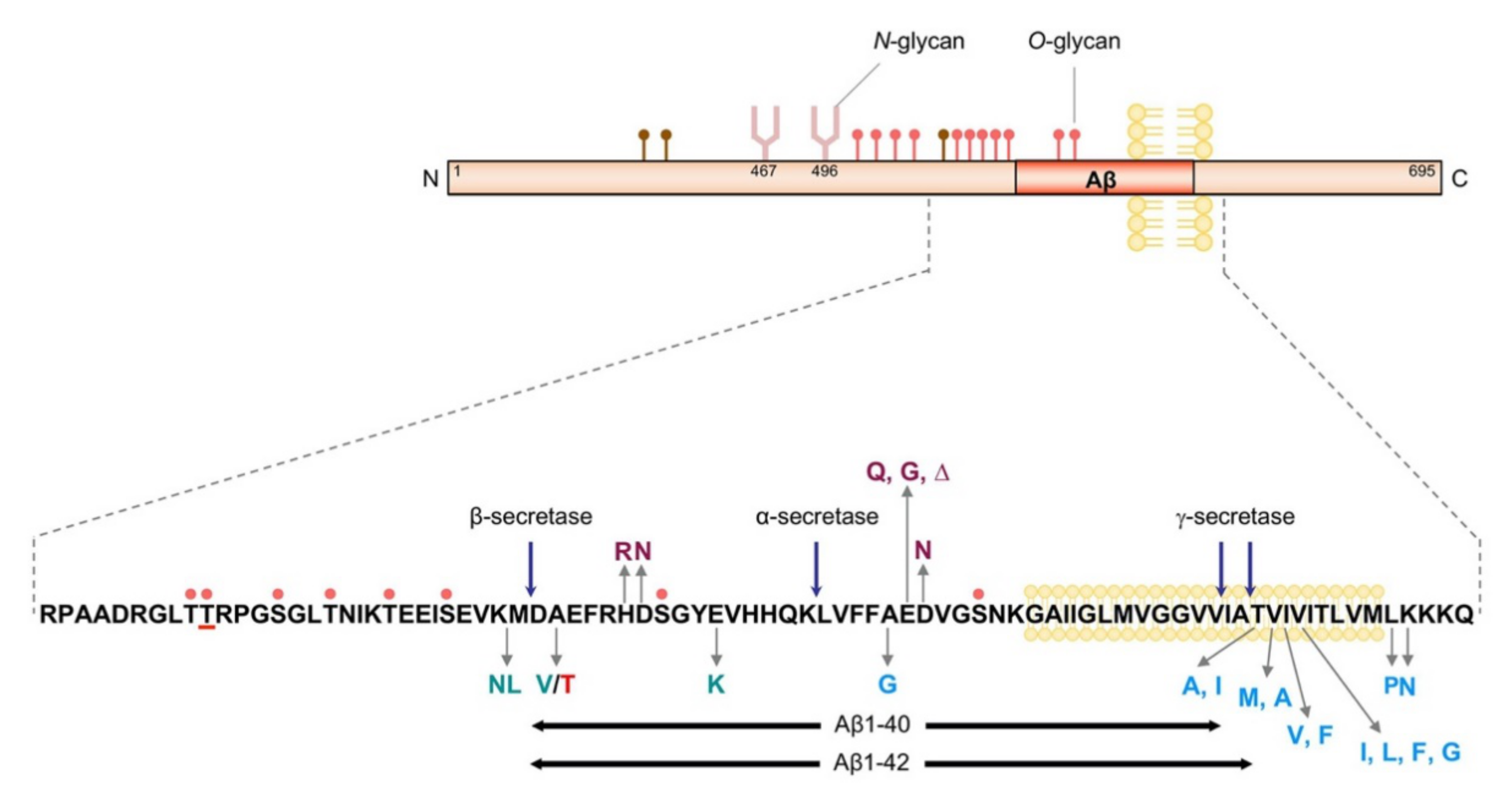
2. APP and Aβ
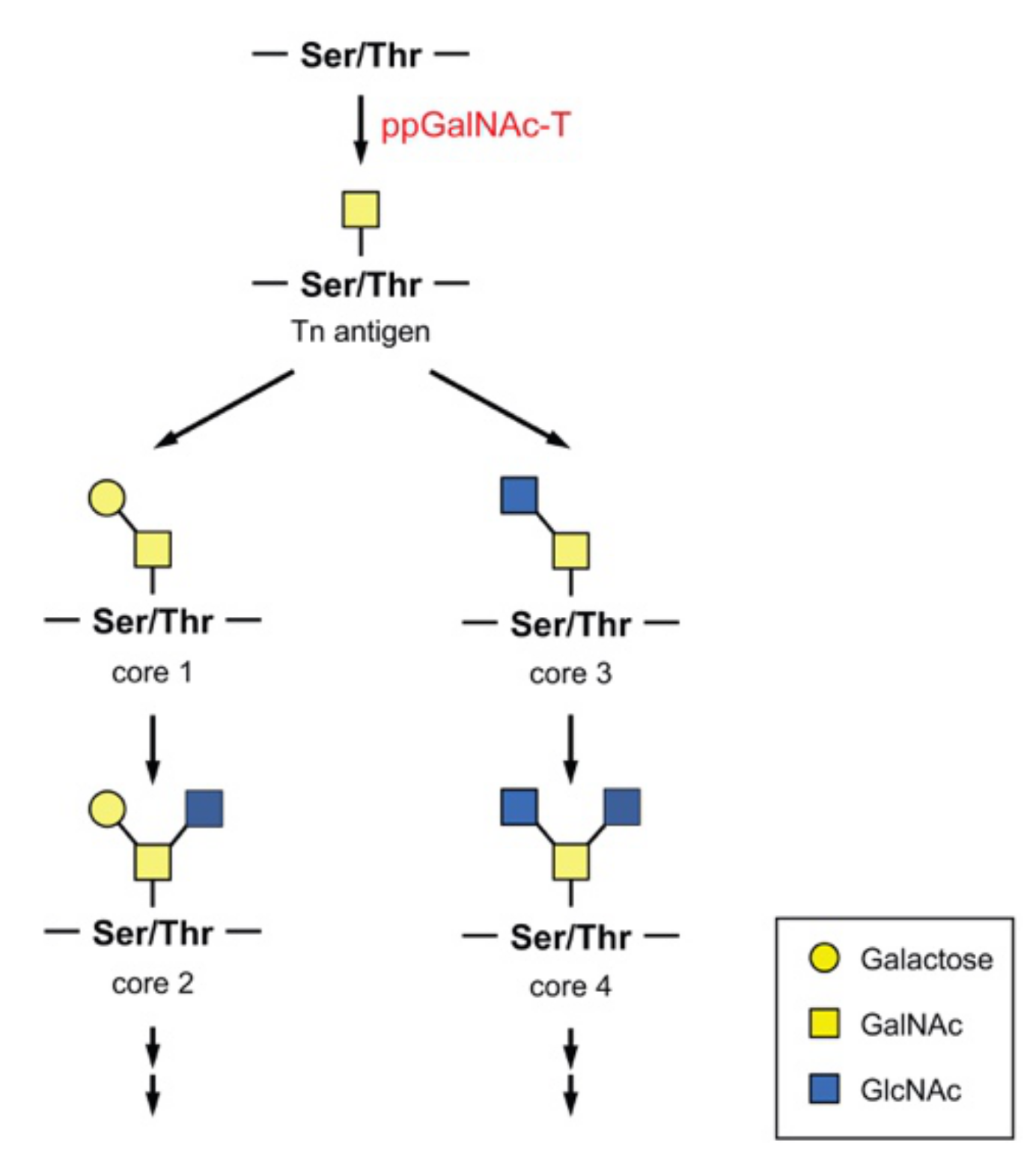
3. O-Glycans and ppGalNAc-T
4. O-Glycosylation and Protein Processing
5. Glycosylation of APP
6. O-Glycosylation and Aβ Production
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeze, H.H.; Chong, J.X.; Bamshad, M.J.; Ng, B.G. Solving glycosylation disorders: Fundamental approaches reveal complicated pathways. Am. J. Hum. Genet. 2014, 94, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeken, J.; Peanne, R. What is new in CDG? J. Inherit. Metab. Dis. 2017, 40, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Manya, H.; Endo, T. Glycosylation with ribitol-phosphate in mammals: New insights into the O-mannosyl glycan. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, Y.; Ise, W.; Harazono, A.; Takakura, D.; Fukuyama, H.; Baba, Y.; Narazaki, M.; Shoda, H.; Takahashi, N.; Ohkawa, Y.; et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat. Commun. 2016, 7, 11205. [Google Scholar] [CrossRef] [Green Version]
- Knoppova, B.; Reily, C.; Maillard, N.; Rizk, D.V.; Moldoveanu, Z.; Mestecky, J.; Raska, M.; Renfrow, M.B.; Julian, B.A.; Novak, J. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front. Immunol. 2016, 7, 117. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, J.I.; Hanamatsu, H.; Nishikaze, T.; Manya, H.; Miura, N.; Yagi, H.; Yokota, I.; Akasaka-Manya, K.; Endo, T.; Kanagawa, M.; et al. Lactone-driven ester-to-amide derivatization for sialic acid linkage-specific alkylamidation (LEAD-SALSA). Anal. Chem. 2020. [Google Scholar] [CrossRef]
- Han, J.; Pan, Y.; Gu, Y.; Xu, X.; Zhao, R.; Sha, J.; Zhang, R.; Gu, J.; Ren, S. Profiling of IgG N-glycome during mouse aging: Fucosylated diantennary glycans containing one Neu5Gc-linked LacNAc are associated with age. J. Proteom. 2020, 229, 103966. [Google Scholar] [CrossRef]
- Sato, Y.; Endo, T. Alteration of brain glycoproteins during aging. Geriatr. Gerontol. Int. 2010, 10, S32–S40. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the beta-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Belyaev, N.D.; Kellett, K.A.; Beckett, C.; Makova, N.Z.; Revett, T.J.; Nalivaeva, N.N.; Hooper, N.M.; Turner, A.J. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a {beta}-secretase-dependent pathway. J. Biol. Chem. 2010, 285, 41443–41454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.; Hussain, I.; Karran, E.H.; Turner, A.J.; Hooper, N.M. Cleavage of Alzheimer’s amyloid precursor protein by alpha-secretase occurs at the surface of neuronal cells. Biochemistry 1999, 38, 9728–9734. [Google Scholar] [CrossRef]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar]
- Haass, C.; Koo, E.H.; Mellon, A.; Hung, A.Y.; Selkoe, D.J. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: Alternative processing into amyloid-bearing fragments. Nature 1992, 357, 500–503. [Google Scholar] [CrossRef]
- Yamazaki, T.; Koo, E.H.; Selkoe, D.J. Trafficking of cell-surface amyloid beta-protein precursor. II. Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J. Cell Sci. 1996, 109, 999–1008. [Google Scholar]
- Zhang, X.; Song, W. The role of APP and BACE1 trafficking in APP processing and amyloid-beta generation. Alzheimers Res. Ther. 2013, 5, 46. [Google Scholar] [CrossRef]
- Rottger, S.; White, J.; Wandall, H.H.; Olivo, J.C.; Stark, A.; Bennett, E.P.; Whitehouse, C.; Berger, E.G.; Clausen, H.; Nilsson, T. Localization of three human polypeptide GalNAc-transferases in HeLa cells suggests initiation of O-linked glycosylation throughout the Golgi apparatus. J. Cell Sci. 1998, 111, 45–60. [Google Scholar]
- Gerken, T.A.; Ten Hagen, K.G.; Jamison, O. Conservation of peptide acceptor preferences between Drosophila and mammalian polypeptide-GalNAc transferase ortholog pairs. Glycobiology 2008, 18, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schjoldager, K.T.; Clausen, H. Site-specific protein O-glycosylation modulates proprotein processing—Deciphering specific functions of the large polypeptide GalNAc-transferase gene family. Biochim. Biophys. Acta 2012, 1820, 2079–2094. [Google Scholar] [CrossRef] [PubMed]
- Young, W.W., Jr.; Holcomb, D.R.; Ten Hagen, K.G.; Tabak, L.A. Expression of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase isoforms in murine tissues determined by real-time PCR: A new view of a large family. Glycobiology 2003, 13, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Hagen, K.G.; Fritz, T.A.; Tabak, L.A. All in the family: The UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 2003, 13, 1R–16R. [Google Scholar] [CrossRef] [Green Version]
- Raman, J.; Guan, Y.; Perrine, C.L.; Gerken, T.A.; Tabak, L.A. UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferases: Completion of the family tree. Glycobiology 2012, 22, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, J.; Li, W.; Xu, Y.; Shao, D.; Xie, Y.; Xie, W.; Kubota, T.; Narimatsu, H.; Zhang, Y. Characterization of ppGalNAc-T18, a member of the vertebrate-specific Y subfamily of UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 2012, 22, 602–615. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, Y.; Nakamura, N.; Kawai, T.; Kaneda, E.; Takahashi, Y.; Miyake, A.; Itoh, N.; Kurosaka, A. Identification and expression analysis of zebrafish polypeptide alpha-N-acetylgalactosaminyltransferase Y-subfamily genes during embryonic development. Gene. Expr. Patterns 2014, 16, 1–7. [Google Scholar] [CrossRef]
- Shan, A.; Lu, J.; Xu, Z.; Li, X.; Xu, Y.; Li, W.; Liu, F.; Yang, F.; Sato, T.; Narimatsu, H.; et al. Polypeptide N-acetylgalactosaminyltransferase 18 non-catalytically regulates the ER homeostasis and O-glycosylation. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 870–882. [Google Scholar] [CrossRef]
- Perrine, C.L.; Ganguli, A.; Wu, P.; Bertozzi, C.R.; Fritz, T.A.; Raman, J.; Tabak, L.A.; Gerken, T.A. Glycopeptide-preferring polypeptide GalNAc transferase 10 (ppGalNAc T10), involved in mucin-type O-glycosylation, has a unique GalNAc-O-Ser/Thr-binding site in its catalytic domain not found in ppGalNAc T1 or T2. J. Biol. Chem. 2009, 284, 20387–20397. [Google Scholar] [CrossRef] [Green Version]
- Gerken, T.A.; Jamison, O.; Perrine, C.L.; Collette, J.C.; Moinova, H.; Ravi, L.; Markowitz, S.D.; Shen, W.; Patel, H.; Tabak, L.A. Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J. Biol. Chem. 2011, 286, 14493–14507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, J.W.; Bennett, E.P.; Schjoldager, K.T.; Meldal, M.; Holmer, A.P.; Blixt, O.; Clo, E.; Levery, S.B.; Clausen, H.; Wandall, H.H. Lectin domains of polypeptide GalNAc transferases exhibit glycopeptide binding specificity. J. Biol. Chem. 2011, 286, 32684–32696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul Daniel, E.J.; de las Rivas, M.; Lira-Navarrete, E.; Garcia-Garcia, A.; Hurtado-Guerrero, R.; Clausen, H.; Gerken, T.A. Ser and Thr Acceptor Preferences of the GalNAc-Ts Vary Among Isoenzymes to Modulate Mucin Type O-Glycosylation. Glycobiology 2020. [Google Scholar] [CrossRef]
- Revoredo, L.; Wang, S.; Bennett, E.P.; Clausen, H.; Moremen, K.W.; Jarvis, D.L.; Ten Hagen, K.G.; Tabak, L.A.; Gerken, T.A. Mucin-type O-glycosylation is controlled by short- and long-range glycopeptide substrate recognition that varies among members of the polypeptide GalNAc transferase family. Glycobiology 2016, 26, 360–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.L.; Tran, D.T.; Tabak, L.A. Members of the GalNAc-T family of enzymes utilize distinct Golgi localization mechanisms. Glycobiology 2018, 28, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Lackman, J.J.; Goth, C.K.; Halim, A.; Vakhrushev, S.Y.; Clausen, H.; Petaja-Repo, U.E. Site-specific O-glycosylation of N-terminal serine residues by polypeptide GalNAc-transferase 2 modulates human delta-opioid receptor turnover at the plasma membrane. Cell Signal. 2018, 42, 184–193. [Google Scholar] [CrossRef]
- Pedersen, N.B.; Wang, S.; Narimatsu, Y.; Yang, Z.; Halim, A.; Schjoldager, K.T.; Madsen, T.D.; Seidah, N.G.; Bennett, E.P.; Levery, S.B.; et al. Low density lipoprotein receptor class A repeats are O-glycosylated in linker regions. J. Biol. Chem. 2014, 289, 17312–17324. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Mao, Y.; Narimatsu, Y.; Ye, Z.; Tian, W.; Goth, C.K.; Lira-Navarrete, E.; Pedersen, N.B.; Benito-Vicente, A.; Martin, C.; et al. Site-specific O-glycosylation of members of the low-density lipoprotein receptor superfamily enhances ligand interactions. J. Biol. Chem. 2018, 293, 7408–7422. [Google Scholar] [CrossRef] [Green Version]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Khetarpal, S.A.; Schjoldager, K.T.; Christoffersen, C.; Raghavan, A.; Edmondson, A.C.; Reutter, H.M.; Ahmed, B.; Ouazzani, R.; Peloso, G.M.; Vitali, C.; et al. Loss of Function of GALNT2 Lowers High-Density Lipoproteins in Humans, Nonhuman Primates, and Rodents. Cell Metab. 2016, 24, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Zilmer, M.; Edmondson, A.C.; Khetarpal, S.A.; Alesi, V.; Zaki, M.S.; Rostasy, K.; Madsen, C.G.; Lepri, F.R.; Sinibaldi, L.; Cusmai, R.; et al. Novel congenital disorder of O-linked glycosylation caused by GALNT2 loss of function. Brain 2020, 143, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tran, D.T.; Ten Hagen, K.G. An O-glycosyltransferase promotes cell adhesion during development by influencing secretion of an extracellular matrix integrin ligand. J. Biol. Chem. 2010, 285, 19491–19501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, E.; Wang, S.; Zhang, L.; Zhang, Y.; Malicdan, M.C.; Mao, Y.; Christoffersen, C.; Tabak, L.A.; Schjoldager, K.T.; Ten Hagen, K.G. Galnt11 regulates kidney function by glycosylating the endocytosis receptor megalin to modulate ligand binding. Proc. Natl. Acad. Sci. USA 2019, 116, 25196–25202. [Google Scholar] [CrossRef]
- Boskovski, M.T.; Yuan, S.; Pedersen, N.B.; Goth, C.K.; Makova, S.; Clausen, H.; Brueckner, M.; Khokha, M.K. The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature 2013, 504, 456–459. [Google Scholar] [CrossRef] [Green Version]
- Frishberg, Y.; Ito, N.; Rinat, C.; Yamazaki, Y.; Feinstein, S.; Urakawa, I.; Navon-Elkan, P.; Becker-Cohen, R.; Yamashita, T.; Araya, K.; et al. Hyperostosis-hyperphosphatemia syndrome: A congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J. Bone Miner. Res. 2007, 22, 235–242. [Google Scholar] [CrossRef]
- Kato, K.; Jeanneau, C.; Tarp, M.A.; Benet-Pages, A.; Lorenz-Depiereux, B.; Bennett, E.P.; Mandel, U.; Strom, T.M.; Clausen, H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 2006, 281, 18370–18377. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.J.; Chia, J.; Senewiratne, J.; Bard, F. Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J. Cell Biol. 2010, 189, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.J.; Tham, K.M.; Chia, J.; Wang, S.C.; Steentoft, C.; Clausen, H.; Bard-Chapeau, E.A.; Bard, F.A. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc. Natl. Acad. Sci. USA 2013, 110, E3152–E3161. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Chia, J.; Ros, M.; Hui, K.M.; Saltel, F.; Bard, F. Organelle Specific O-Glycosylation Drives MMP14 Activation, Tumor Growth, and Metastasis. Cancer Cell 2017, 32, 639–653.e636. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.J.; Hu, R.H.; Chou, C.H.; Hsu, C.L.; Liu, Y.W.; Huang, J.; Hung, J.S.; Lai, I.R.; Juan, H.F.; Yu, S.L.; et al. Knockdown of GALNT1 suppresses malignant phenotype of hepatocellular carcinoma by suppressing EGFR signaling. Oncotarget 2015, 6, 5650–5665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Nishidate, T.; Kijima, K.; Ohashi, T.; Takegawa, K.; Fujikane, T.; Hirata, K.; Nakamura, Y.; Katagiri, T. Critical roles of mucin 1 glycosylation by transactivated polypeptide N-acetylgalactosaminyltransferase 6 in mammary carcinogenesis. Cancer Res. 2010, 70, 2759–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.Y.; Shun, C.T.; Hung, K.Y.; Juan, H.F.; Hsu, C.L.; Huang, M.C.; Lai, I.R. Mucin glycosylating enzyme GALNT2 suppresses malignancy in gastric adenocarcinoma by reducing MET phosphorylation. Oncotarget 2016, 7, 11251–11262. [Google Scholar] [CrossRef] [PubMed]
- Berois, N.; Mazal, D.; Ubillos, L.; Trajtenberg, F.; Nicolas, A.; Sastre-Garau, X.; Magdelenat, H.; Osinaga, E. UDP-N-acetyl-D-galactosamine: Polypeptide N-acetylgalactosaminyltransferase-6 as a new immunohistochemical breast cancer marker. J. Histochem. Cytochem. 2006, 54, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patani, N.; Jiang, W.; Mokbel, K. Prognostic utility of glycosyltransferase expression in breast cancer. Cancer Genom. Proteom. 2008, 5, 333–340. [Google Scholar]
- Goth, C.K.; Halim, A.; Khetarpal, S.A.; Rader, D.J.; Clausen, H.; Schjoldager, K.T. A systematic study of modulation of ADAM-mediated ectodomain shedding by site-specific O-glycosylation. Proc. Natl. Acad. Sci. USA 2015, 112, 14623–14628. [Google Scholar] [CrossRef] [Green Version]
- Shirakabe, K.; Omura, T.; Shibagaki, Y.; Mihara, E.; Homma, K.; Kato, Y.; Yoshimura, A.; Murakami, Y.; Takagi, J.; Hattori, S.; et al. Mechanistic insights into ectodomain shedding: Susceptibility of CADM1 adhesion molecule is determined by alternative splicing and O-glycosylation. Sci. Rep. 2017, 7, 46174. [Google Scholar] [CrossRef]
- De Las Rivas, M.; Paul Daniel, E.J.; Narimatsu, Y.; Companon, I.; Kato, K.; Hermosilla, P.; Thureau, A.; Ceballos-Laita, L.; Coelho, H.; Bernado, P.; et al. Molecular basis for fibroblast growth factor 23 O-glycosylation by GalNAc-T3. Nat. Chem. Biol. 2020, 16, 351–360. [Google Scholar] [CrossRef]
- Schjoldager, K.T.; Vester-Christensen, M.B.; Bennett, E.P.; Levery, S.B.; Schwientek, T.; Yin, W.; Blixt, O.; Clausen, H. O-glycosylation modulates proprotein convertase activation of angiopoietin-like protein 3: Possible role of polypeptide GalNAc-transferase-2 in regulation of concentrations of plasma lipids. J. Biol. Chem. 2010, 285, 36293–36303. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, A.; Konig, G.; Bunke, D.; Fischer, P.; Salbaum, J.M.; Masters, C.L.; Beyreuther, K. Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell 1989, 57, 115–126. [Google Scholar] [CrossRef]
- Tomita, S.; Kirino, Y.; Suzuki, T. Cleavage of Alzheimer’s amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J. Biol. Chem. 1998, 273, 6277–6284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Liu, C.; Wojczyk, B.S.; Kobata, A.; Spitalnik, S.L.; Endo, T. Study of the sugar chains of recombinant human amyloid precursor protein produced by Chinese hamster ovary cells. Biochim. Biophys. Acta 1999, 1472, 344–358. [Google Scholar] [CrossRef]
- Pahlsson, P.; Spitalnik, S.L. The role of glycosylation in synthesis and secretion of beta-amyloid precursor protein by Chinese hamster ovary cells. Arch. Biochem. Biophys. 1996, 331, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Tagawa, K.; Maruyama, K.; Sorimachi, H.; Tsuchiya, T.; Ishiura, S.; Suzuki, K. Mutation of potential N-linked glycosylation sites in the Alzheimer’s disease amyloid precursor protein (APP). Neurosci. Lett. 1996, 221, 57–60. [Google Scholar] [CrossRef]
- Nakagawa, K.; Kitazume, S.; Oka, R.; Maruyama, K.; Saido, T.C.; Sato, Y.; Endo, T.; Hashimoto, Y. Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J. Neurochem. 2006, 96, 924–933. [Google Scholar] [CrossRef]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Spitalnik, S.L.; Endo, T. Increased bisecting and core-fucosylated N-glycans on mutant human amyloid precursor proteins. Glycoconj. J. 2008, 25, 775–786. [Google Scholar] [CrossRef]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Kozutsumi, Y.; Saito, Y.; Taniguchi, N.; Murayama, S.; Spitalnik, S.L.; Endo, T. Protective effect of N-glycan bisecting GlcNAc residues on beta-amyloid production in Alzheimer’s disease. Glycobiology 2010, 20, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Kizuka, Y.; Nakano, M.; Kitazume, S.; Saito, T.; Saido, T.C.; Taniguchi, N. Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem. J. 2016, 473, 21–30. [Google Scholar] [CrossRef]
- Perdivara, I.; Deterding, L.J.; Cozma, C.; Tomer, K.B.; Przybylski, M. Glycosylation profiles of epitope-specific anti-beta-amyloid antibodies revealed by liquid chromatography-mass spectrometry. Glycobiology 2009, 19, 958–970. [Google Scholar] [CrossRef] [Green Version]
- Halim, A.; Brinkmalm, G.; Ruetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef] [Green Version]
- Brinkmalm, G.; Portelius, E.; Ohrfelt, A.; Mattsson, N.; Persson, R.; Gustavsson, M.K.; Vite, C.H.; Gobom, J.; Mansson, J.E.; Nilsson, J.; et al. An online nano-LC-ESI-FTICR-MS method for comprehensive characterization of endogenous fragments from amyloid beta and amyloid precursor protein in human and cat cerebrospinal fluid. J. Mass Spectrom. 2012, 47, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Tachida, Y.; Kato, M.; Yamaguchi, Y.; Honda, T.; Hashimoto, Y.; Wada, Y.; Saito, T.; Iwata, N.; Saido, T.; et al. Brain endothelial cells produce amyloid {beta} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 2010, 285, 40097–40103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Xu, K.; Xu, Z.; de Las Rivas, M.; Wang, C.; Li, X.; Lu, J.; Zhou, Y.; Delso, I.; Merino, P.; et al. The small molecule luteolin inhibits N-acetyl-alpha-galactosaminyltransferases and reduces mucin-type O-glycosylation of amyloid precursor protein. J. Biol. Chem. 2017, 292, 21304–21319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schjoldager, K.T.; Vakhrushev, S.Y.; Kong, Y.; Steentoft, C.; Nudelman, A.S.; Pedersen, N.B.; Wandall, H.H.; Mandel, U.; Bennett, E.P.; Levery, S.B.; et al. Probing isoform-specific functions of polypeptide GalNAc-transferases using zinc finger nuclease glycoengineered SimpleCells. Proc. Natl. Acad. Sci. USA 2012, 109, 9893–9898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akasaka-Manya, K.; Kawamura, M.; Tsumoto, H.; Saito, Y.; Tachida, Y.; Kitazume, S.; Hatsuta, H.; Miura, Y.; Hisanaga, S.I.; Murayama, S.; et al. Excess APP O-glycosylation by GalNAc-T6 decreases Abeta production. J. Biochem. 2017, 161, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, L.; Vosseller, K.; Hart, G.W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001, 291, 2376–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [Green Version]
- Griffith, L.S.; Mathes, M.; Schmitz, B. Beta-amyloid precursor protein is modified with O-linked N-acetylglucosamine. J. Neurosci. Res. 1995, 41, 270–278. [Google Scholar] [CrossRef]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-beta precursor protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef]
- Chun, Y.S.; Park, Y.; Oh, H.G.; Kim, T.W.; Yang, H.O.; Park, M.K.; Chung, S. O-GlcNAcylation promotes non-amyloidogenic processing of amyloid-beta protein precursor via inhibition of endocytosis from the plasma membrane. J. Alzheimers. Dis. 2015, 44, 261–275. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kwon, O.H.; Chung, S. O-GlcNAcylation of amyloid-beta precursor protein at threonine 576 residue regulates trafficking and processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Sakaidani, Y.; Nomura, T.; Matsuura, A.; Ito, M.; Suzuki, E.; Murakami, K.; Nadano, D.; Matsuda, T.; Furukawa, K.; Okajima, T. O-linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nat. Commun. 2011, 2, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezai-Zadeh, K.; Douglas Shytle, R.; Bai, Y.; Tian, J.; Hou, H.; Mori, T.; Zeng, J.; Obregon, D.; Town, T.; Tan, J. Flavonoid-mediated presenilin-1 phosphorylation reduces Alzheimer’s disease beta-amyloid production. J. Cell. Mol. Med. 2009, 13, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Kwon, O.H.; Oh, H.G.; Kim, T.W.; McIntire, L.B.; Park, M.K.; Chung, S. Threonine 576 residue of amyloid-beta precursor protein regulates its trafficking and processing. Biochem. Biophys. Res. Commun. 2015, 467, 955–960. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Bamne, M.N.; Demirci, F.Y.; Berman, S.; Snitz, B.E.; Rosenthal, S.L.; Wang, X.; Lopez, O.L.; Kamboh, M.I. Investigation of an amyloid precursor protein protective mutation (A673T) in a North American case-control sample of late-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1779.e15–1779.e16. [Google Scholar] [CrossRef] [Green Version]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Maloney, J.A.; Bainbridge, T.; Gustafson, A.; Zhang, S.; Kyauk, R.; Steiner, P.; van der Brug, M.; Liu, Y.; Ernst, J.A.; Watts, R.J.; et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J. Biol. Chem. 2014, 289, 30990–31000. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Gallardo, R.; Ungureanu, A.A.; Castillo Cano, V.; Snellinx, A.; Ramakers, M.; Bartic, C.; Rousseau, F.; Schymkowitz, J.; De Strooper, B. The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-beta (Abeta) aggregation. J. Biol. Chem. 2014, 289, 30977–30989. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Hata, S.; Suzuki, T. Alternative Selection of beta-Site APP-Cleaving Enzyme 1 (BACE1) Cleavage Sites in Amyloid beta-Protein Precursor (APP) Harboring Protective and Pathogenic Mutations within the Abeta Sequence. J. Biol. Chem. 2016, 291, 24041–24053. [Google Scholar] [CrossRef] [Green Version]
- Das, U.; Wang, L.; Ganguly, A.; Saikia, J.M.; Wagner, S.L.; Koo, E.H.; Roy, S. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 2016, 19, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Li, Z.; Xu, L.; Shi, Y.; Zhang, X.; Shi, S.; Hou, K.; Fan, Y.; Li, C.; Wang, X.; et al. GALNT6 promotes breast cancer metastasis by increasing mucin-type O-glycosylation of alpha2M. Aging 2020, 12, 11794–11811. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Chen, L.; Gao, H.; Zhen, T.; Li, H.; Liang, J.; Zhang, F.; Shi, H.; Han, A. GALNT6 suppresses progression of colorectal cancer. Am. J. Cancer Res. 2018, 8, 2419–2435. [Google Scholar] [PubMed]
- Sun, Z.; Xue, H.; Wei, Y.; Wang, C.; Yu, R.; Wang, C.; Wang, S.; Xu, J.; Qian, M.; Meng, Q.; et al. Mucin O-glycosylating enzyme GALNT2 facilitates the malignant character of glioma by activating the EGFR/PI3K/Akt/mTOR axis. Clin. Sci. 2019, 133, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.T.; Yeh, C.C.; Liu, S.Y.; Huang, M.C.; Lai, I.R. The O-glycosylating enzyme GALNT2 suppresses the malignancy of gastric adenocarcinoma by reducing EGFR activities. Am. J. Cancer Res. 2018, 8, 1739–1751. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akasaka-Manya, K.; Manya, H. The Role of APP O-Glycosylation in Alzheimer’s Disease. Biomolecules 2020, 10, 1569. https://doi.org/10.3390/biom10111569
Akasaka-Manya K, Manya H. The Role of APP O-Glycosylation in Alzheimer’s Disease. Biomolecules. 2020; 10(11):1569. https://doi.org/10.3390/biom10111569
Chicago/Turabian StyleAkasaka-Manya, Keiko, and Hiroshi Manya. 2020. "The Role of APP O-Glycosylation in Alzheimer’s Disease" Biomolecules 10, no. 11: 1569. https://doi.org/10.3390/biom10111569
APA StyleAkasaka-Manya, K., & Manya, H. (2020). The Role of APP O-Glycosylation in Alzheimer’s Disease. Biomolecules, 10(11), 1569. https://doi.org/10.3390/biom10111569