The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Causes of Syndromic TAAD
2.1. Marfan Syndrome (MFS)
2.2. Loeys–Dietz Syndrome (LDS)
2.3. Ehlers-Danlos Syndrome (EDS)
2.4. Other Causes of Syndromic TAAD
3. Causes of Non-Syndromic TAAD
3.1. ACTA2
3.2. MYLK
3.3. MYH11
3.4. PRKG1
3.5. Other Genes Associated with Familial TAAD
3.6. Genetics of Sporadic TAAD
4. Genetic Testing—Past and Future
5. Conclusions
Conflicts of Interest
References
- Kuzmik, G.A.; Sang, A.X.; Elefteriades, J.A. Natural history of thoracic aortic aneurysms. J. Vasc. Surg. 2012, 56, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elefteriades, J.A.; Sang, A.; Kuzmik, G.; Hornick, M. Guilt by association: Paradigm for detecting a silent killer (thoracic aortic aneurysm). Open Heart 2015, 2, e000169. [Google Scholar] [CrossRef] [PubMed]
- Web-based Injury Statistics Query and Reporting System (WISQARS). Available online: www.cdc.gov/injury/wisqars (accessed on 26 August 2019).
- Zhan, S.; Hong, S.; Shan-Shan, L.; Chen-Ling, Y.; Lai, W.; Dong-Wei, S.; Chao-Yang, T.; Xian-Hong, S.; Chun-Sheng, W. Misdiagnosis of aortic dissection: Experience of 361 patients. J. Clin. Hypertens. 2012, 14, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafar, M.A.; Farkas, E.A.; Javier, A.; Anderson, M.; Gilani, O.; Elefteriades, J.A. Are thromboembolic and bleeding complications a drawback for composite aortic root replacement? Ann. Thorac. Surg. 2012, 94, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, B.A.; Rajbanshi, B.G.; Tranquilli, M.; Fang, H.; Rizzo, J.A.; Elefteriades, J.A. Straight deep hypothermic circulatory arrest for cerebral protection during aortic arch surgery: Safe and effective. J. Thorac. Cardiovasc. Surg. 2014, 148, 888–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, G.; Coady, M.A.; Roberts, M.; Davies, R.R.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Familial thoracic aortic aneurysms and dissections--incidence, modes of inheritance, and phenotypic patterns. Ann. Thorac. Surg. 2006, 82, 1400–1405. [Google Scholar] [CrossRef]
- Brownstein, A.J.; Kostiuk, V.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2018 Update and Clinical Implications. Aorta 2018, 6, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Keramati, A.R.; Sadeghpour, A.; Farahani, M.M.; Chandok, G.; Mani, A. The non-syndromic familial thoracic aortic aneurysms and dissections maps to 15q21 locus. BMC Med. Genet. 2010, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Teixidó-Tura, G.; Franken, R.; Galuppo, V.; Gutiérrez García-Moreno, L.; Borregan, M.; Mulder, B.J.; García-Dorado, D.; Evangelista, A. Heterogeneity of aortic disease severity in patients with Loeys-Dietz syndrome. Heart 2016, 102, 626–632. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Dietz, H.C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011, 473, 308–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, L.; Collod-Beroud, G.; Adès, L.; Arbustini, E.; Child, A.; Callewaert, B.L.; Loeys, B.; Binquet, C.; Gautier, E.; Mayer, K.; et al. The new Ghent criteria for Marfan syndrome: What do they change? Clin. Genet. 2012, 81, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Iams, H.D. Diagnosis and management of Marfan syndrome. Curr. Sports Med. Rep. 2010, 9, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e369. [Google Scholar]
- Milewicz, D.M.; Michael, K.; Fisher, N.; Coselli, J.S.; Markello, T.; Biddinger, A. Fibrillin-1 (FBN1) mutations in patients with thoracic aortic aneurysms. Circulation 1996, 94, 2708–2711. [Google Scholar] [CrossRef]
- Biddinger, A.; Rocklin, M.; Coselli, J.; Milewicz, D.M. Familial thoracic aortic dilatations and dissections: A case control study. J. Vasc. Surg. 1997, 25, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Coady, M.A.; Davies, R.R.; Roberts, M.; Goldstein, L.J.; Rogalski, M.J.; Rizzo, J.A.; Hammond, G.L.; Kopf, G.S.; Elefteriades, J.A. Familial patterns of thoracic aortic aneurysms. Arch. Surg. 1999, 134, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Hasham, S.N.; Lewin, M.R.; Tran, V.T.; Pannu, H.; Muilenburg, A.; Willing, M.; Milewicz, D.M. Nonsyndromic genetic predisposition to aortic dissection: A newly recognized, diagnosable, and preventable occurrence in families. Ann. Emerg. Med. 2004, 43, 79–82. [Google Scholar] [CrossRef]
- LeMaire, S.A.; McDonald, M.L.; Guo, D.C.; Russell, L.; Miller, C.C.; Johnson, R.J.; Bekheirnia, M.R.; Franco, L.M.; Nguyen, M.; Pyeritz, R.E.; et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat. Genet. 2011, 43, 996–1000. [Google Scholar] [CrossRef]
- Mimler, T.; Nebert, C.; Eichmair, E.; Winter, B.; Aschacher, T.; Stelzmueller, M.E.; Andreas, M.; Ehrlich, M.; Laufer, G.; Messner, B. Extracellular matrix in ascending aortic aneurysms and dissections—What we learn from decellularization and scanning electron microscopy. PLoS ONE 2019, 14, e0213794. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Trybus, K.M.; Guo, D.C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, J.B.; Ikonomidis, J.S.; Jones, J.A. Connective tissue disorders and cardiovascular complications: The indomitable role of transforming growth factor-beta signaling. Adv. Exp. Med. Biol. 2014, 802, 107–127. [Google Scholar]
- Eckersley, A.; Mellody, K.T.; Pilkington, S.; Griffiths, C.E.M.; Watson, R.E.B.; O’Cualain, R.; Baldock, C.; Knight, D.; Sherratt, M.J. Structural and compositional diversity of fibrillin microfibrils in human tissues. J. Biol. Chem. 2018, 293, 5117–5133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, L.Y.; Keene, D.R.; Engvall, E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J. Cell. Biol. 1986, 103, 2499–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.R.; Summers, K.M. Structure and function of the mammalian fibrillin gene family: Implications for human connective tissue diseases. Mol. Genet. Metab. 2012, 107, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, P.; Hanna, N.; Aubart, M.; Leheup, B.; Dupuis-Girod, S.; Naudion, S.; Lacombe, D.; Milleron, O.; Odent, S.; Faivre, L.; et al. Homozygous and compound heterozygous mutations in the FBN1 gene: Unexpected findings in molecular diagnosis of Marfan syndrome. J. Med. Genet. 2017, 54, 100–103. [Google Scholar] [CrossRef]
- Ziganshin, B.A.; Bailey, A.E.; Coons, C.; Dykas, D.; Charilaou, P.; Tanriverdi, L.H.; Liu, L.; Tranquilli, M.; Bale, A.E.; Elefteriades, J.A. Routine Genetic Testing for Thoracic Aortic Aneurysm and Dissection in a Clinical Setting. Ann. Thorac. Surg. 2015, 100, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Elefteriades, J.A.; Brownstein, A.J.; Ziganshin, B.A. Clinical and molecular genetics of aortic dissection. In Aortic Dissection Patients: True Stories and the Innovations that Saved Their Lives; Melissano, G., Chiesa, R., Eds.; Edi. Ermes: Milano, Italy, 2016; pp. 49–80. ISBN1 978-88-7051-565-7. (hardcover); ISBN2 978-88-7051-566-4. (digital edition). [Google Scholar]
- Pyeritz, R.E. The Marfan syndrome. Annu. Rev. Med. 2000, 51, 481–510. [Google Scholar] [CrossRef] [Green Version]
- Judge, D.P.; Biery, N.J.; Keene, D.R.; Geubtner, J.; Myers, L.; Huso, D.L.; Sakai, L.Y.; Dietz, H.C. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Investig. 2004, 114, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.H.; Kim, Y.M.; Kang, E.; Kim, G.H.; Seo, E.J.; Lee, B.H.; Choi, J.H.; Yoo, H.W. The phenotypic heterogeneity of patients with Marfan-related disorders and their variant spectrums. Medicine 2018, 97, e10767. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Groenink, M.; de Waard, V.; Feenstra, H.M.; Scholte, A.J.; van den Berg, M.P.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J. Genotype impacts survival in Marfan syndrome. Eur. Heart J. 2016, 37, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Vollbrandt, T.; Tiedemann, K.; El-Hallous, E.; Lin, G.; Brinckmann, J.; John, H.; Bätge, B.; Notbohm, H.; Reinhardt, D.P. Consequences of cysteine mutations in calcium-binding epidermal growth factor modules of fibrillin-1. J. Biol. Chem. 2004, 279, 32924–32931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, L.; Collod-Beroud, G.; Loeys, B.L.; Child, A.; Binquet, C.; Gautier, E.; Callewaert, B.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: An international study. Am. J. Hum. Genet. 2007, 81, 454–466. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, D.P.; Mechling, D.E.; Boswell, B.A.; Keene, D.R.; Sakai, L.Y.; Bächinger, H.P. Calcium determines the shape of fibrillin. J. Biol. Chem. 1997, 272, 7368–7373. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Li, Z.; Zhou, C.; Cao, Y.; Zhang, L.; Li, X.; Cianflone, K.; Wang, Y.; Wang, D.W. FBN1 mutations largely contribute to sporadic non-syndromic aortic dissection. Hum. Mol. Genet. 2017, 26, 4814–4822. [Google Scholar] [CrossRef]
- Chen, J.Z.; Sawada, H.; Moorleghen, J.J.; Weiland, M.; Daugherty, A.; Sheppard, M.B. Aortic Strain Correlates with Elastin Fragmentation in Fibrillin-1 Hypomorphic Mice. Circ. Rep. 2019, 1, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Kaartinen, V.; Warburton, D. Fibrillin controls TGF-beta activation. Nat. Genet. 2003, 33, 331–332. [Google Scholar] [CrossRef]
- Jones, K.B.; Myers, L.; Judge, D.P.; Kirby, P.A.; Dietz, H.C.; Sponseller, P.D. Toward an understanding of dural ectasia: A light microscopy study in a murine model of Marfan syndrome. Spine 2005, 30, 291–293. [Google Scholar] [CrossRef]
- Takeda, N.; Hara, H.; Fujiwara, T.; Kanaya, T.; Maemura, S.; Komuro, I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int. J. Mol. Sci. 2018, 19, 2125. [Google Scholar] [CrossRef] [Green Version]
- Weinsaft, J.W.; Devereux, R.B.; Preiss, L.R.; Feher, A.; Roman, M.J.; Basson, C.T.; Geevarghese, A.; Ravekes, W.; Dietz, H.C.; Holmes, K.; et al. Aortic Dissection in Patients With Genetically Mediated Aneurysms: Incidence and Predictors in the GenTAC Registry. J. Am. Coll. Cardiol. 2016, 67, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Saeyeldin, A.; Zafar, M.A.; Velasquez, C.A.; Ip, K.; Gryaznov, A.; Brownstein, A.J.; Li, Y.; Rizzo, J.A.; Erben, Y.; Ziganshin, B.A.; et al. Natural history of aortic root aneurysms in Marfan syndrome. Ann. Cardiothorac. Surg. 2017, 6, 625–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, M.J.; Pugh, N.L.; Hendershot, T.P.; Devereux, R.B.; Dietz, H.; Holmes, K.; Eagle, K.A.; LeMaire, S.A.; Milewicz, D.M.; Morris, S.A.; et al. Aortic Complications Associated With Pregnancy in Marfan Syndrome: The NHLBI National Registry of Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions (GenTAC). J. Am. Heart Assoc. 2016, 5, e004052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef]
- Van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef]
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell. Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Daugherty, A.; Chen, Z.; Sawada, H.; Rateri, D.L.; Sheppard, M.B. Transforming Growth Factor-β in Thoracic Aortic Aneurysms: Good, Bad, or Irrelevant? J. Am. Heart Assoc. 2017, 6, e00521. [Google Scholar] [CrossRef]
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef]
- Akhurst, R.J. The paradoxical TGF-β vasculopathies. Nat. Genet. 2012, 44, 838–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, D.P. Ehlers-Danlos syndrome type IV. Orphanet J. Rare Dis. 2007, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Byers, P. Vascular Ehlers-Danlos Syndrome. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Carter, J.; Fenves, A.Z. Understanding vascular-type Ehlers-Danlos syndrome and avoiding vascular complications. Bayl. Univ. Med. Cent. Proc. 2017, 30, 52–53. [Google Scholar] [CrossRef] [Green Version]
- Legrand, A.; Devriese, M.; Dupuis-Girod, S.; Simian, C.; Venisse, A.; Mazzella, J.M.; Auribault, K.; Adham, S.; Frank, M.; Albuisson, J.; et al. Frequency of de novo variants and parental mosaicism in vascular Ehlers-Danlos syndrome. Genet. Med. 2019, 21, 1568–1575. [Google Scholar] [CrossRef]
- Shalhub, S.; Black, J.H.; Cecchi, A.C.; Xu, Z.; Griswold, B.F.; Safi, H.J.; Milewicz, D.M.; McDonnell, N.B. Molecular diagnosis in vascular Ehlers-Danlos syndrome predicts pattern of arterial involvement and outcomes. J. Vasc. Surg. 2014, 60, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Cortini, F.; Marinelli, B.; Seia, M.; De Giorgio, B.; Pesatori, A.C.; Montano, N.; Bassotti, A. Next-generation sequencing and a novel COL3A1 mutation associated with vascular Ehlers-Danlos syndrome with severe intestinal involvement: A case report. J Med. Case Rep. 2016, 10, 303. [Google Scholar] [CrossRef] [Green Version]
- Schievink, W.I. Cerebrovascular Involvement in Ehlers-Danlos Syndrome. Curr. Treat. Options Cardiovasc. Med. 2004, 6, 231–236. [Google Scholar] [CrossRef]
- North, K.N.; Whiteman, D.A.; Pepin, M.G.; Byers, P.H. Cerebrovascular complications in Ehlers-Danlos syndrome type IV. Ann. Neurol. 1995, 38, 960–964. [Google Scholar] [CrossRef]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Persikov, A.V.; Pillitteri, R.J.; Amin, P.; Schwarze, U.; Byers, P.H.; Brodsky, B. Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum. Mutat. 2004, 24, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, U.; Schievink, W.I.; Petty, E.; Jaff, M.R.; Babovic-Vuksanovic, D.; Cherry, K.J.; Pepin, M.; Byers, P.H. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am. J. Hum. Genet. 2001, 69, 989–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, H.; Byrne, M.; Krane, S.; Jaenisch, R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 1852–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prockop, D.J.; Kivirikko, K.I. Heritable diseases of collagen. N. Engl. J. Med. 1984, 311, 376–386. [Google Scholar] [CrossRef]
- El Masri, H.; Loong, T.H.; Meurette, G.; Podevin, J.; Zinzindohoue, F.; Lehur, P.A. Bowel perforation in type IV vascular Ehlers-Danlos syndrome. A systematic review. Tech. Coloproctol. 2018, 22, 333–341. [Google Scholar] [CrossRef]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.M.; Bal-Theoleyre, L.; Fauret, A.L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664. [Google Scholar] [CrossRef]
- Leistritz, D.F.; Pepin, M.G.; Schwarze, U.; Byers, P.H. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet. Med. 2011, 13, 717–722. [Google Scholar] [CrossRef]
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Bartolomeo, R.D.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar]
- Meester, J.A.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2017, 19, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Davis, E.C.; Chapman, S.L.; Budatha, M.; Marmorstein, L.Y.; Word, R.A.; Yanagisawa, H. Fibulin-4 deficiency results in ascending aortic aneurysms: A potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 2010, 106, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Igoucheva, O.; Alexeev, V.; Halabi, C.M.; Adams, S.M.; Stoilov, I.; Sasaki, T.; Arita, M.; Donahue, A.; Mecham, R.P.; Birk, D.E.; et al. Fibulin-4 E57K Knock-in Mice Recapitulate Cutaneous, Vascular and Skeletal Defects of Recessive Cutis Laxa 1B with both Elastic Fiber and Collagen Fibril Abnormalities. J. Biol. Chem. 2015, 290, 21443–21459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callewaert, B.; Renard, M.; Hucthagowder, V.; Albrecht, B.; Hausser, I.; Blair, E.; Dias, C.; Albino, A.; Wachi, H.; Sato, F.; et al. New insights into the pathogenesis of autosomal-dominant cutis laxa with report of five ELN mutations. Hum. Mutat. 2011, 32, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callewaert, B.L.; Willaert, A.; Kerstjens-Frederikse, W.S.; De Backer, J.; Devriendt, K.; Albrecht, B.; Ramos-Arroyo, M.A.; Doco-Fenzy, M.; Hennekam, R.C.; Pyeritz, R.E.; et al. Arterial tortuosity syndrome: Clinical and molecular findings in 12 newly identified families. Hum. Mutat. 2008, 29, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.G.; Chou, A.S.; Mok, S.C.M.; Ziganshin, B.A.; Charilaou, P.; Zafar, M.A.; Sieller, R.S.; Tranquilli, M.; Rizzo, J.A.; Elefteriades, J.A. Positive family history of aortic dissection dramatically increases dissection risk in family members. Int. J. Cardiol. 2017, 240, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Elefteriades, J.A.; Farkas, E.A. Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J. Am. Coll. Cardiol. 2010, 55, 841–857. [Google Scholar] [CrossRef]
- Guo, D.C.; Hostetler, E.M.; Fan, Y.; Kulmacz, R.J.; Zhang, D.; Nickerson, D.A.; Leal, S.M.; LeMaire, S.A.; Regalado, E.S.; Milewicz, D.M. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. J. Am. Coll. Cardiol. 2017, 70, 2728–2730. [Google Scholar] [CrossRef]
- Luyckx, I.; Loeys, B.L. The genetic architecture of non-syndromic thoracic aortic aneurysm. Heart 2015, 101, 1678–1684. [Google Scholar] [CrossRef]
- Sherrah, A.G.; Andvik, S.; van der Linde, D.; Davies, L.; Bannon, P.G.; Padang, R.; Vallely, M.P.; Wilson, M.K.; Keech, A.C.; Jeremy, R.W. Nonsyndromic Thoracic Aortic Aneurysm and Dissection: Outcomes With Marfan Syndrome Versus Bicuspid Aortic Valve Aneurysm. J. Am. Coll. Cardiol. 2016, 67, 618–626. [Google Scholar] [CrossRef]
- Bradley, T.J.; Bowdin, S.C.; Morel, C.F.; Pyeritz, R.E. The Expanding Clinical Spectrum of Extracardiovascular and Cardiovascular Manifestations of Heritable Thoracic Aortic Aneurysm and Dissection. Can. J. Cardiol. 2016, 32, 86–99. [Google Scholar] [CrossRef]
- Guo, D.C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef]
- Morisaki, H.; Akutsu, K.; Ogino, H.; Kondo, N.; Yamanaka, I.; Tsutsumi, Y.; Yoshimuta, T.; Okajima, T.; Matsuda, H.; Minatoya, K.; et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum. Mutat. 2009, 30, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Andelfinger, G.; Loeys, B.; Dietz, H. A Decade of Discovery in the Genetic Understanding of Thoracic Aortic Disease. Can. J. Cardiol. 2016, 32, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Ince, H.; Nienaber, C.A. Etiology, pathogenesis and management of thoracic aortic aneurysm. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 418–427. [Google Scholar] [CrossRef]
- Karimi, A.; Milewicz, D.M. Structure of the Elastin-Contractile Units in the Thoracic Aorta and How Genes That Cause Thoracic Aortic Aneurysms and Dissections Disrupt This Structure. Can. J. Cardiol. 2016, 32, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disabella, E.; Grasso, M.; Gambarin, F.I.; Narula, N.; Dore, R.; Favalli, V.; Serio, A.; Antoniazzi, E.; Mosconi, M.; Pasotti, M.; et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha-actin 2 (ACTA2). Heart 2011, 97, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regalado, E.S.; Guo, D.C.; Prakash, S.; Bensend, T.A.; Flynn, K.; Estrera, A.; Safi, H.; Liang, D.; Hyland, J.; Child, A.; et al. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circ. Cardiovasc. Genet. 2015, 8, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Renard, M.; Callewaert, B.; Baetens, M.; Campens, L.; MacDermot, K.; Fryns, J.P.; Bonduelle, M.; Dietz, H.C.; Gaspar, I.M.; Cavaco, D.; et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFβ signaling in FTAAD. Int. J. Cardiol. 2013, 165, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Milewicz, D.M.; Guo, D.C.; Tran-Fadulu, V.; Lafont, A.L.; Papke, C.L.; Inamoto, S.; Kwartler, C.S.; Pannu, H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genomics Hum. Genet. 2008, 9, 283–302. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Ostergaard, J.R.; Ala-Kokko, L.M.; Khan, N.; Grange, D.K.; Mendoza-Londono, R.; Bradley, T.J.; Olney, A.H.; Ades, L.; Maher, J.F.; et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J. Med. Genet. A 2010, 152, 2437–2443. [Google Scholar] [CrossRef] [Green Version]
- El-Hamamsy, I.; Yacoub, M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat. Rev. Cardiol. 2009, 6, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Bellini, C.; Wang, S.; Milewicz, D.M.; Humphrey, J.D. Myh11(R247C/R247C) mutations increase thoracic aorta vulnerability to intramural damage despite a general biomechanical adaptivity. J. Biomech. 2015, 48, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Zhou, X.; Jiang, X.; Sun, T. Deletion of ACTA2 in mice promotes angiotensin II induced pathogenesis of thoracic aortic aneurysms and dissections. J. Thorac. Dis. 2018, 10, 4733–4740. [Google Scholar] [CrossRef]
- Regalado, E.S.; Guo, D.C.; Estrera, A.L.; Buja, L.M.; Milewicz, D.M. Acute aortic dissections with pregnancy in women with ACTA2 mutations. Am J. Med. Genet. A 2014, 164, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Guo, D.C.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.Q.; Zhu, M.S.; et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 2010, 87, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stull, J.T.; Tansey, M.G.; Tang, D.C.; Word, R.A.; Kamm, K.E. Phosphorylation of myosin light chain kinase: a cellular mechanism for Ca2+ desensitization. Mol. Cell. Biochem. 1993, 127–128, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Huang, J.; He, W.; Zhu, M.; Kamm, K.E.; Stull, J.T. Signaling through myosin light chain kinase in smooth muscles. J. Biol. Chem. 2013, 288, 7596–7605. [Google Scholar] [CrossRef] [Green Version]
- Wallace, S.E.; Regalado, E.S.; Gong, L.; Janda, A.L.; Guo, D.C.; Russo, C.F.; Kulmacz, R.J.; Hanna, N.; Jondeau, G.; Boileau, C.; et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet. Med. 2019, 21, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Shalata, A.; Mahroom, M.; Milewicz, D.M.; Limin, G.; Kassum, F.; Badarna, K.; Tarabeih, N.; Assy, N.; Fell, R.; Cohen, H.; et al. Fatal thoracic aortic aneurysm and dissection in a large family with a novel MYLK gene mutation: Delineation of the clinical phenotype. Orphanet J. Rare Dis. 2018, 13, 41. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Vranckx, R.; Khau Van Kien, P.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef]
- Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Scherer, S.; Liu, Y.; Presley, C.; Guo, D.; Estrera, A.L.; Safi, H.J.; Brasier, A.R.; et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007, 16, 2453–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harakalova, M.; van der Smagt, J.; de Kovel, C.G.; Van’t Slot, R.; Poot, M.; Nijman, I.J.; Medic, J.; Joziasse, I.; Deckers, J.; Roos-Hesselink, J.W.; et al. Incomplete segregation of MYH11 variants with thoracic aortic aneurysms and dissections and patent ductus arteriosus. Eur. J. Hum. Genet. 2013, 21, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.; Guo, D.C.; Estrera, A.L.; Safi, H.J.; Huynh, T.T.; Yin, Z.; Cao, S.N.; Lin, J.; Kurian, T.; Buja, L.M.; et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J. Thorac. Cardiovasc. Surg. 2006, 131, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, S.H.; Corbin, J.D. Cyclic nucleotide-dependent protein kinases: Intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab. Sci. 1999, 36, 275–328. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet. 2013, 93, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Han, Q.; Liu, Z.; Zhou, W.; Cao, Q. Exome sequencing reveals a de novo PRKG1 mutation in a sporadic patient with aortic dissection. BMC Med. Genet. 2018, 19, 218. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.Q.; Medina-Martinez, O.; Guo, D.C.; Gong, L.; Regalado, E.S.; Reynolds, C.L.; Boileau, C.; Jondeau, G.; Prakash, S.K.; Kwartler, C.S.; et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J. Clin. Investig. 2016, 126, 948–961. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.S.; Halabi, C.M.; Hoffman, E.P.; Carmichael, N.; Leshchiner, I.; Lian, C.G.; Bierhals, A.J.; Vuzman, D.; Mecham, R.P.; Frank, N.Y.; et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc. Natl. Acad. Sci. USA 2016, 113, 8759–8764. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.C.; Gong, L.; Regalado, E.S.; Santos-Cortez, R.L.; Zhao, R.; Cai, B.; Veeraraghavan, S.; Prakash, S.K.; Johnson, R.J.; Muilenburg, A.; et al. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am. J. Hum. Genet. 2015, 96, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Combs, M.D.; Knutsen, R.H.; Broekelmann, T.J.; Toennies, H.M.; Brett, T.J.; Miller, C.A.; Kober, D.L.; Craft, C.S.; Atkinson, J.J.; Shipley, J.M.; et al. Microfibril-associated glycoprotein 2 (MAGP2) loss of function has pleiotropic effects in vivo. J. Biol. Chem. 2013, 288, 28869–28880. [Google Scholar] [CrossRef] [Green Version]
- Koenig, S.N.; LaHaye, S.; Feller, J.D.; Rowland, P.; Hor, K.N.; Trask, A.J.; Janssen, P.M.; Radtke, F.; Lilly, B.; Garg, V. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. JCI Insight 2017, 2, 91353. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.A.; Aziz, H.; Woods, C.E.; Seman-Senderos, M.A.; Sparks, E.; Preuss, C.; Wünnemann, F.; Bedja, D.; Moats, C.R.; McClymont, S.A.; et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat. Genet. 2019, 51, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.L.; Haelterman, N.A.; Kwartler, C.S.; Regalado, E.S.; Lee, P.T.; Nagarkar-Jaiswal, S.; Guo, D.C.; Duraine, L.; Wangler, M.F.; Bamshad, M.J.; et al. Ari-1 Regulates Myonuclear Organization Together with Parkin and Is Associated with Aortic Aneurysms. Dev. Cell 2018, 45, 226–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiñones-Pérez, B.; VanNoy, G.E.; Towne, M.C.; Shen, Y.; Singh, M.N.; Agrawal, P.B.; Smith, S.E. Three-generation family with novel contiguous gene deletion on chromosome 2p22 associated with thoracic aortic aneurysm syndrome. Am J. Med. Genet. A 2018, 176, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilberberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2018, 102, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.C.; Grove, M.L.; Prakash, S.K.; Eriksson, P.; Hostetler, E.M.; LeMaire, S.A.; Body, S.C.; Shalhub, S.; Estrera, A.L.; Safi, H.J.; et al. Genetic Variants in LRP1 and ULK4 Are Associated with Acute Aortic Dissections. Am. J. Hum. Genet. 2016, 99, 762–769. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Chen, S.; Zhang, H.; Zou, Y.; Zhao, J.; Yu, J.; Le, S.; Cui, J.; Jiang, L.; Wu, J.; et al. Network-based analysis reveals novel gene signatures in the peripheral blood of patients with sporadic nonsyndromic thoracic aortic aneurysm. J. Cell. Physiol. 2020, 235, 2478–2491. [Google Scholar] [CrossRef]
- Kwartler, C.S.; Gong, L.; Chen, J.; Wang, S.; Kulmacz, R.; Duan, X.Y.; Janda, A.; Huang, J.; Kamm, K.E.; Stull, J.T.; et al. Variants of Unknown Significance in Genes Associated with Heritable Thoracic Aortic Disease Can Be Low Penetrant Risk Variants. Am. J. Hum. Genet. 2018, 103, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Johansson, G.; Markstrom, U.; Swedenborg, J. Ruptured thoracic aortic aneurysms: A study of incidence and mortality rates. J. Vasc. Surg. 1995, 21, 985–988. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, C.J.; Casey, M.; He, J.; Veugelers, M.; Henderson, K.; Guo, D.; Campagna, R.; Roman, M.J.; Milewicz, D.M.; Devereux, R.B.; et al. Identification of a chromosome 11q23.2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation 2001, 103, 2469–2475. [Google Scholar] [CrossRef] [Green Version]
- Weerakkody, R.; Ross, D.; Parry, D.A.; Ziganshin, B.; Vandrovcova, J.; Gampawar, P.; Abdullah, A.; Biggs, J.; Dumfarth, J.; Ibrahim, Y.; et al. Targeted genetic analysis in a large cohort of familial and sporadic cases of aneurysm or dissection of the thoracic aorta. Genet. Med. 2018, 20, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Regalado, E.S.; Shendure, J.; Nickerson, D.A.; Guo, D.C. Successes and challenges of using whole exome sequencing to identify novel genes underlying an inherited predisposition for thoracic aortic aneurysms and acute aortic dissections. Trends Cardiovasc. Med. 2014, 24, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proost, D.; Vandeweyer, G.; Meester, J.A.; Salemink, S.; Kempers, M.; Ingram, C.; Peeters, N.; Saenen, J.; Vrints, C.; Lacro, R.V.; et al. Performant Mutation Identification Using Targeted Next-Generation Sequencing of 14 Thoracic Aortic Aneurysm Genes. Hum. Mutat. 2015, 36, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Landis, B.J.; Schubert, J.A.; Lai, D.; Jegga, A.G.; Shikany, A.R.; Foroud, T.; Ware, S.M.; Hinton, R.B. Exome Sequencing Identifies Candidate Genetic Modifiers of Syndromic and Familial Thoracic Aortic Aneurysm Severity. J. Cardiovasc. Transl. Res. 2017, 10, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Yu, C.; Chen, S.; Xiong, W.; Li, X.; Zeng, R.; Zhuang, J.; Fan, R. Identification of Novel Clinically Relevant Variants in 70 Southern Chinese patients with Thoracic Aortic Aneurysm and Dissection by Next-generation Sequencing. Sci. Rep. 2017, 7, 10035. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Carlson, A.A.; Regalado, E.S. Genetic testing in aortic aneurysm disease: PRO. Cardiol. Clin. 2010, 28, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Campens, L.; Callewaert, B.; Muino Mosquera, L.; Renard, M.; Symoens, S.; De Paepe, A.; Coucke, P.; De Backer, J. Gene panel sequencing in heritable thoracic aortic disorders and related entities - results of comprehensive testing in a cohort of 264 patients. Orphanet J. Rare Dis. 2015, 10, 9. [Google Scholar] [CrossRef]
- Arnaud, P.; Hanna, N.; Benarroch, L.; Aubart, M.; Bal, L.; Bouvagnet, P.; Busa, T.; Dulac, Y.; Dupuis-Girod, S.; Edouard, T.; et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet. Med. 2019, 21, 2015–2024. [Google Scholar] [CrossRef]
- Lawal, T.A.; Lewis, K.L.; Johnston, J.J.; Heidlebaugh, A.R.; Ng, D.; Gaston-Johansson, F.G.; Klein, W.M.P.; Biesecker, B.B.; Biesecker, L.G. Disclosure of cardiac variants of uncertain significance results in an exome cohort. Clin. Genet. 2018, 93, 1022–1029. [Google Scholar] [CrossRef]
- Wang, Y.; Barbacioru, C.C.; Shiffman, D.; Balasubramanian, S.; Iakoubova, O.; Tranquilli, M.; Albornoz, G.; Blake, J.; Mehmet, N.N.; Ngadimo, D.; et al. Gene expression signature in peripheral blood detects thoracic aortic aneurysm. PLoS ONE 2007, 2, e1050. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, S.; Sangiorgi, G.; Sang, X.; Rampoldi, V.; Suzuki, T.; Eagle, K.A.; Elefteriades, J.A. In search of blood tests for thoracic aortic diseases. Ann. Thorac. Surg. 2010, 90, 1735–1742. [Google Scholar] [CrossRef]
- Boileau, A.; Lino Cardenas, C.L.; Courtois, A.; Zhang, L.; Rodosthenous, R.S.; Das, S.; Sakalihasan, N.; Michel, J.B.; Lindsay, M.E.; Devaux, Y. MiR-574-5p: A Circulating Marker of Thoracic Aortic Aneurysm. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasiulė, S.; Stankevičius, V.; Patamsytė, V.; Ražanskas, R.; Žukovas, G.; Kapustina, Ž.; Žaliaduonytė, D.; Benetis, R.; Lesauskaitė, V.; Vilkaitis, G. Tissue-Specific miRNAs Regulate the Development of Thoracic Aortic Aneurysm: The Emerging Role of KLF4 Network. J. Clin. Med. 2019, 8, 1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariscalco, G.; Debiec, R.; Elefteriades, J.A.; Samani, N.J.; Murphy, G.J. Systematic Review of Studies That Have Evaluated Screening Tests in Relatives of Patients Affected by Nonsyndromic Thoracic Aortic Disease. J. Am. Heart Assoc. 2018, 7, e009302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolford, B.N.; Hornsby, W.E.; Guo, D.; Zhou, W.; Lin, M.; Farhat, L.; McNamara, J.; Driscoll, A.; Wu, X.; Schmidt, E.M.; et al. Clinical Implications of Identifying Pathogenic Variants in Individuals With Thoracic Aortic Dissection. Circ. Genomic Precis. Med. 2019, 12, e002476. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.S.; Ostergren, J. Direct-to-Consumer Genetic Testing and Personal Genomics Services: A Review of Recent Empirical Studies. Curr. Genet. Med. Rep. 2013, 1, 182–200. [Google Scholar] [CrossRef] [Green Version]
- Takayama, T.; Miyata, T.; Nagawa, H. True abdominal aortic aneurysm in Marfan syndrome. J. Vasc. Surg. 2009, 49, 1162–1165. [Google Scholar] [CrossRef] [Green Version]
- Remus, E.W.; O’Donnell, R.E.; Rafferty, K.; Weiss, D.; Joseph, G.; Csiszar, K.; Fong, S.F.; Taylor, W.R. The role of lysyl oxidase family members in the stabilization of abdominal aortic aneurysms. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1067–H1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micha, D.; Guo, D.C.; Hilhorst-Hofstee, Y.; van Kooten, F.; Atmaja, D.; Overwater, E.; Cayami, F.K.; Regalado, E.S.; van Uffelen, R.; Venselaar, H.; et al. SMAD2 Mutations Are Associated with Arterial Aneurysms and Dissections. Hum. Mutat. 2015, 36, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Danyi, P.; Elefteriades, J.A.; Jovin, I.S. Medical therapy of thoracic aortic aneurysms: Are we there yet? Circulation 2011, 124, 1469–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milewicz, D.; Hostetler, E.; Wallace, S.; Mellor-Crummey, L.; Gong, L.; Pannu, H.; Guo, D.C.; Regalado, E. Precision medical and surgical management for thoracic aortic aneurysms and acute aortic dissections based on the causative mutant gene. J. Cardiovasc. Surg. 2016, 57, 172–177. [Google Scholar]

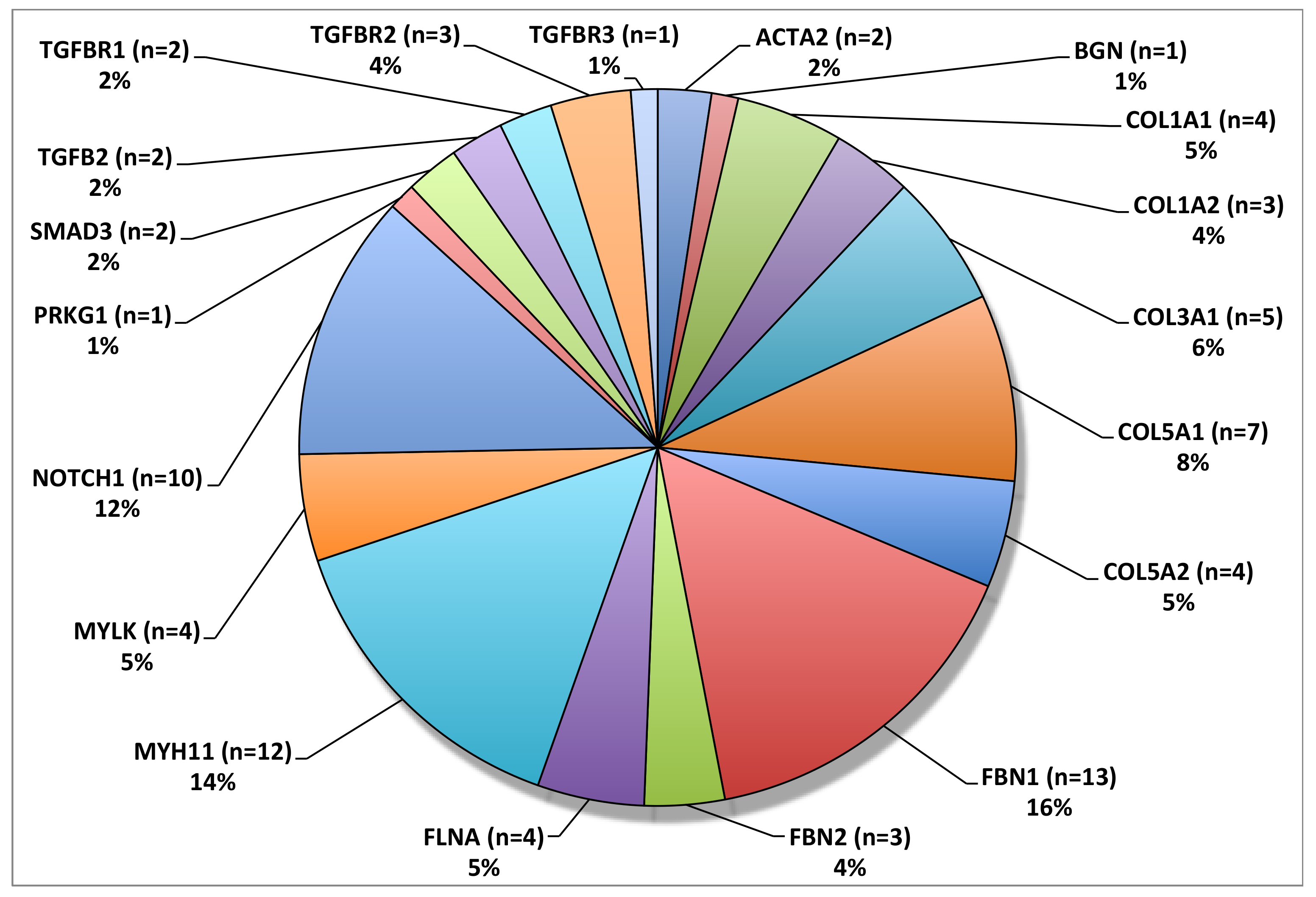


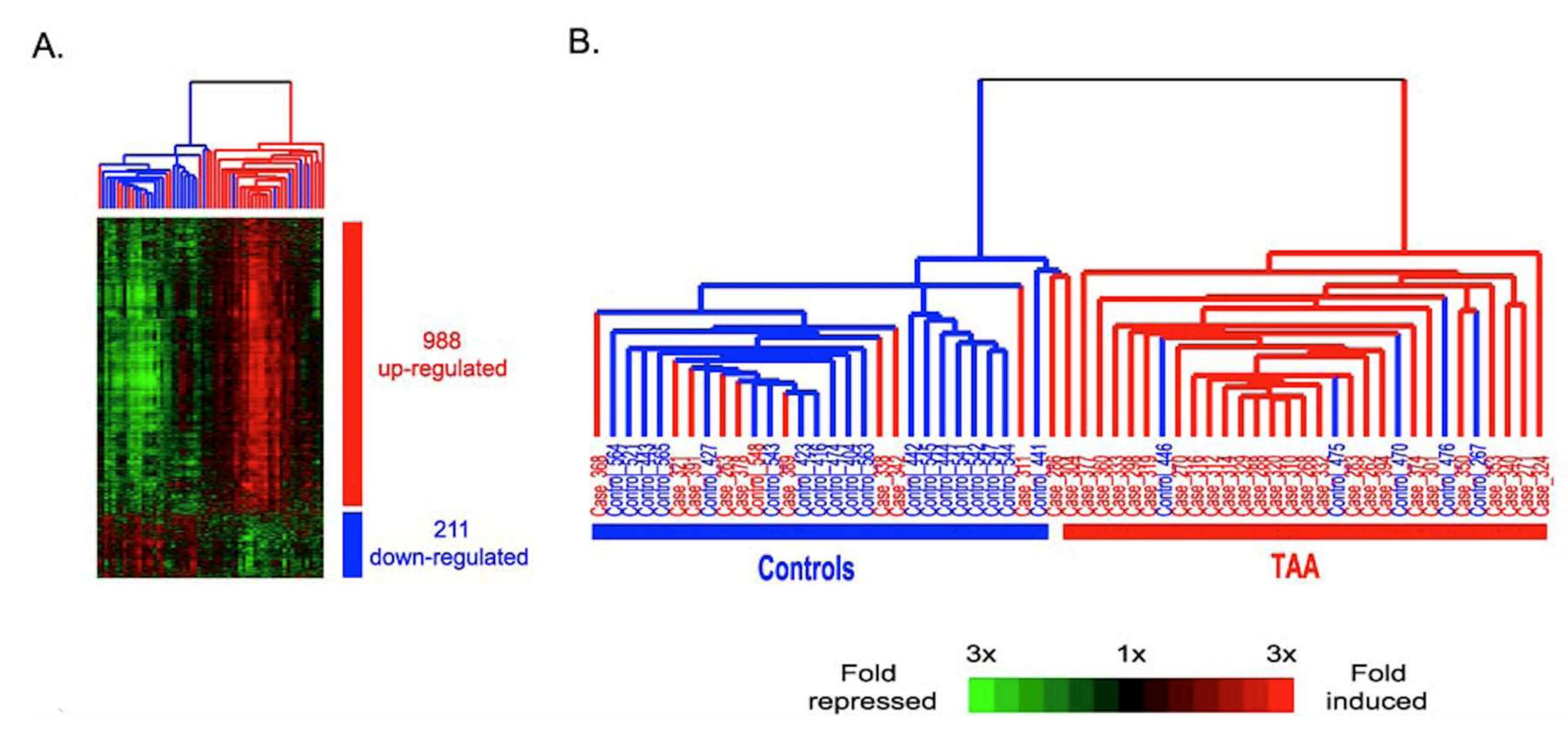

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182. https://doi.org/10.3390/biom10020182
Ostberg NP, Zafar MA, Ziganshin BA, Elefteriades JA. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules. 2020; 10(2):182. https://doi.org/10.3390/biom10020182
Chicago/Turabian StyleOstberg, Nicolai P., Mohammad A. Zafar, Bulat A. Ziganshin, and John A. Elefteriades. 2020. "The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective" Biomolecules 10, no. 2: 182. https://doi.org/10.3390/biom10020182
APA StyleOstberg, N. P., Zafar, M. A., Ziganshin, B. A., & Elefteriades, J. A. (2020). The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules, 10(2), 182. https://doi.org/10.3390/biom10020182