hNGF Peptides Elicit the NGF-TrkA Signalling Pathway in Cholinergic Neurons and Retain Full Neurotrophic Activity in the DRG Assay

 ,
,  , ,
, ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. hNGF Peptides Synthesis
2.2. hNGF Peptides and NGF Purification
2.3. Reagents and Antibodies
2.4. Dorsal Root Ganglion Dissociated Culture
2.5. Cholinergic Neurons Culture
2.6. Western Blotting
2.7. Immunofluorescence Labelling and Microscopy
2.8. Neuronal Counts
2.9. Analysis of DRG Bioassay
2.10. Electrophysiology
2.11. Viability Assay
2.12. Statistical Analysis
3. Results
3.1. hNGF1–14 Peptides Activate both TrkA-Shc and TrkA-PLC-γ Signalling Pathways in Cholinergic Neurons
3.2. hNGF1–14 Peptides Administration Increases the Number of Cholinergic Neurons Expressing pAKT and Nuclear pCREB
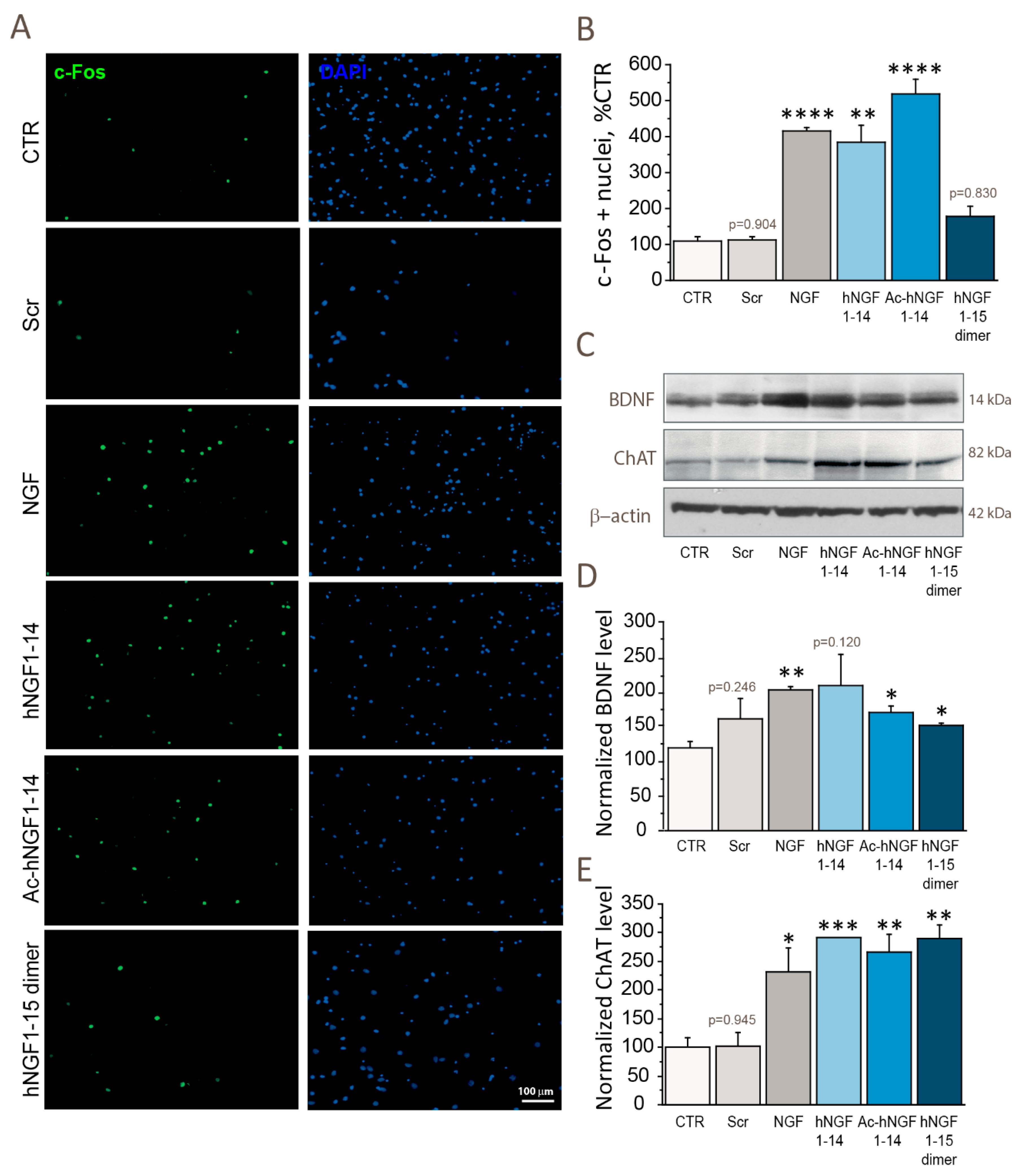
3.3. hNGF1–14 Peptides Show NGF-like Activity in Stimulating Neuronal Activity (c-Fos), Cholinergic Functions (ChAT) and Neuronal Plasticity (BDNF)
3.4. The Neurotrophic Effect of hNGF1–14 Peptides on Cholinergic Neurons is Lost upon TrkA Inhibition
3.5. Ac-hNGF1–14 Peptide Administration Induces an NGF-like Electrophysiological Activity in Primary Cholinergic Neurons
3.6. hNGF1–14 Peptides Sustain the Survival and Promote Neurite Outgrowth of Dissociated Dorsal Root Ganglia Neurons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levi-Montalcini, R. The nerve growth factor 35 years later. Science 1987, 237, 1154–1162. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Howe, C.L.; Mobley, W.C. Nerve growth factor signaling, neuroprotection, and neural repair. Annu. Rev. Neurosci. 2001, 24, 1217–1281. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, R.B.; Martynowski, C. Nerve growth factor induces Fos-like immunoreactivity within identified cholinergic neurons in the adult rat basal forebrain. Brain Res. 1997, 753, 141–151. [Google Scholar] [CrossRef]
- Bradshaw, R.A.; Pundavela, J.; Biarc, J.; Chalkley, R.J.; Burlingame, A.L.; Hondermarck, H. NGF and ProNGF: Regulation of neuronal and neoplastic responses through receptor signaling. Adv. Biol. Regul. 2015, 58, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.W.; Yung, K.K.L.; Chan, Y.S.; Shum, D.K.Y.; Bolam, J.P. The proNGF-p75NTR-sortilin signalling complex as new target for the therapeutic treatment of Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2008, 7, 512–523. [Google Scholar] [CrossRef]
- Faiq, M.A.; Wollstein, G.; Schuman, J.S.; Chan, K.C. Cholinergic nervous system and glaucoma: From basic science to clinical applications. Prog. Retin. Eye Res. 2019. [Google Scholar] [CrossRef]
- Fischer, W.; Wictorin, K.; Björklund, A.; Williams, L.R.; Varon, S.; Gage, F.H. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 1987, 329, 65–68. [Google Scholar] [CrossRef]
- Kromer, L.F. Nerve growth factor treatment after brain injury prevents neuronal death. Science 1987, 235, 214–216. [Google Scholar] [CrossRef]
- Longo, F.M.; Manthorpe, M.; Xie, Y.M.; Varon, S. Synthetic NGF peptide derivatives prevent neuronal death via a p75 receptor-dependent mechanism. J. Neurosci. Res. 1997, 48, 1–17. [Google Scholar] [CrossRef]
- Maliartchouk, S.; Feng, Y.; Ivanisevic, L.; Debeir, T.; Cuello, A.C.; Burgess, K.; Saragovi, H.U. A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol. Pharmacol. 2000, 57, 385–391. [Google Scholar]
- Peleshok, J.; Saragovi, H.U. Functional mimetics of neurotrophins and their receptors. Biochem. Soc. Trans. 2006, 34, 612–617. [Google Scholar] [CrossRef] [PubMed]
- McDonald, N.Q.; Lapatto, R.; Murray-Rust, J.; Gunning, J.; Wlodawer, A.; Blundell, T.L. New protein fold revealed by a 2.3-A resolution crystal structure of nerve growth factor. Nature 1991, 354, 411–414. [Google Scholar] [CrossRef] [PubMed]
- McDonald, N.Q.; Chao, M. V Structural determinants of neurotrophin action. J. Biol. Chem. 1995, 270, 19669–19672. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.B.; Timm, D.E.; Neet, K.E. Alteration of NH2-terminal residues of nerve growth factor affects activity and Trk binding without affecting stability or conformation. J. Biol. Chem. 1995, 270, 6278–6285. [Google Scholar] [CrossRef] [Green Version]
- Krüttgen, A.; Heymach, J.V.; Kahle, P.J.; Shooter, E.M. The role of the nerve growth factor carboxyl terminus in receptor binding and conformational stability. J. Biol. Chem. 1997, 272, 29222–29228. [Google Scholar] [CrossRef] [Green Version]
- Kullander, K.; Kaplan, D.; Ebendal, T. Two restricted sites on the surface of the nerve growth factor molecule independently determine specific TrkA receptor binding and activation. J. Biol. Chem. 1997, 272, 9300–9307. [Google Scholar] [CrossRef] [Green Version]
- Rydén, M.; Ibáñez, C.F. A second determinant of binding to the p75 neurotrophin receptor revealed by alanine-scanning mutagenesis of a conserved loop in nerve growth factor. J. Biol. Chem. 1997, 272, 33085–33091. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, R.A.; Murray-Rust, J.; Blundell, T.L.; Mcdonald, N.Q.; Lapatto, R.; Ibáñez, C.F. Nerve growth factor: Structure/function relationships. Protein Sci. 1994, 3, 1901–1913. [Google Scholar] [CrossRef] [Green Version]
- Urfer, R.; Tsoulfas, P.; O’Connell, L.; Hongo, J.A.; Zhao, W.; Presta, L.G. High resolution mapping of the binding site of TrkA for nerve growth factor and TrkC for neurotrophin-3 on the second immunoglobulin-like domain of the Trk receptors. J. Biol. Chem. 1998, 273, 5829–5840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibáñez, C.F.; Ebendal, T.; Barbany, G.; Murray-Rust, J.; Blundell, T.L.; Persson, H. Disruption of the low affinity receptor-binding site in NGF allows neuronal survival and differentiation by binding to the trk gene product. Cell 1992, 69, 329–341. [Google Scholar] [CrossRef]
- Kahle, P.; Burton, L.E.; Schmelzer, C.H.; Hertel, C. The amino terminus of nerve growth factor is involved in the interaction with the receptor tyrosine kinase p140trkA. J. Biol. Chem. 1992, 267, 22707–22710. [Google Scholar] [PubMed]
- Drinkwater, C.C.; Barker, P.A.; Suter, U.; Shooter, E.M. The carboxyl terminus of nerve growth factor is required for biological activity. J. Biol. Chem. 1993, 268, 23202–23207. [Google Scholar] [PubMed]
- Shih, A.; Laramee, G.R.; Schmelzer, C.H.; Burton, L.E.; Winslow, J.W. Mutagenesis identifies amino-terminal residues of nerve growth factor necessary for Trk receptor binding and biological activity. J. Biol. Chem. 1994, 269, 27679–27686. [Google Scholar]
- Wiesmann, C.; Ultsch, M.H.; Bass, S.H.; de Vos, A.M. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 1999, 401, 184–188. [Google Scholar] [CrossRef]
- Travaglia, A.; Pietropaolo, A.; Di Martino, R.; Nicoletti, V.G.; La Mendola, D.; Calissano, P.; Rizzarelli, E. A small linear peptide encompassing the NGF N-terminus partly mimics the biological activities of the entire neurotrophin in PC12 cells. ACS Chem. Neurosci. 2015, 6, 1379–1392. [Google Scholar] [CrossRef]
- Naletova, I.; Satriano, C.; Pietropaolo, A.; Gianì, F.; Pandini, G.; Triaca, V.; Amadoro, G.; Latina, V.; Calissano, P.; Travaglia, A.; et al. The Copper(II)-Assisted Connection between NGF and BDNF by Means of Nerve Growth Factor-Mimicking Short Peptides. Cells 2019, 8, 301. [Google Scholar] [CrossRef] [Green Version]
- Pérez, P.; Coll, P.M.; Hempstead, B.L.; Martín-Zanca, D.; Chao, M. V NGF binding to the trk tyrosine kinase receptor requires the extracellular immunoglobulin-like domains. Mol. Cell. Neurosci. 1995, 6, 97–105. [Google Scholar] [CrossRef]
- Urfer, R.; Tsoulfas, P.; O’Connell, L.; Shelton, D.L.; Parada, L.F.; Presta, L.G. An immunoglobulin-like domain determines the specificity of neurotrophin receptors. EMBO J. 1995, 14, 2795–2805. [Google Scholar] [CrossRef]
- Wehrman, T.; He, X.; Raab, B.; Dukipatti, A.; Blau, H.; Garcia, K.C. Structural and Mechanistic Insights into Nerve Growth Factor Interactions with the TrkA and p75 Receptors. Neuron 2007, 53, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banfield, M.J.; Naylor, R.L.; Robertson, A.G.; Allen, S.J.; Dawbarn, D.; Brady, R.L. Specificity in Trk Receptor:Neurotrophin Interactions. Structure 2001, 9, 1191–1199. [Google Scholar] [CrossRef]
- Robertson, A.G.S.; Allen, S.J.; Mason, G.G.F.; Tyler, S.J.; Naylor, R.L.; Dawbarn, D.; Banfield, M.J.; Dando, J.A.; Clarke, A.R.; Brady, R.L.; et al. Identification and structure of the nerve growth factor binding site on TrkA. Biochem. Biophys. Res. Commun. 2001, 282, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Ivanisevic, L.; Zheng, W.H.; Woo, S.B.; Neet, K.E.; Saragovi, H.U. TrkA receptor “hot spots” for binding of NT-3 as a heterologous ligand. J. Biol. Chem. 2007, 282, 16754–16763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawbarn, D.; Fahey, M.; Watson, J.; Tyler, S.; Shoemark, D.; Sessions, R.; Zhang, R.; Brady, L.; Willis, C.; Allen, S.J. NGF receptor TrkAd5: therapeutic agent and drug design target. Biochem. Soc. Trans. 2006, 34, 587–590. [Google Scholar] [CrossRef]
- Pandini, G.; Satriano, C.; Pietropaolo, A.; Gianì, F.; Travaglia, A.; La Mendola, D.; Nicoletti, V.G.; Rizzarelli, E. The Inorganic Side of NGF: Copper(II) and Zinc(II) Affect the NGF Mimicking Signaling of the N-Terminus Peptides Encompassing the Recognition Domain of TrkA Receptor. Front. Neurosci. 2016, 10, 569. [Google Scholar] [CrossRef]
- Travaglia, A.; Arena, G.; Fattorusso, R.; Isernia, C.; La Mendola, D.; Malgieri, G.; Nicoletti, V.G.; Rizzarelli, E. The inorganic perspective of nerve growth factor: interactions of Cu2+ and Zn2+ with the N-terminus fragment of nerve growth factor encompassing the recognition domain of the TrkA receptor. Chemistry 2011, 17, 3726–3738. [Google Scholar] [CrossRef]
- Bocchini, V.; Angeletti, P. The nerve growth factor: purification as a 30,000-molecular-weight protein. Proc. Natl. Acad. Sci. USA 1969, 64, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Triaca, V.; Sposato, V.; Bolasco, G.; Ciotti, M.T.; Pelicci, P.; Bruni, A.C.; Cupidi, C.; Maletta, R.; Feligioni, M.; Nisticò, R.; et al. NGF controls APP cleavage by downregulating APP phosphorylation at Thr668: relevance for Alzheimer’s disease. Aging Cell 2016, 15, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Sposato, V.; Canu, N.; Fico, E.; Fusco, S.; Bolasco, G.; Ciotti, M.T.; Spinelli, M.; Mercanti, D.; Grassi, C.; Triaca, V.; et al. The Medial Septum Is Insulin Resistant in the AD Presymptomatic Phase: Rescue by Nerve Growth Factor-Driven IRS1 Activation. Mol. Neurobiol. 2018. [Google Scholar]
- Bonnington, J.K.; McNaughton, P.A. Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J. Physiol. 2003, 551, 433–446. [Google Scholar] [CrossRef]
- Taneda, K.; Tominaga, M.; Tengara, S.; Ogawa, H.; Takamori, K. Neurotropin inhibits both capsaicin-induced substance P release and nerve growth factor-induced neurite outgrowth in cultured rat dorsal root ganglion neurones. Clin. Exp. Dermatol. 2010, 35, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.A. An analysis of critical factors for quantitative immunoblotting. Sci. Signal. 2015, 8, rs2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melli, G.; Höke, A. Dorsal root ganglia sensory neuronal cultures: a tool for drug discovery for peripheral neuropathies. Expert Opin. Drug Discov. 2009, 4, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Hartikka, J.; Hefti, F. Development of septal cholinergic neurons in culture: plating density and glial cells modulate effects of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. J. Neurosci. 1988, 8, 2967–2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, C.Y.L.; Danik, M.; Manseau, F.; Trudeau, L.-E.; Williams, S. Chronic Exposure to Nerve Growth Factor Increases Acetylcholine and Glutamate Release from Cholinergic Neurons of the Rat Medial Septum and Diagonal Band of Broca via Mechanisms Mediated by p75NTR. J. Neurosci. 2008, 28, 1404–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Sensory neurons in culture: changing requirements for survival factors during embryonic development. Proc. Natl. Acad. Sci. USA 1980, 77, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Yip, H.K.; Rich, K.M.; Lampe, P.A.; Johnson, E.M. The effects of nerve growth factor and its antiserum on the postnatal development and survival after injury of sensory neurons in rat dorsal root ganglia. J. Neurosci. 1984, 4, 2986–2992. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Shen, H.; Taglialatela, G.; Chung, J.M.; Chung, K. Expression of nerve growth factor in the dorsal root ganglion after peripheral nerve injury. Brain Res. 1998, 796, 99–106. [Google Scholar] [CrossRef]
- Eichler, M.E.; Rich, K.M. Death of sensory ganglion neurons after acute withdrawal of nerve growth factor in dissociated cell cultures. Brain Res. 1989, 482, 340–346. [Google Scholar] [CrossRef]
- Chang, J.H.; Mellon, E.; Schanen, N.C.; Twiss, J.L. Persistent TrkA Activity Is Necessary to Maintain Transcription in Neuronally Differentiated PC12 Cells. J. Biol. Chem. 2003, 278, 42877–42885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrera, M.; Cattaneo, A.; Carloni, P. Molecular simulation of the binding of nerve growth factor peptide mimics to the receptor tyrosine kinase A. Biophys. J. 2006, 91, 2063–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, F.M.; Massa, S.M. Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Scarpi, D.; Cirelli, D.; Matrone, C.; Castronovo, G.; Rosini, P.; Occhiato, E.G.; Romano, F.; Bartali, L.; Clemente, A.M.; Bottegoni, G.; et al. Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis. 2012, 3, e339-13. [Google Scholar] [CrossRef]
- Finkbeiner, S.; Tavazoie, S.F.; Maloratsky, A.; Jacobs, K.M.; Harris, K.M.; Greenberg, M.E. CREB: A major mediator of neuronal neurotrophin responses. Neuron 1997, 19, 1031–1047. [Google Scholar] [CrossRef] [Green Version]
- Bullitt, E. Expression of C-fos-like protein as a marker for neuronal activity following noxious stimulation in the rat. J. Comp. Neurol. 1990, 296, 517–530. [Google Scholar] [CrossRef]
- Shen, M.; Greenberg, M. The Regulation and Function of c-fos and Other Immediate in the Nervous System. Cell 1990, 4, 477–485. [Google Scholar]
- Curran, T.; Morgan, J.I. Superinduction of c-fos by nerve growth factor in the presence of peripherally active benzodiazepines. Science (80-.) 1985, 229, 1265–1268. [Google Scholar] [CrossRef]
- Milbrandt, J. Nerve growth factor rapidly induces c-fos mRNA in PC12 rat pheochromocytoma cells. Proc. Natl. Acad. Sci. USA 2006, 83, 4789–4793. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Ji, Y.; Wang, S.; Sun, Y.; Lu, B. Neuronal activity alters BDNF–TrkB signaling kinetics and downstream functions. J. Cell Sci. 2014, 127, 2249–2260. [Google Scholar] [CrossRef] [Green Version]
- Pongrac, J.L.; Rylett, R.J. Molecular Mechanisms Regulating NGF-Mediated Enhancement of Cholinergic Neuronal Phenotype: c-Fos Trans-Activation of the Choline Acetyltransferase Gene. J. Mol. Neurosci. 2003, 11, 79–94. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Berse, B. The cholinergic neuronal phenotype in Alzheimer’s disease. Metab. Brain Dis. 2000, 15, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Barrantes, F.J. Deficits in cholinergic neurotransmission and their clinical correlates in Parkinson’s disease. npj Park. Dis. 2016, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi, A.; Wesson Ashford, J.; Mufson, J.E. Editorial (Thematic Issue: The Link between Alzheimer’s Disease and Down Syndrome. A Historical Perspective). Curr. Alzheimer Res. 2015, 13, 2–6. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Michael, G.J.; Averill, S.; Nitkunan, A.; Rattray, M.; Bennett, D.L.; Yan, Q.; Priestley, J. V Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J. Neurosci. 1997, 17, 8476–8490. [Google Scholar] [CrossRef]
- María Frade, J.; Rodríguez-Tébar, A.; Barde, Y.-A. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 1996, 383, 166–168. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Galletly, N.P.; Isacson, O.; Svendsen, C.N. Survival of adult basal forebrain cholinergic neurons after loss of target neurons. Science 1990, 247, 338–342. [Google Scholar] [CrossRef]
- Latina, V.; Caioli, S.; Zona, C.; Ciotti, M.T.; Amadoro, G.; Calissano, P. Impaired NGF/TrkA Signaling Causes Early AD-Linked Presynaptic Dysfunction in Cholinergic Primary Neurons. Front. Cell. Neurosci. 2017, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.K.; Yeh, H.H. Nerve growth factor rapidly increases muscarinic tone in mouse medial septum/diagonal band of Broca. J. Neurosci. 2005, 25, 4232–4242. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H. Dimerization of cell surface receptors in signal transduction. Cell 1995, 80, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Tisi, M.A.; Yeo, T.T.; Longo, F.M. Nerve Growth Factor (NGF) Loop 4 Dimeric Mimetics Activate ERK and AKT and Promote NGF-like Neurotrophic Effects. J. Biol. Chem. 2000, 275, 29868–29874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahimi, F.; Liu, J.; Malakhov, A.; Chowdhury, S.; Purisima, E.O.; Ivanisevic, L.; Caron, A.; Burgess, K.; Saragovi, H.U. A monovalent agonist of TrkA tyrosine kinase receptors can be converted into a bivalent antagonist. Biochim. Biophys. Acta 2010, 1800, 1018–1026. [Google Scholar] [CrossRef] [Green Version]
- Hanson, L.R.; Frey, W.H. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008, 9, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freiherr, J.; Hallschmid, M.; Frey, W.H.; Brünner, Y.F.; Chapman, C.D.; Hölscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirassa, P. The nerve growth factor administrated as eye drops activates mature and precursor cells in subventricular zone of adult rats. Arch. Ital. Biol. 2011, 149, 205–213. [Google Scholar] [PubMed]
- Lambiase, A.; Coassin, M.; Sposato, V.; Micera, A.; Sacchetti, M.; Bonini, S.; Aloe, L. NGF topical application in patients with corneal ulcer does not generate circulating NGF antibodies. Pharmacol. Res. 2007, 56, 65–69. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Triaca, V.; Fico, E.; Sposato, V.; Caioli, S.; Ciotti, M.T.; Zona, C.; Mercanti, D.; La Mendola, D.; Satriano, C.; Rizzarelli, E.; et al. hNGF Peptides Elicit the NGF-TrkA Signalling Pathway in Cholinergic Neurons and Retain Full Neurotrophic Activity in the DRG Assay. Biomolecules 2020, 10, 216. https://doi.org/10.3390/biom10020216
Triaca V, Fico E, Sposato V, Caioli S, Ciotti MT, Zona C, Mercanti D, La Mendola D, Satriano C, Rizzarelli E, et al. hNGF Peptides Elicit the NGF-TrkA Signalling Pathway in Cholinergic Neurons and Retain Full Neurotrophic Activity in the DRG Assay. Biomolecules. 2020; 10(2):216. https://doi.org/10.3390/biom10020216
Chicago/Turabian StyleTriaca, Viviana, Elena Fico, Valentina Sposato, Silvia Caioli, Maria Teresa Ciotti, Cristina Zona, Delio Mercanti, Diego La Mendola, Cristina Satriano, Enrico Rizzarelli, and et al. 2020. "hNGF Peptides Elicit the NGF-TrkA Signalling Pathway in Cholinergic Neurons and Retain Full Neurotrophic Activity in the DRG Assay" Biomolecules 10, no. 2: 216. https://doi.org/10.3390/biom10020216
APA StyleTriaca, V., Fico, E., Sposato, V., Caioli, S., Ciotti, M. T., Zona, C., Mercanti, D., La Mendola, D., Satriano, C., Rizzarelli, E., Tirassa, P., & Calissano, P. (2020). hNGF Peptides Elicit the NGF-TrkA Signalling Pathway in Cholinergic Neurons and Retain Full Neurotrophic Activity in the DRG Assay. Biomolecules, 10(2), 216. https://doi.org/10.3390/biom10020216