Pharmacological Inhibition of Caspase-8 Suppresses Inflammation-Induced Angiogenesis in the Cornea

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Mouse Model of Corneal AB
2.3. Observation and Quantification of CNV
2.4. Histological Assessment of Mouse Eyes
2.5. Immunofluorescence
2.6. Cell Culture and Treatment
2.7. Real-Time Quantitative PCR
2.8. Caspase-8 and Caspase-1 Activity
2.9. Statistical Analysis
3. Results
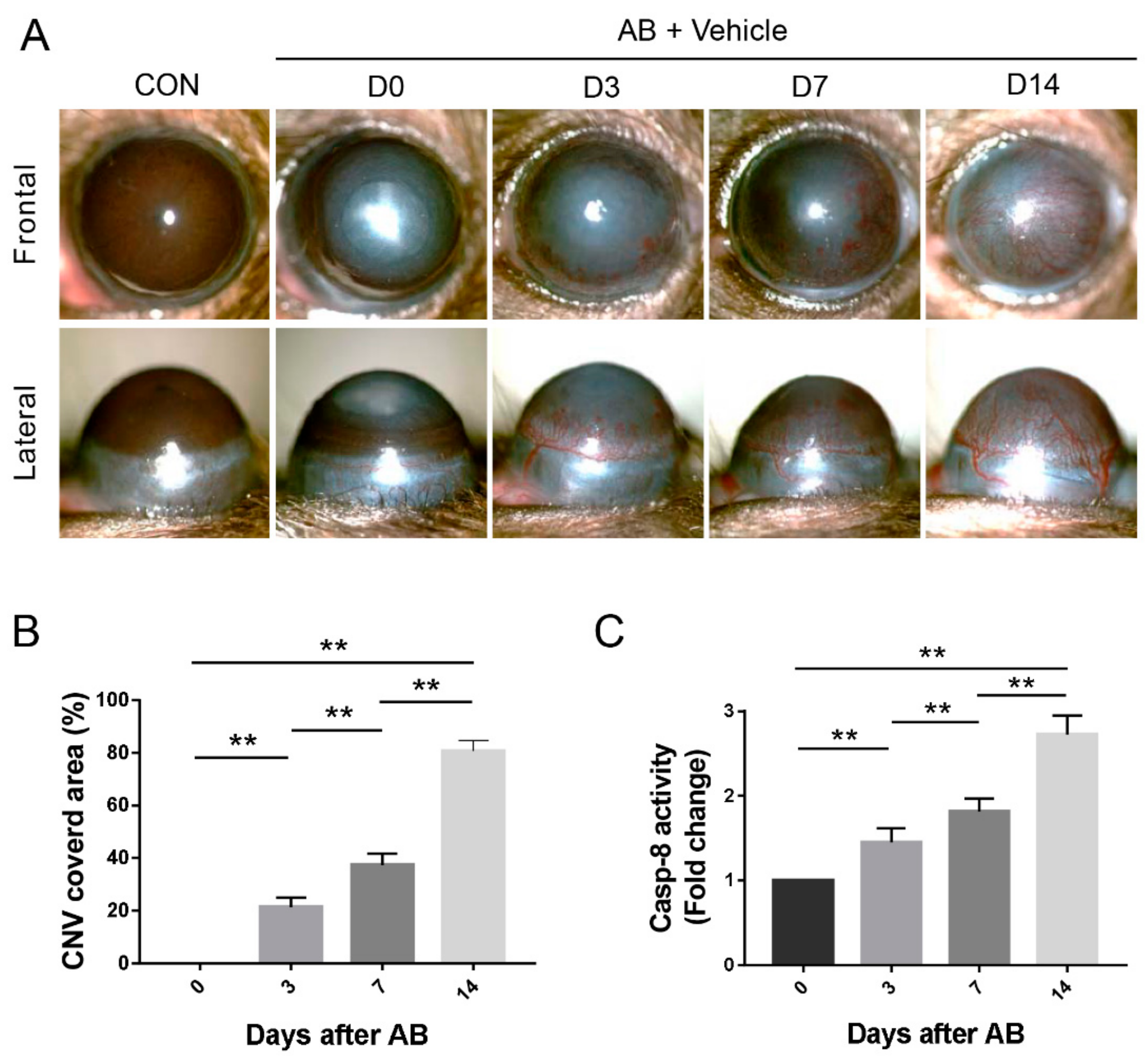
3.1. Corneal AB Induced CNV and Increased Caspase-8 Activity
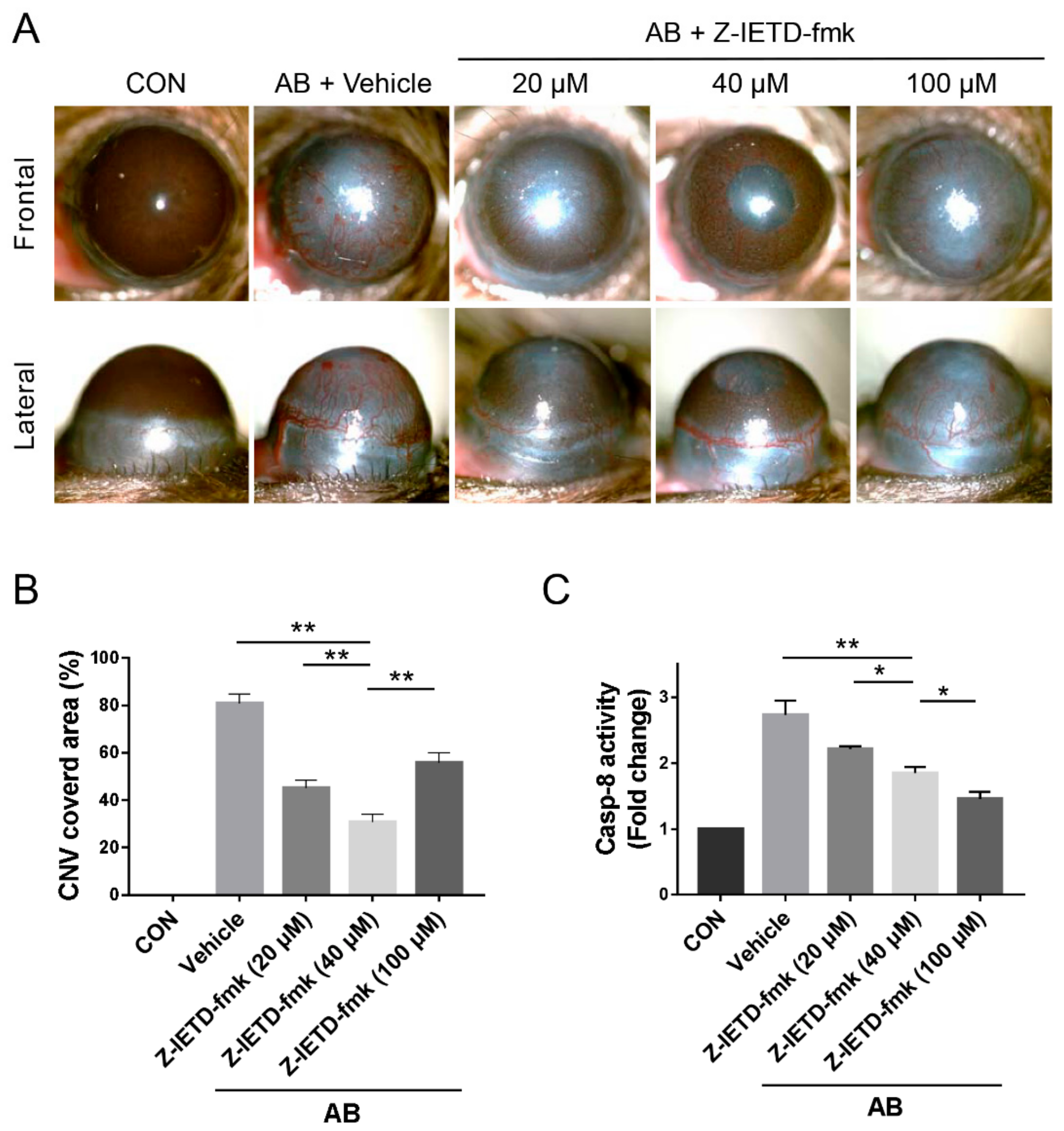
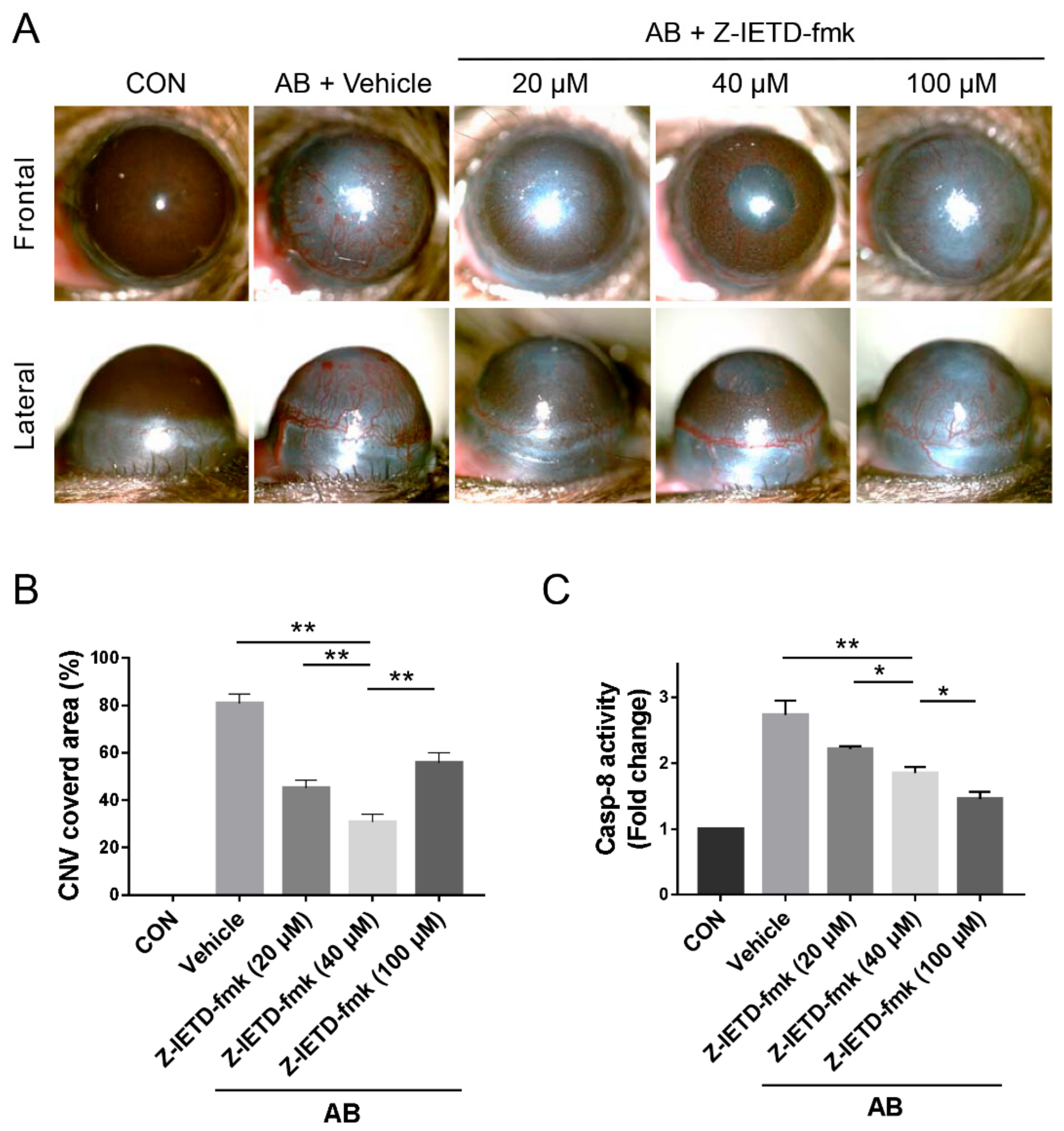
3.2. Pharmacological Inhibition of Caspase-8 Suppressed CNV after Corneal AB
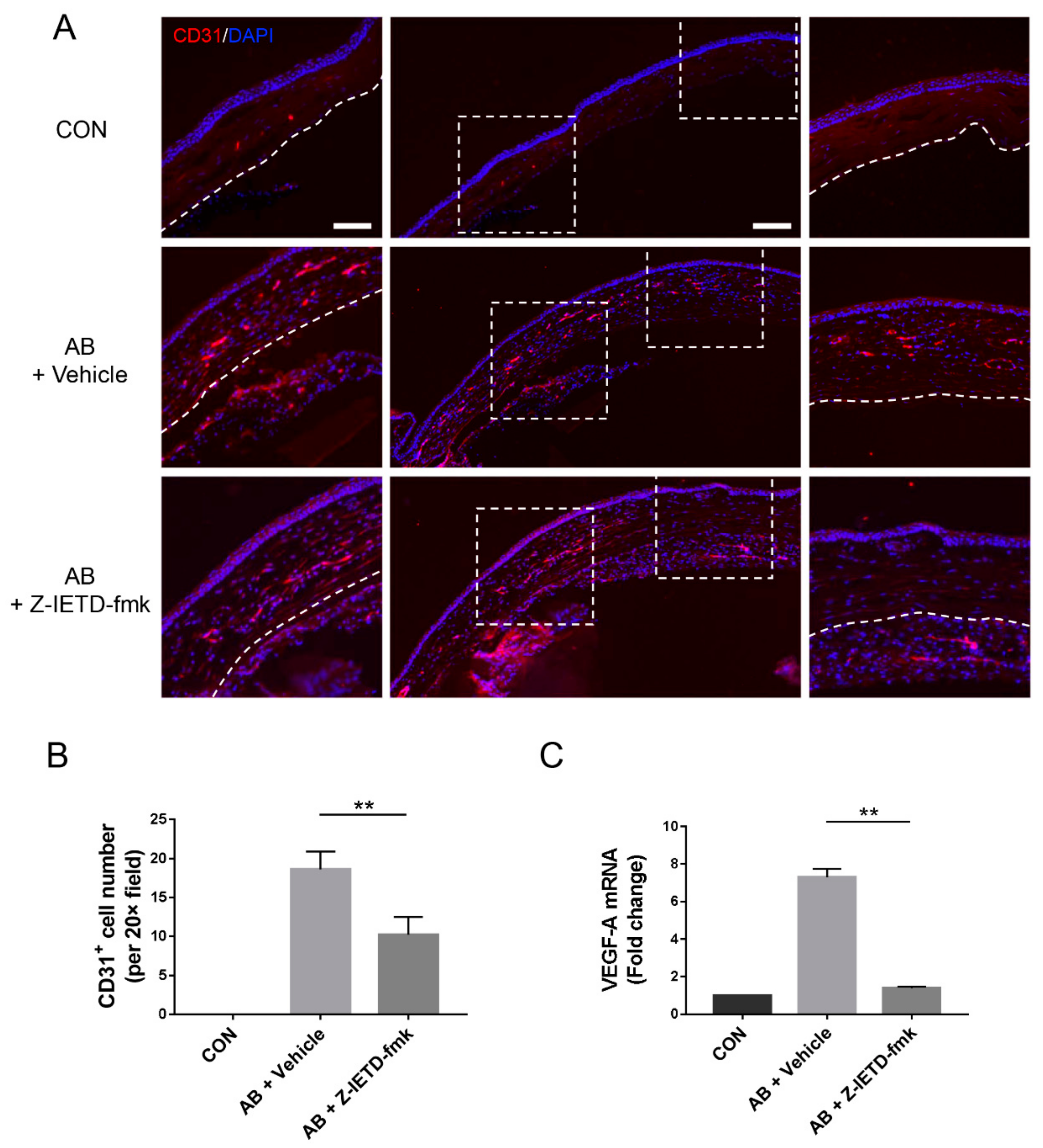
3.3. Pharmacological Inhibition of Caspase-8 Suppressed CD31 and VEGF-A Expression after Corneal AB
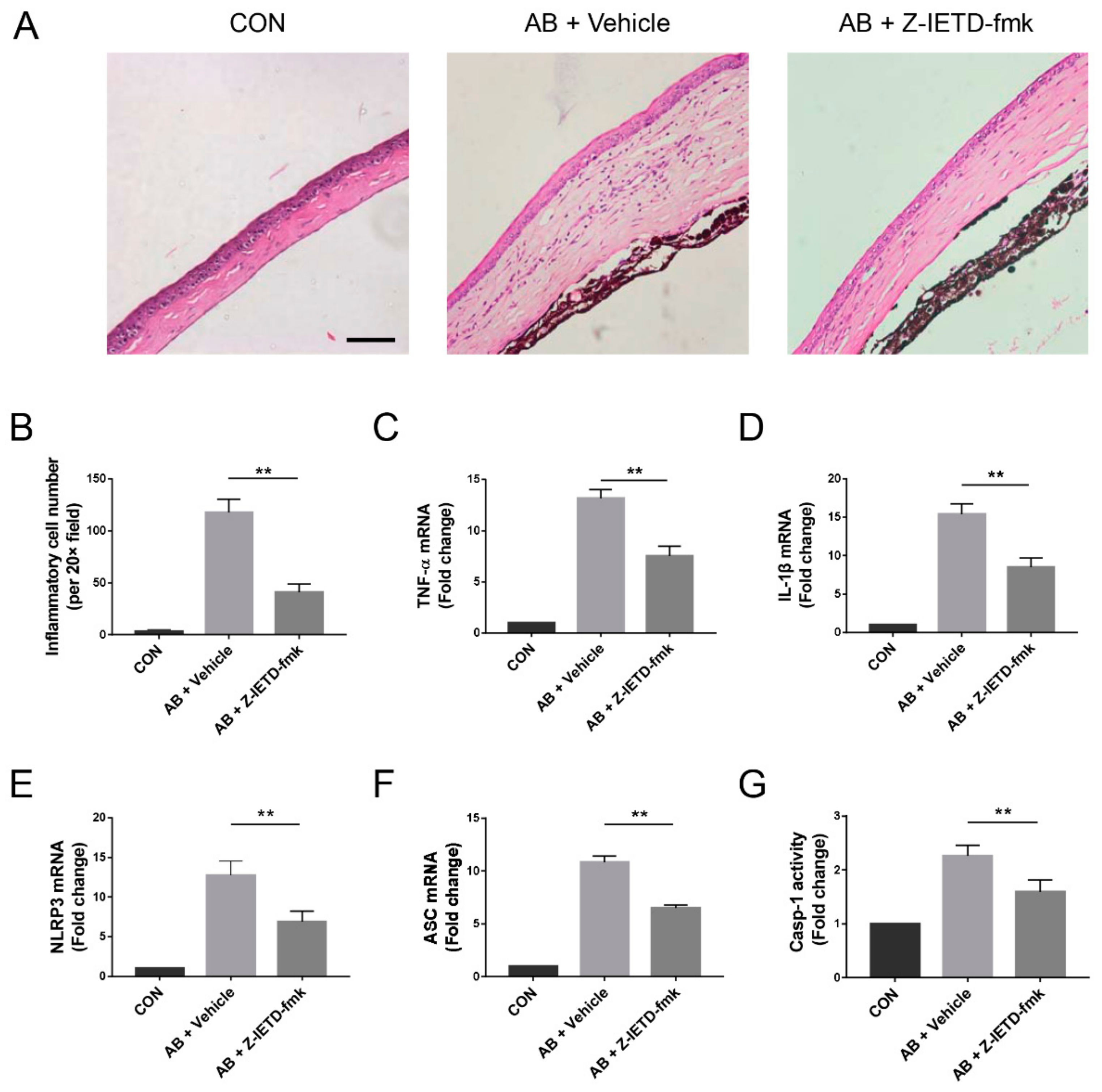
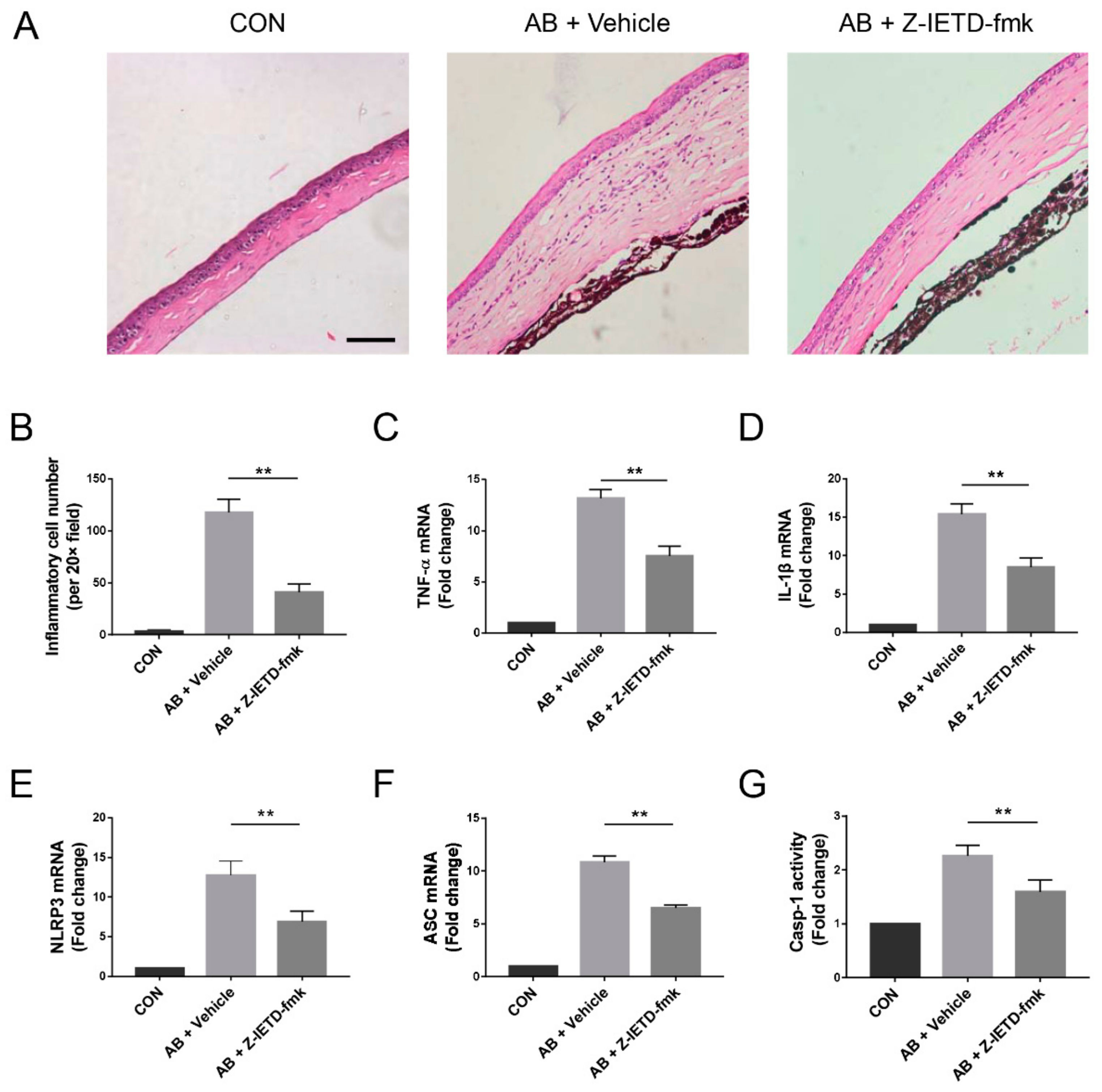
3.4. Pharmacological Inhibition of Caspase-8 Suppressed Alkali-Induced Inflammation
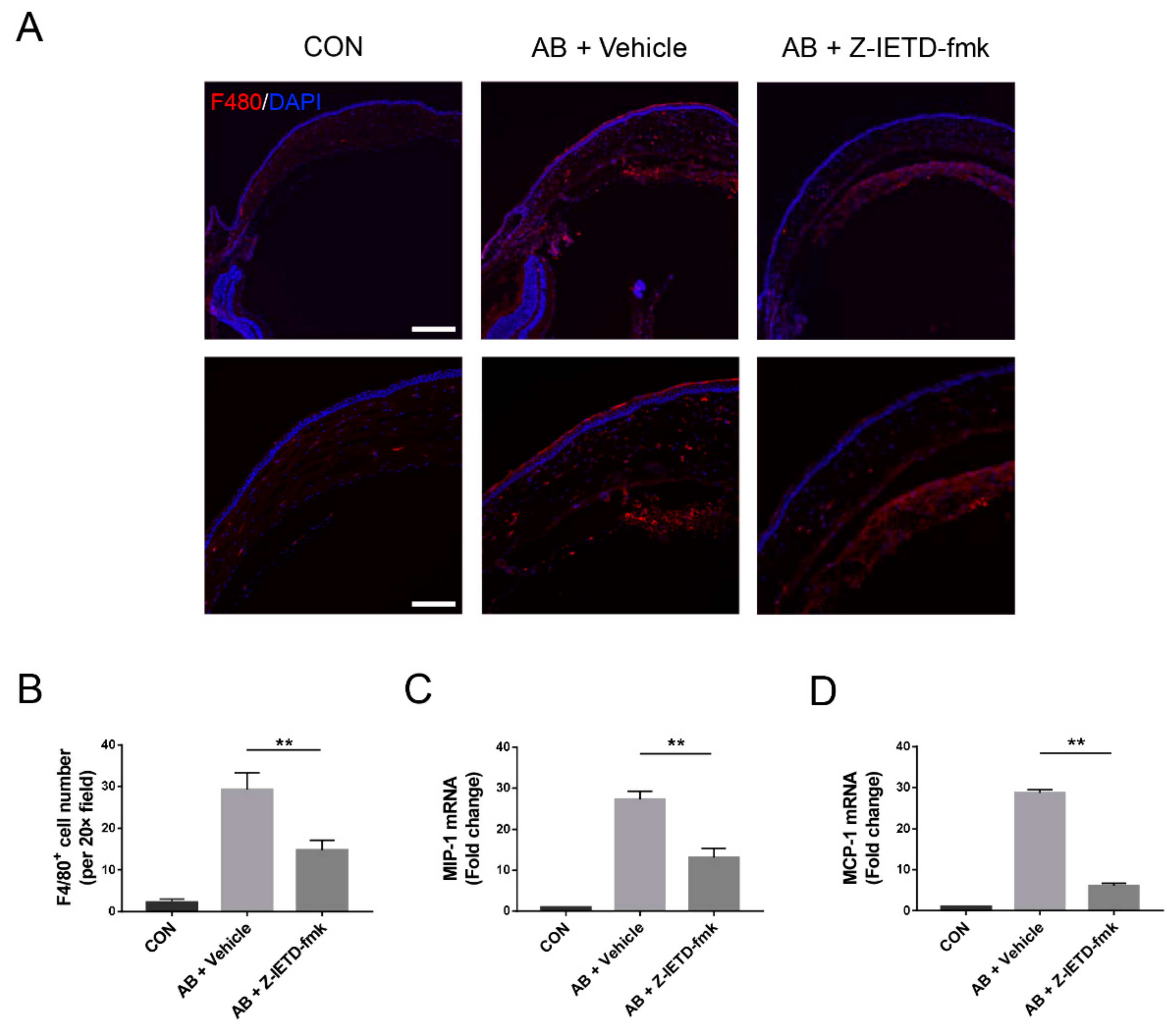
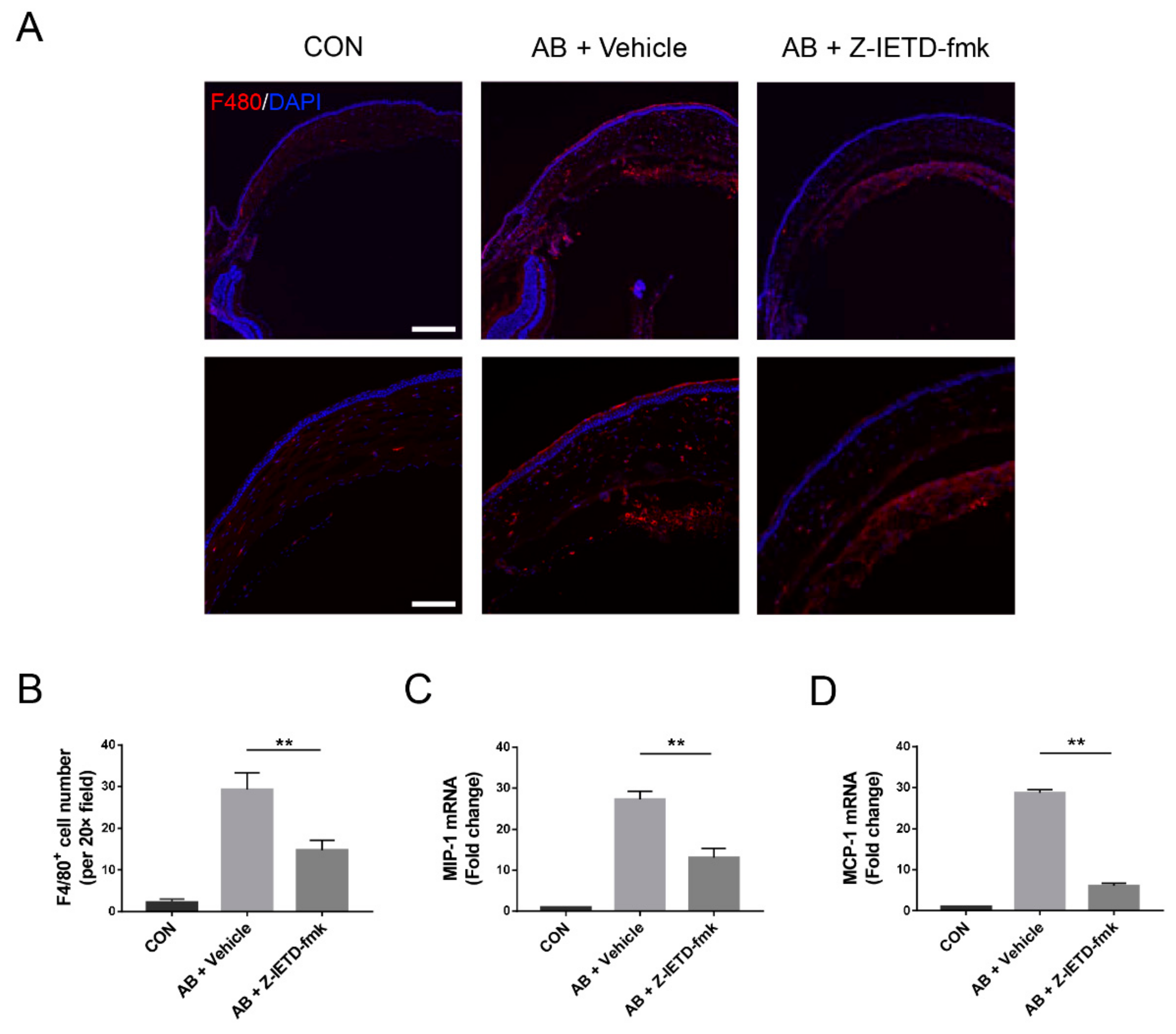
3.5. Pharmacological Inhibition of Caspase-8 Suppressed Macrophage Recruitment
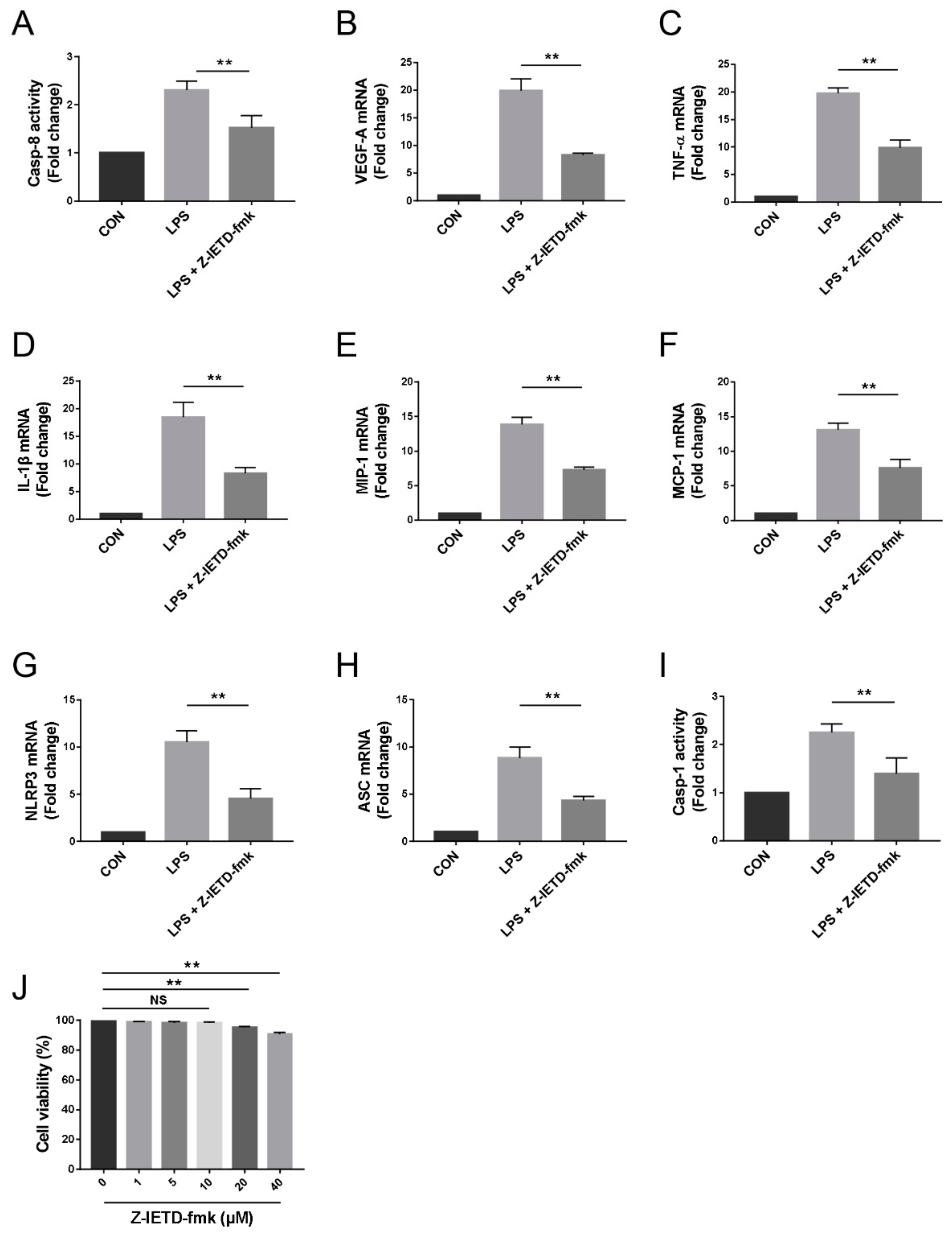
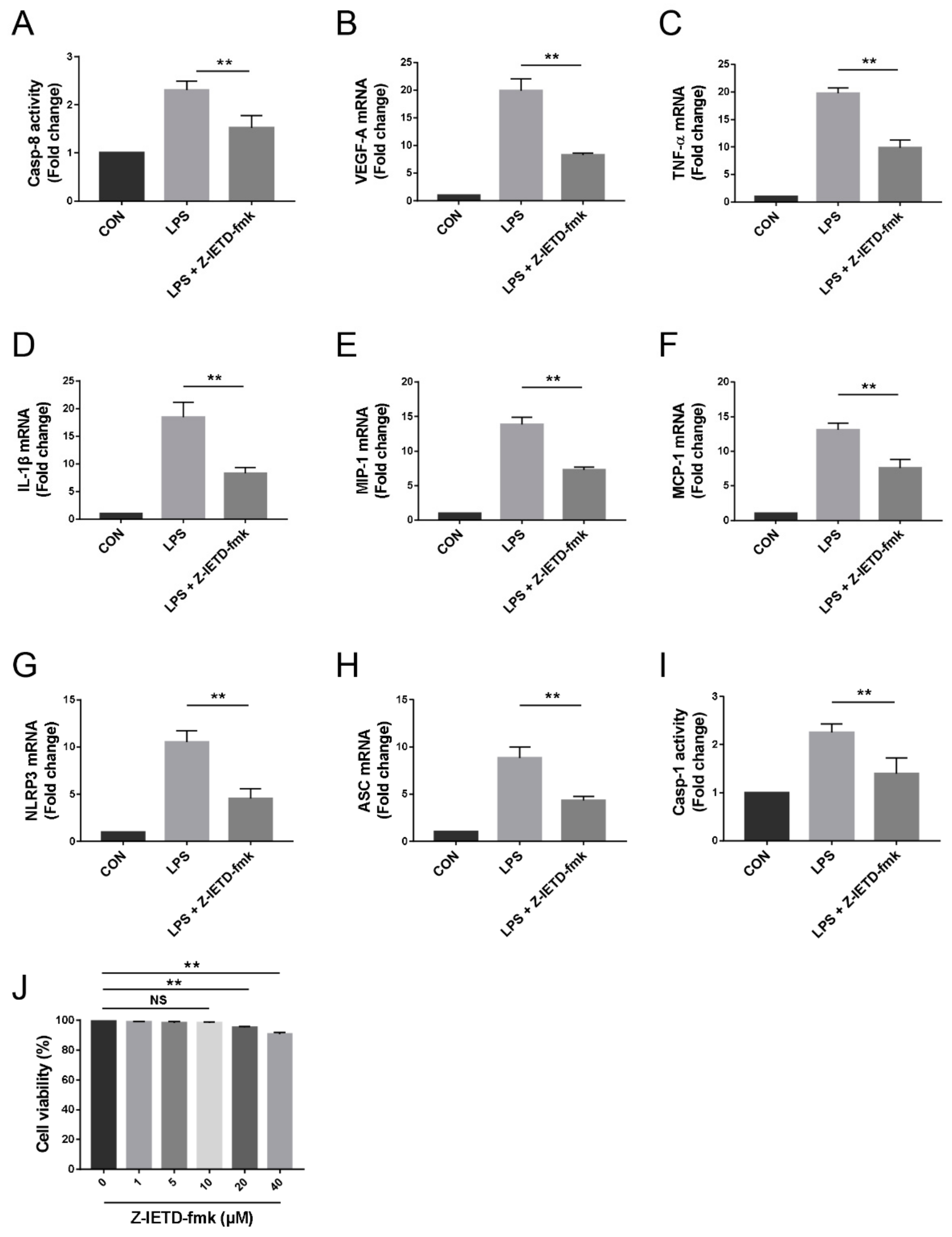
3.6. Pharmacological Inhibition of Caspase-8 Suppressed the Inflammatory Profiles of Macrophage In Vitro
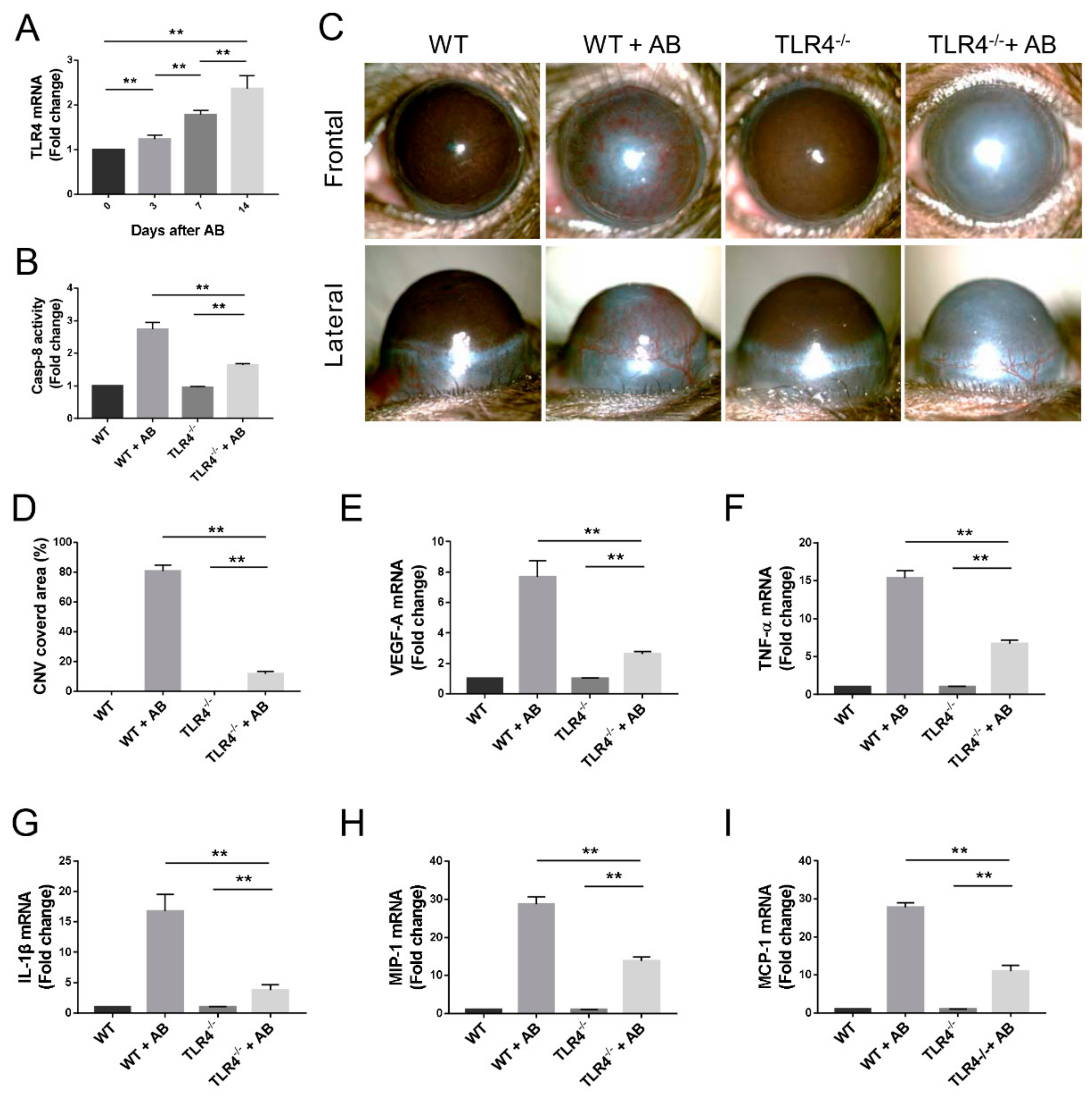
3.7. Caspase-8 Activity was Partially Regulated by TLR4 Signaling
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AB | alkali burn |
| CNV | corneal neovascularization |
| NLRP3 | Nod-like receptor family pyrin domain-containing 3 |
| ASC | apoptosis-associated speck-like protein containing CARD |
| WT | wild-type |
| TLR4 | toll like receptor 4 |
| SPF | specific pathogen-free |
| PFA | paraformaldehyde |
| BSA | bovine serum albumin |
| TBS | Tris-buffered saline |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | fetal bovine serum |
| LPS | lipopolysaccharide |
| VEGF-A | vascular endothelial growth factor-A |
| TNF-α | tumor necrosis factor alpha |
| IL-1β | interleukin-1 beta |
| MIP-1 | macrophage inflammatory protein-1 |
| MCP-1 | monocyte chemoattractant protein-1 |
| CON | control |
| Casp | caspase |
References
- Szade, A.; Grochot-Przeczek, A.; Florczyk, U.; Jozkowicz, A.; Dulak, J. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life 2015, 67, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Khurana, R.; Simons, M.; Martin, J.F.; Zachary, I.C. Role of angiogenesis in cardiovascular disease: a critical appraisal. Circulation 2005, 112, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [Green Version]
- Witmer, A.N.; Vrensen, G.F.; Van Noorden, C.J.; Schlingemann, R.O. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin Eye Res. 2003, 22, 1–29. [Google Scholar] [CrossRef]
- Gariano, R.F.; Gardner, T.W. Retinal angiogenesis in development and disease. Nature 2005, 438, 960–966. [Google Scholar] [CrossRef]
- Ellenberg, D.; Azar, D.T.; Hallak, J.A.; Tobaigy, F.; Han, K.Y.; Jain, S.; Zhou, Z.; Chang, J.H. Novel aspects of corneal angiogenic and lymphangiogenic privilege. Prog. Retin Eye Res. 2010, 29, 208–248. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El–Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. [Google Scholar] [CrossRef]
- Doyle, B.; Caplice, N. Plaque neovascularization and antiangiogenic therapy for atherosclerosis. J. Am. Coll. Cardiol. 2007, 49, 2073–2080. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Li, Y.; Lin, T.; Yuan, L.; Li, Y.; Wu, S.; Xia, L.; Shen, H.; Lu, J. IL–35 Inhibits Angiogenesis through VEGF/Ang2/Tie2 Pathway in Rheumatoid Arthritis. Cell Physiol. Biochem. 2016, 40, 1105–1116. [Google Scholar] [CrossRef]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Wong, T.Y.; Cheung, C.M.; Larsen, M.; Sharma, S.; Simó, R. Diabetic retinopathy. Nat. Rev. Dis. Primers 2016, 2, 16012. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Garg, N.K.; Lunde, E.; Han, K.Y.; Jain, S.; Azar, D.T. Corneal neovascularization: an anti-VEGF therapy review. Surv. Ophthalmol. 2012, 57, 415–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Kanneganti, T.D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef]
- Feltham, R.; Vince, J.E.; Lawlor, K.E. Caspase-8: not so silently deadly. Clin. Transl. Immunol. 2017, 6, e124. [Google Scholar] [CrossRef]
- Crowder, R.N.; El–Deiry, W.S. Caspase-8 regulation of TRAIL-mediated cell death. Exp. Oncol. 2012, 34, 160–164. [Google Scholar]
- Tezel, G.; Wax, M.B. Inhibition of caspase activity in retinal cell apoptosis induced by various stimuli in vitro. Invest. Ophthalmol. Vis. Sci. 1999, 40, 2660–2667. [Google Scholar]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; Garcia-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef]
- Gao, Y.; Tang, J.; Chen, W.; Li, Q.; Nie, J.; Lin, F.; Wu, Q.; Chen, Z.; Gao, Z.; Fan, H.; et al. Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc. Natl. Acad. Sci. USA 2015, 112, E3246–E3254. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.B.; Kim, H.R.; Park, M.C.; Cho, S.; Goughnour, P.C.; Han, D.; Yoon, I.; Kim, Y.; Kang, T.; Song, E. Caspase-8 controls the secretion of inflammatory lysyl-tRNA synthetase in exosomes from cancer cells. J. Cell Biol. 2017, 216, 2201–2216. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Gurung, P.; Malireddi, R.K.; Karmaus, P.W.; Sharma, D.; Vogel, P.; Chi, H.; Green, D.R.; Kanneganti, T.D. Critical role of caspase-8-mediated IL-1 signaling in promoting Th2 responses during asthma pathogenesis. Mucosal Immunol. 2017, 10, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, C.; Russo, H.M.; El Sanadi, C.; Martin, B.N.; Li, X.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J. Biol. Chem. 2015, 290, 20167–20184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Han, L.; Chen, X.; Yu, J.; Cheng, X.; Huang, J.; Xiao, Y.; Tian, Y.; Olsen, N.; Zheng, S.G.; et al. Pharmacological inhibition of caspase-8 suppresses inflammation-induced lymphangiogenesis and allograft rejection in the cornea. J. Allergy Clin. Immunol. 2018, 142, 290–294.e9. [Google Scholar] [CrossRef] [Green Version]
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; Del Bufalo, D.; Cinà, I.; Di Benedetto, A.; Mottolese, M.; et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. eLife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Liu, X.; Wu, J.; Li, J.; Dong, C.; Wu, X.; Xiao, X.; Yu, F.X. CXCL10 suppression of hem- and lymph-angiogenesis in inflamed corneas through MMP13. Angiogenesis 2017, 20, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Lennikov, A.; Mirabelli, P.; Mukwaya, A.; Schaupper, M.; Thangavelu, M.; Lachota, M.; Ali, Z.; Jensen, L.; Lagali, N. Selective IKK2 inhibitor IMD0354 disrupts NF-κB signaling to suppress corneal inflammation and angiogenesis. Angiogenesis 2018, 21, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Baradaran-Rafii, A.; Eslani, M.; Haq, Z.; Shirzadeh, E.; Huvard, M.J.; Djalilian, A.R. Current and Upcoming Therapies for Ocular Surface Chemical Injuries. Ocul. Surf. 2017, 15, 48–64. [Google Scholar] [CrossRef] [Green Version]
- Mulik, S.; Xu, J.; Reddy, P.B.; Rajasagi, N.K.; Gimenez, F.; Sharma, S.; Lu, P.Y.; Rouse, B.T. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am. J. Pathol. 2012, 181, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Suryawanshi, A.; Veiga-Parga, T.; Reddy, P.B.; Rajasagi, N.K.; Rouse, B.T. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J. Immunol. 2012, 188, 3434–3446. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.W.; Yan, L.; Jiang, D.; Qin, P.; Tse, H.F.; Wong, I.Y.; Wong, D.S.; Tergaonkar, V.; Lian, Q. Inhibition of RAP1 enhances corneal recovery following alkali injury. Invest. Ophthalmol. Vis. Sci. 2015, 56, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.; Choi, J.S.; Rho, C.R.; Joo, C.K.; Lee, S.K. MicroRNA miR-466 inhibits Lymphangiogenesis by targeting prospero-related homeobox 1 in the alkali burn corneal injury model. J. Biomed. Sci. 2015, 22, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.; Zhou, Q.; Wang, S. An alkali-burn injury model of corneal neovascularization in the mouse. J. Vis. Exp. 2014, 86. [Google Scholar] [CrossRef]
- Hua, X.; Yuan, X.; Li, Y.; Chen, H.; Yuan, J.; Tanumiharjo, S.; Bian, F.; Su, L.; Hong, Y.; Liu, Y.; et al. Desiccating stress worsens alkali burn injury by magnifying caspase-8-induced imbalance of NLRP3 and NLRP6. J. Allergy Clin. Immunol 2017, 140, 1172–1176.e3. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Li, Z.; Li, Y.; Lin, M.; Yao, L.; Liu, Y.; He, Z.; Wu, C.; Liang, D. Doxycycline enhances the inhibitory effects of bevacizumab on corneal neovascularization and prevents its side effects. Invest. Ophthalmol. Vis. Sci. 2011, 52, 9108–9115. [Google Scholar] [CrossRef] [Green Version]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef] [Green Version]
- Sunderkötter, C.; Steinbrink, K.; Goebeler, M.; Bhardwaj, R.; Sorg, C. Macrophages and angiogenesis. J. Leukoc. Biol. 1994, 55, 410–422. [Google Scholar] [CrossRef]
- Lamagna, C.; Aurrand-Lions, M.; Imhof, B.A. Dual role of macrophages in tumor growth and angiogenesis. J. Leukoc. Biol. 2006, 80, 705–713. [Google Scholar] [CrossRef]
- David Dong, Z.M.; Aplin, A.C.; Nicosia, R.F. Regulation of angiogenesis by macrophages, dendritic cells, and circulating myelomonocytic cells. Curr. Pharm. Des. 2009, 15, 365–379. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [Green Version]
- Gillitzer, R.; Goebeler, M. Chemokines in cutaneous wound healing. J. Leukoc. Biol. 2001, 69, 513–521. [Google Scholar]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Tohme, S.; Billiar, T.R. High-mobility group box-1 in sterile inflammation. J. Intern. Med. 2014, 276, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Yang, X.P.; Fang, D.; Ren, X.; Zhou, H.; Fang, J.; Liu, X.; Zhou, S.; Wen, F.; Yao, X.; et al. High-mobility group box-1 mediates toll-like receptor 4-dependent angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Park, J.S.; Strassheim, D.; Douglas, I.; Diaz del Valle, F.; Asehnoune, K.; Mitra, S.; Kwak, S.H.; Yamada, S.; Maruyama, I.; et al. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L958–L965. [Google Scholar] [CrossRef] [Green Version]
- Tsung, A.; Zheng, N.; Jeyabalan, G.; Izuishi, K.; Klune, J.R.; Geller, D.A.; Lotze, M.T.; Lu, L.; Billiar, T.R. Increasing numbers of hepatic dendritic cells promote HMGB1-mediated ischemia-reperfusion injury. J. Leukoc. Biol. 2007, 81, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Shenderov, K.; Riteau, N.; Yip, R.; Mayer-Barber, K.D.; Oland, S.; Hieny, S.; Fitzgerald, P.; Oberst, A.; Dillon, C.P.; Green, D.R.; et al. Cutting edge: Endoplasmic reticulum stress licenses macrophages to produce mature IL-1β in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J. Immunol. 2014, 192, 2029–2033. [Google Scholar] [CrossRef]
- Philip, N.H.; DeLaney, A.; Peterson, L.W.; Santos-Marrero, M.; Grier, J.T.; Sun, Y.; Wynosky-Dolfi, M.A.; Zwack, E.E.; Hu, B.; Olsen, T.M.; et al. Activity of Uncleaved Caspase-8 Controls Anti-bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog. 2016, 12, e1005910. [Google Scholar] [CrossRef]
- Nogueira-Machado, J.A.; Volpe, C.M.; Veloso, C.A.; Chaves, M.M. HMGB1, TLR and RAGE: A functional tripod that leads to diabetic inflammation. Expert Opin. Ther. Targets 2011, 15, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Peng, S.; Liu, X.; Han, C.; Wang, X.; Jin, T.; Liu, S.; Wang, W.; Xie, X.; He, X.; et al. Glycyrrhizin, a Direct HMGB1 Antagonist, Ameliorates Inflammatory Infiltration in a Model of Autoimmune Thyroiditis via Inhibition of TLR2-HMGB1 Signaling. Thyroid 2017, 27, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-κB pathway in acute glaucoma. J. Neuroinflammation 2015, 12, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueta, T.; Ishihara, K.; Notomi, S.; Lee, J.J.; Maidana, D.E.; Efstathiou, N.E.; Murakami, Y.; Hasegawa, E.; Azuma, K.; Toyono, T.; et al. RIP1 kinase mediates angiogenesis by modulating macrophages in experimental neovascularization. Proc. Natl. Acad. Sci. USA 2019, 116, 23705–23713. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Li, H.; Liu, X.; Xie, L.; Huang, Z.; Li, W.; Li, Z.; Pan, Y.; Chen, X.; Su, W. Pharmacological Inhibition of Caspase-8 Suppresses Inflammation-Induced Angiogenesis in the Cornea. Biomolecules 2020, 10, 210. https://doi.org/10.3390/biom10020210
Tian Y, Li H, Liu X, Xie L, Huang Z, Li W, Li Z, Pan Y, Chen X, Su W. Pharmacological Inhibition of Caspase-8 Suppresses Inflammation-Induced Angiogenesis in the Cornea. Biomolecules. 2020; 10(2):210. https://doi.org/10.3390/biom10020210
Chicago/Turabian StyleTian, Yunzhe, He Li, Xiuxing Liu, Lihui Xie, Zhaohao Huang, Weihua Li, Zhuang Li, Yuan Pan, Xiaoqing Chen, and Wenru Su. 2020. "Pharmacological Inhibition of Caspase-8 Suppresses Inflammation-Induced Angiogenesis in the Cornea" Biomolecules 10, no. 2: 210. https://doi.org/10.3390/biom10020210
APA StyleTian, Y., Li, H., Liu, X., Xie, L., Huang, Z., Li, W., Li, Z., Pan, Y., Chen, X., & Su, W. (2020). Pharmacological Inhibition of Caspase-8 Suppresses Inflammation-Induced Angiogenesis in the Cornea. Biomolecules, 10(2), 210. https://doi.org/10.3390/biom10020210