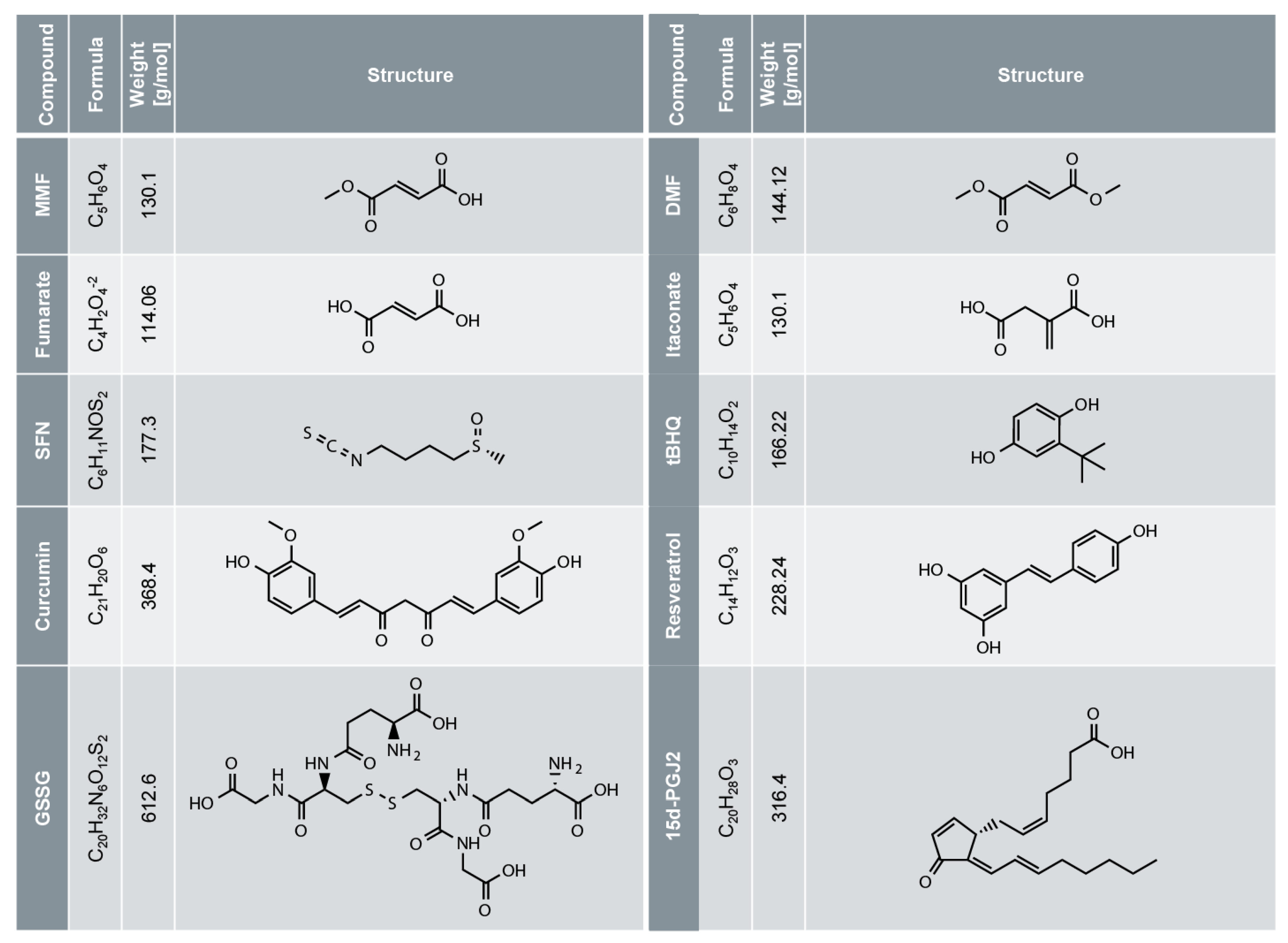
Electrophiles against (Skin) Diseases: More Than Nrf2



Abstract
:1. Introduction
2. Redox Sensing and Redox Regulation
3. NRF2 is Activated by Electrophiles upon Cysteine Oxidation of its Inhibitor KEAP1
4. DMF for the Treatment of Psoriasis and Multiple Sclerosis (MS)
5. DMF and Other Electrophiles Inhibit the NF-κB Pathway
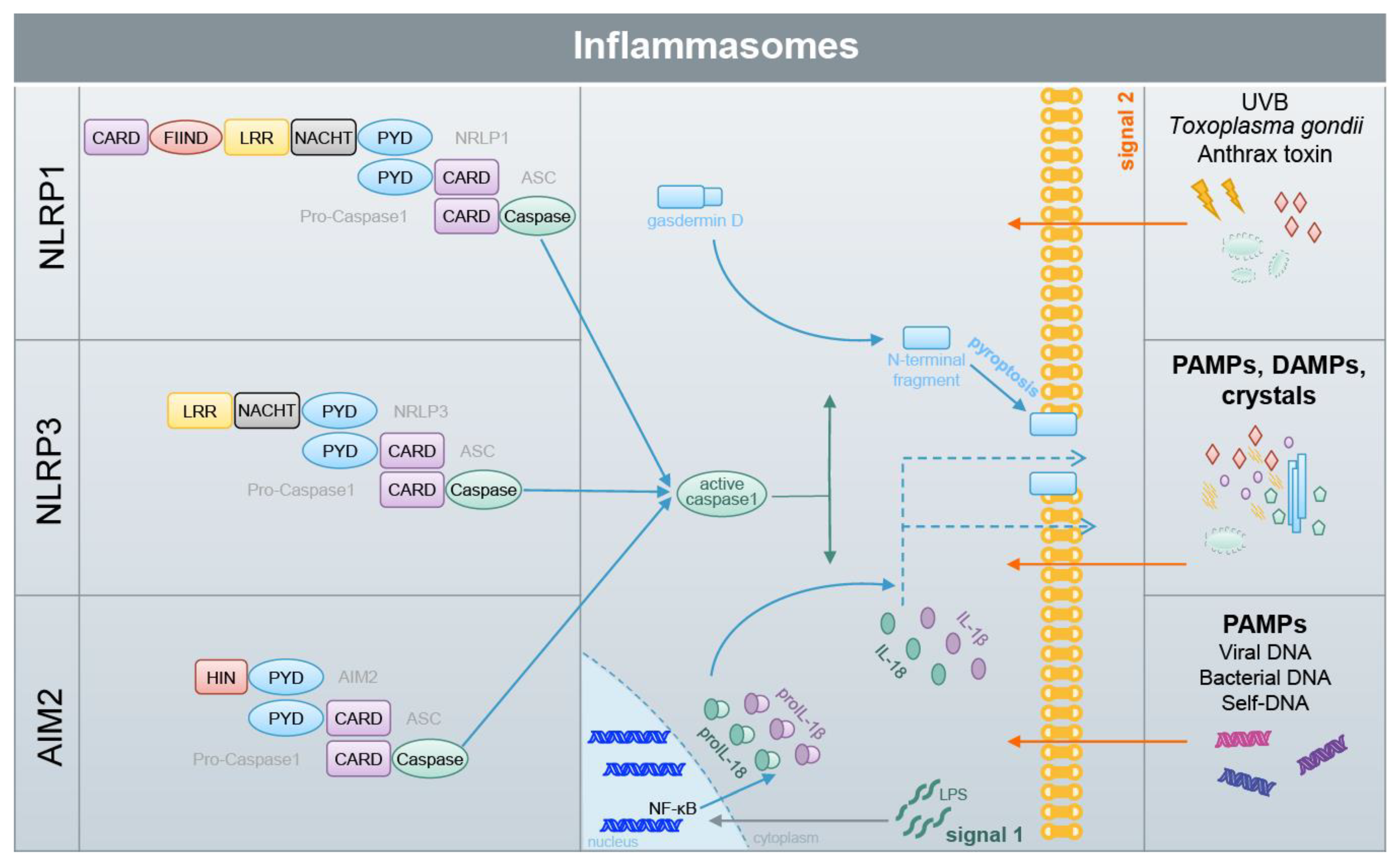
6. DMF and Other Electrophiles Inhibit Inflammasome Activation
7. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fuchs, E.; Raghavan, S. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet. 2002, 3, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Haase, I.; Nestle, F.O. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 2014, 14, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The keap1-nrf2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiebert, P.; Werner, S. Regulation of wound healing by the nrf2 transcription factor-more than cytoprotection. Int. J. Mol. Sci. 2019, 20, 856. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.; Dutsch, S.; auf dem Keller, U.; Werner, S. Nrf2: A central regulator of uv protection in the epidermis. Cell Cycle 2010, 9, 2917–2918. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 2016, 7, 11624. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Molecular basis of the keap1-nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Feldmeyer, L.; Werner, S.; French, L.E.; Beer, H.D. Interleukin-1, inflammasomes and the skin. Eur. J. Cell Biol. 2010, 89, 638–644. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Pasparakis, M. Role of nf-kappab in epithelial biology. Immunol. Rev. 2012, 246, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Wullaert, A.; Bonnet, M.C.; Pasparakis, M. Nf-kappab in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011, 21, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. Nf-kappab signaling in inflammation. Signal Transduct. Target Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennig, P.; Garstkiewicz, M.; Grossi, S.; Di Filippo, M.; French, L.E.; Beer, H.D. The crosstalk between nrf2 and inflammasomes. Int. J. Mol. Sci. 2018, 19, 562. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. Nf-kappab restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. Nf-kappab is a negative regulator of il-1beta secretion as revealed by genetic and pharmacological inhibition of ikkbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.H.; et al. Electrophilic properties of itaconate and derivatives regulate the ikappabzeta-atf3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates nrf2 via alkylation of keap1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the nrf2 and keap1 partnership in chronic diseases. Nat Rev Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Mrowietz, U.; Barker, J.; Boehncke, W.H.; Iversen, L.; Kirby, B.; Naldi, L.; Reich, K.; Tanew, A.; van de Kerkhof, P.C.M.; Warren, R.B. Clinical use of dimethyl fumarate in moderate-to-severe plaque-type psoriasis: A european expert consensus. J. Eur. Acad. Dermatol. Venereol. 2018, 32 Suppl 3, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Ashrafian, H.; Czibik, G.; Bellahcene, M.; Aksentijevic, D.; Smith, A.C.; Mitchell, S.J.; Dodd, M.S.; Kirwan, J.; Byrne, J.J.; Ludwig, C. , et al. Fumarate is cardioprotective via activation of the nrf2 antioxidant pathway. Cell Metab. 2012, 15, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garstkiewicz, M.; Strittmatter, G.E.; Grossi, S.; Sand, J.; Fenini, G.; Werner, S.; French, L.E.; Beer, H.D. Opposing effects of nrf2 and nrf2-activating compounds on the nlrp3 inflammasome independent of nrf2-mediated gene expression. Eur. J. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewe, R.; Holnthoner, W.; Groger, M.; Pillinger, M.; Gruber, F.; Mechtcheriakova, D.; Hofer, E.; Wolff, K.; Petzelbauer, P. Dimethylfumarate inhibits tnf-induced nuclear entry of nf-kappa b/p65 in human endothelial cells. J. Immunol. 2002, 168, 4781–4787. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species--the good, the bad and the ugly. Acta Physiol. (Oxf.) 2015, 214, 329–348. [Google Scholar] [CrossRef]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef]
- McEligot, A.J.; Yang, S.; Meyskens, F.L., Jr. Redox regulation by intrinsic species and extrinsic nutrients in normal and cancer cells. Annu. Rev. Nutr. 2005, 25, 261–295. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Carroll, K.S.; Liebler, D.C. The expanding landscape of the thiol redox proteome. Mol. Cell. Proteomics 2016, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gorelenkova Miller, O.; Mieyal, J.J. Sulfhydryl-mediated redox signaling in inflammation: Role in neurodegenerative diseases. Arch. Toxicol. 2015, 89, 1439–1467. [Google Scholar] [CrossRef]
- Pastore, A.; Piemonte, F. S-glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci. 2012, 46, 279–292. [Google Scholar] [CrossRef]
- Gambhir, L.; Checker, R.; Thoh, M.; Patwardhan, R.S.; Sharma, D.; Kumar, M.; Sandur, S.K. 1,4-naphthoquinone, a pro-oxidant, suppresses immune responses via keap-1 glutathionylation. Biochem. Pharmacol. 2014, 88, 95–105. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, X.; Ma, Z. Pyddt, a novel phase 2 enzymes inducer, activates keap1-nrf2 pathway via depleting the cellular level of glutathione. Toxicol. Lett. 2010, 199, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Murdoch, C.E.; Sano, S.; Ido, Y.; Bachschmid, M.M.; Cohen, R.A.; Matsui, R. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize hif-1alpha and improve limb revascularization. Proc. Natl. Acad. Sci. USA 2016, 113, 6011–6016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and consequences of cysteine s-glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Eriksson, S.; Wang, L. Oxidative stress induced s-glutathionylation and proteolytic degradation of mitochondrial thymidine kinase 2. J. Biol. Chem. 2012, 287, 24304–24312. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P., Jr. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Berk, B.C. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: Key role for glutaredoxin in the death pathway. Circ. Res. 2007, 100, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Canli, O.; Alankus, Y.B.; Grootjans, S.; Vegi, N.; Hultner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016, 127, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol. 2008, 9, 866–872. [Google Scholar] [CrossRef]
- Guglielmo, A.; Sabra, A.; Elbery, M.; Cerveira, M.M.; Ghenov, F.; Sunasee, R.; Ckless, K. A mechanistic insight into curcumin modulation of the il-1beta secretion and nlrp3 s-glutathionylation induced by needle-like cationic cellulose nanocrystals in myeloid cells. Chem. Biol. Interact. 2017, 274, 1–12. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, P.; Zhang, C.; Chiewchengchol, D.; Zhao, F.; Yu, H.; Li, J.; Kambara, H.; Luo, K.Y.; Venkataraman, A.; et al. Positive regulation of interleukin-1beta bioactivity by physiological ros-mediated cysteine s-glutathionylation. Cell Rep. 2017, 20, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, B.; Otten, E.G.; Manni, D.; Stefanatos, R.; Menzies, F.M.; Smith, G.R.; Jurk, D.; Kenneth, N.; Wilkinson, S.; Passos, J.F.; et al. Oxidation of sqstm1/p62 mediates the link between redox state and protein homeostasis. Nat Commun 2018, 9, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, S.; Pfeuffer, S.; Arnold, P.; Treitz, C.; Aden, K.; Ebsen, H.; Falk-Paulsen, M.; Gisch, N.; Fazio, A.; Kuiper, J.; et al. Prdx4 limits caspase-1 activation and restricts inflammasome-mediated signaling by extracellular vesicles. EMBO J. 2019, 38, e101266. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The keap1-nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by nrf2. Antioxid Redox Signal 2017. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the keap1-nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 6379–6384. [Google Scholar] [CrossRef] [Green Version]
- Helou, D.G.; Braham, S.; De Chaisemartin, L.; Granger, V.; Damien, M.H.; Pallardy, M.; Kerdine-Romer, S.; Chollet-Martin, S. Nrf2 downregulates zymosan-induced neutrophil activation and modulates migration. PLoS ONE 2019, 14, e0216465. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.; Werner, S. Nrf2--a regulator of keratinocyte redox signaling. Free Radic. Biol. Med. 2015, 88, 243–252. [Google Scholar] [CrossRef]
- Li, J.; Guo, C.; Wu, J. 15-deoxy--(12,14)-prostaglandin j2 (15d-pgj2), an endogenous ligand of ppar-gamma: Function and mechanism. PPAR Res 2019, 2019, 7242030. [Google Scholar] [CrossRef] [Green Version]
- Hewlings, S.J.; Kalman, D.S. Curcumin: A review of its’ effects on human health. Foods 2017, 6, 92. [Google Scholar] [CrossRef]
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952. [Google Scholar] [CrossRef] [PubMed]
- Smith, D. Fumaric acid esters for psoriasis: A systematic review. Ir. J. Med. Sci. 2017, 186, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral bg-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral bg-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Al-Jaderi, Z.; Maghazachi, A.A. Utilization of dimethyl fumarate and related molecules for treatment of multiple sclerosis, cancer, and other diseases. Front. Immunol. 2016, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Mrowietz, U.; Asadullah, K. Dimethylfumarate for psoriasis: More than a dietary curiosity. Trends Mol. Med. 2005, 11, 43–48. [Google Scholar] [CrossRef]
- Bruck, J.; Dringen, R.; Amasuno, A.; Pau-Charles, I.; Ghoreschi, K. A review of the mechanisms of action of dimethylfumarate in the treatment of psoriasis. Exp. Dermatol. 2018, 27, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Meissner, M.; Valesky, E.M.; Kippenberger, S.; Kaufmann, R. Dimethyl fumarate - only an anti-psoriatic medication? J Dtsch Dermatol Ges 2012, 10, 793–801. [Google Scholar] [CrossRef]
- Mrowietz, U.; Morrison, P.J.; Suhrkamp, I.; Kumanova, M.; Clement, B. The pharmacokinetics of fumaric acid esters reveal their in vivo effects. Trends Pharmacol. Sci. 2018, 39, 1–12. [Google Scholar] [CrossRef]
- Landeck, L.; Asadullah, K.; Amasuno, A.; Pau-Charles, I.; Mrowietz, U. Dimethyl fumarate (dmf) vs. Monoethyl fumarate (mef) salts for the treatment of plaque psoriasis: A review of clinical data. Arch. Dermatol. Res. 2018, 310, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Lu, J.Y.; Zheng, X.; Yang, Y.; Reagan, J.D. The psoriasis drug monomethylfumarate is a potent nicotinic acid receptor agonist. Biochem. Biophys. Res. Commun. 2008, 375, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Assmann, J.C.; Krenz, A.; Rahman, M.; Grimm, M.; Karsten, C.M.; Kohl, J.; Offermanns, S.; Wettschureck, N.; Schwaninger, M. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in eae. J. Clin. Invest. 2014, 124, 2188–2192. [Google Scholar] [CrossRef] [PubMed]
- Scannevin, R.H.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, M.; Ammal Kaidery, N.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced experimental parkinson’s-like disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The absence of the pro-antioxidant transcription factor nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol. Sci. 2010, 114, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. Shared principles in nf-kappab signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Guerau-de-Arellano, M.; Mehta, V.B.; Yang, Y.; Huss, D.J.; Papenfuss, T.L.; Lovett-Racke, A.E.; Racke, M.K. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor kappab (nf-kappab) and extracellular signal-regulated kinase 1 and 2 (erk1/2) and mitogen stress-activated kinase 1 (msk1) signaling. J. Biol. Chem. 2012, 287, 28017–28026. [Google Scholar] [CrossRef] [Green Version]
- Gesser, B.; Johansen, C.; Rasmussen, M.K.; Funding, A.T.; Otkjaer, K.; Kjellerup, R.B.; Kragballe, K.; Iversen, L. Dimethylfumarate specifically inhibits the mitogen and stress-activated kinases 1 and 2 (msk1/2): Possible role for its anti-psoriatic effect. J. Invest. Dermatol. 2007, 127, 2129–2137. [Google Scholar] [CrossRef] [Green Version]
- Seidel, P.; Merfort, I.; Hughes, J.M.; Oliver, B.G.; Tamm, M.; Roth, M. Dimethylfumarate inhibits nf-{kappa}b function at multiple levels to limit airway smooth muscle cell cytokine secretion. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L326–L339. [Google Scholar] [CrossRef] [PubMed]
- Seidel, P.; Merfort, I.; Tamm, M.; Roth, M. Inhibition of nf-kappab and ap-1 by dimethylfumarate correlates with down-regulated il-6 secretion and proliferation in human lung fibroblasts. Swiss Med. Wkly. 2010, 140, w13132. [Google Scholar] [PubMed]
- Kastrati, I.; Siklos, M.I.; Calderon-Gierszal, E.L.; El-Shennawy, L.; Georgieva, G.; Thayer, E.N.; Thatcher, G.R.; Frasor, J. Dimethyl fumarate inhibits the nuclear factor kappab pathway in breast cancer cells by covalent modification of p65 protein. J. Biol. Chem. 2016, 291, 3639–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, A.; Warnken, U.; Roth, D.; Klika, K.D.; Vobis, D.; Barnert, A.; Bujupi, F.; Oberacker, T.; Schnolzer, M.; Nicolay, J.P.; et al. Targeting thioredoxin-1 by dimethyl fumarate induces ripoptosome-mediated cell death. Sci. Rep. 2017, 7, 43168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolay, J.P.; Muller-Decker, K.; Schroeder, A.; Brechmann, M.; Mobs, M.; Geraud, C.; Assaf, C.; Goerdt, S.; Krammer, P.H.; Gulow, K. Dimethyl fumarate restores apoptosis sensitivity and inhibits tumor growth and metastasis in ctcl by targeting nf-kappab. Blood 2016, 128, 805–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebold, M.; Sievers, C.; Bantug, G.; Sanderson, N.; Kappos, L.; Kuhle, J.; Lindberg, R.L.P.; Derfuss, T. Dimethyl fumarate influences innate and adaptive immunity in multiple sclerosis. J. Autoimmun. 2018, 86, 39–50. [Google Scholar] [CrossRef]
- Gillard, G.O.; Collette, B.; Anderson, J.; Chao, J.; Scannevin, R.H.; Huss, D.J.; Fontenot, J.D. Dmf, but not other fumarates, inhibits nf-kappab activity in vitro in an nrf2-independent manner. J. Neuroimmunol. 2015, 283, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.Y.; Bach, F.H. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of nf-kappab signaling by keap1 regulation of ikkbeta activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 2010, 22, 1645–1654. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. Keap1 e3 ligase-mediated downregulation of nf-kappab signaling by targeting ikkbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The neuroprotective effect of dimethyl fumarate in an mptp-mouse model of parkinson’s disease: Involvement of reactive oxygen species/nuclear factor-kappab/nuclear transcription factor related to nf-e2. Antioxid Redox Signal 2017, 27, 453–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid beta-induced neurotoxicity in human neuronal cells. J. Cell. Mol. Med. 2018, 22, 1081–1094. [Google Scholar] [PubMed]
- Allen, E.M.; Mieyal, J.J. Protein-thiol oxidation and cell death: Regulatory role of glutaredoxins. Antioxid Redox Signal 2012, 17, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Qanungo, S.; Starke, D.W.; Pai, H.V.; Mieyal, J.J.; Nieminen, A.L. Glutathione supplementation potentiates hypoxic apoptosis by s-glutathionylation of p65-nfkappab. J. Biol. Chem. 2007, 282, 18427–18436. [Google Scholar] [CrossRef] [Green Version]
- Kil, I.S.; Kim, S.Y.; Park, J.W. Glutathionylation regulates ikappab. Biochem. Biophys. Res. Commun. 2008, 373, 169–173. [Google Scholar] [CrossRef]
- Cheung, K.L.; Kong, A.N. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010, 12, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Grundemann, C.; Huber, R. Chemoprevention with isothiocyanates - from bench to bedside. Cancer Lett. 2018, 414, 26–33. [Google Scholar] [CrossRef]
- Xu, C.; Shen, G.; Chen, C.; Gelinas, C.; Kong, A.N. Suppression of nf-kappab and nf-kappab-regulated gene expression by sulforaphane and peitc through ikappabalpha, ikk pathway in human prostate cancer pc-3 cells. Oncogene 2005, 24, 4486–4495. [Google Scholar] [CrossRef] [Green Version]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhauser, C. Nuclear factor kappa b is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.E.; Ernst, I.; Iori, R.; Desel, C.; Rimbach, G. Sulforaphane but not ascorbigen, indole-3-carbinole and ascorbic acid activates the transcription factor nrf2 and induces phase-2 and antioxidant enzymes in human keratinocytes in culture. Exp. Dermatol. 2010, 19, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.; Gerhauser, C. Time-dependent modulation of thioredoxin reductase activity might contribute to sulforaphane-mediated inhibition of nf-kappab binding to DNA. Antioxid Redox Signal 2005, 7, 1601–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, J.; Cook, J.A.; O’Connor, M.; Zingarelli, B. Peroxisome proliferator-activated receptor gamma is required for the inhibitory effect of ciglitazone but not 15-deoxy-delta 12,14-prostaglandin j2 on the nfkappab pathway in human endothelial cells. Shock 2007, 28, 722–726. [Google Scholar] [PubMed]
- Li, Z.; Jansen, M.; Ogburn, K.; Salvatierra, L.; Hunter, L.; Mathew, S.; Figueiredo-Pereira, M.E. Neurotoxic prostaglandin j2 enhances cyclooxygenase-2 expression in neuronal cells through the p38mapk pathway: A death wish? J. Neurosci. Res. 2004, 78, 824–836. [Google Scholar] [CrossRef]
- Rossi, A.; Kapahi, P.; Natoli, G.; Takahashi, T.; Chen, Y.; Karin, M.; Santoro, M.G. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of ikappab kinase. Nature 2000, 403, 103–108. [Google Scholar] [CrossRef]
- Cernuda-Morollon, E.; Pineda-Molina, E.; Canada, F.J.; Perez-Sala, D. 15-deoxy-delta 12,14-prostaglandin j2 inhibition of nf-kappab-DNA binding through covalent modification of the p50 subunit. J. Biol. Chem. 2001, 276, 35530–35536. [Google Scholar] [CrossRef] [Green Version]
- Straus, D.S.; Pascual, G.; Li, M.; Welch, J.S.; Ricote, M.; Hsiang, C.H.; Sengchanthalangsy, L.L.; Ghosh, G.; Glass, C.K. 15-deoxy-delta 12,14-prostaglandin j2 inhibits multiple steps in the nf-kappa b signaling pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 4844–4849. [Google Scholar] [CrossRef] [Green Version]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Hengartner, M.O. Programmed cell death: Alive and well in the new millennium. Trends Cell Biol. 2001, 11, 526–534. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. Nlrp3 inflammasome activation: The convergence of multiple signalling pathways on ros production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Aglietti, R.A.; Dueber, E.C. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 2017, 38, 261–271. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human monocytes engage an alternative inflammasome pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Gross, C.J.; Sotomayor, F.V.; Stacey, K.J.; Tschopp, J.; Sweet, M.J.; Schroder, K. The neutrophil nlrc4 inflammasome selectively promotes il-1beta maturation without pyroptosis during acute salmonella challenge. Cell Rep. 2014, 8, 570–582. [Google Scholar] [CrossRef] [Green Version]
- Conos, S.A.; Lawlor, K.E.; Vaux, D.L.; Vince, J.E.; Lindqvist, L.M. Cell death is not essential for caspase-1-mediated interleukin-1beta activation and secretion. Cell Death Differ. 2016, 23, 1827–1838. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the nlrp3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the nlrp3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 2018, 17, 688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Place, D.E.; Kanneganti, T.D. Recent advances in inflammasome biology. Curr. Opin. Immunol. 2017, 50, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ros drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in nlrp3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor mavs promotes nlrp3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of nlrp3 inflammasomes: Ros as trigger or effector? Antioxid Redox Signal 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of nrf2, a key inducer of antioxidant defenses, attenuates apoe-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogawa, Y.; Nagasu, H.; Iwase, S.; Ihoriya, C.; Itano, S.; Uchida, A.; Kidokoro, K.; Taniguchi, S.; Takahashi, M.; Satoh, M.; et al. Infiltration of m1, but not m2, macrophages is impaired after unilateral ureter obstruction in nrf2-deficient mice. Sci. Rep. 2017, 7, 8801. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Gillette, D.D.; Li, X.; Zhang, Z.; Wen, H. Nuclear factor e2-related factor-2 (nrf2) is required for nlrp3 and aim2 inflammasome activation. J. Biol. Chem. 2014, 289, 17020–17029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhang, J.J.; Cheng, Y.T.; Ho, C.Y.; Yen, G.C. Monosodium urate crystals trigger nrf2- and heme oxygenase-1-dependent inflammation in thp-1 cells. Cell. Mol. Immunol. 2015, 12, 424–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; Huang, M.; Schneider, M.; Miller, S.D.; Ting, J.P. Nlrp3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating th1 and th17 responses. J. Immunol. 2010, 185, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Williams, K.L.; Gunn, M.D.; Shinohara, M.L. Nlrp3 inflammasome induces chemotactic immune cell migration to the cns in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2012, 109, 10480–10485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Williams, K.L.; Oliver, T.; Vandenabeele, P.; Rajan, J.V.; Miao, E.A.; Shinohara, M.L. Interferon-beta therapy against eae is effective only when development of the disease depends on the nlrp3 inflammasome. Sci Signal 2012, 5, ra38. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Li, Z.; He, X.; Yu, H.; Feng, J. Ghrelin attenuates neuroinflammation and demyelination in experimental autoimmune encephalomyelitis involving nlrp3 inflammasome signaling pathway and pyroptosis. Front. Pharmacol. 2019, 10, 1320. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Fernandez, A.; Skouras, D.B.; Dinarello, C.A.; Lopez-Vales, R. Olt1177 (dapansutrile), a selective nlrp3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front. Immunol. 2019, 10, 2578. [Google Scholar] [CrossRef]
- Barclay, W.; Shinohara, M.L. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (eae). Brain Pathol. 2017, 27, 213–219. [Google Scholar] [CrossRef]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Spritz, R.A. Genetic variations in nalp1 are associated with generalized vitiligo in a romanian population. J. Invest. Dermatol. 2007, 127, 2558–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekman, A.K.; Verma, D.; Fredrikson, M.; Bivik, C.; Enerback, C. Genetic variations of nlrp1: Susceptibility in psoriasis. Br. J. Dermatol. 2014, 171, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.L.; Mamai, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverenyi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline nlrp1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 2016, 167, 187–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenini, G.; Grossi, S.; Contassot, E.; Biedermann, T.; Reichmann, E.; French, L.E.; Beer, H.D. Genome editing of human primary keratinocytes by crispr/cas9 reveals an essential role of the nlrp1 inflammasome in uvb sensing. J. Invest. Dermatol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Goss, C.; Anz, D.; Simanski, M.; Glaser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef] [Green Version]
- Carlstrom, M.; Ekman, A.K.; Petersson, S.; Soderkvist, P.; Enerback, C. Genetic support for the role of the nlrp3 inflammasome in psoriasis susceptibility. Exp. Dermatol. 2012, 21, 932–937. [Google Scholar] [CrossRef] [Green Version]
- Tervaniemi, M.H.; Katayama, S.; Skoog, T.; Siitonen, H.A.; Vuola, J.; Nuutila, K.; Sormunen, R.; Johnsson, A.; Linnarsson, S.; Suomela, S.; et al. Nod-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci. Rep. 2016, 6, 22745. [Google Scholar] [CrossRef]
- Aira, L.E.; Goncalves, D.; Bossowski, J.P.; Rubio-Patino, C.; Chiche, J.; Paul-Bellon, R.; Mondragon, L.; Gesson, M.; Lecucq-Ottavi, P.; Obba, S.; et al. Caspase 1/11 deficiency or pharmacological inhibition mitigates psoriasis-like phenotype in mice. J. Invest. Dermatol. 2019, 139, 1306–1317. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.; Chen, W.; Wang, P.; Zhan, T.; Zheng, W.; Gu, Z.; Wang, X.; Ji, X.; Sun, Y. Inhibition of nlrp3 inflammasome-mediated pyroptosis in macrophage by cycloastragenol contributes to amelioration of imiquimod-induced psoriasis-like skin inflammation in mice. Int. Immunopharmacol. 2019, 74, 105682. [Google Scholar] [CrossRef]
- Sand, J.; Haertel, E.; Biedermann, T.; Contassot, E.; Reichmann, E.; French, L.E.; Werner, S.; Beer, H.D. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis. 2018, 9, 24. [Google Scholar] [CrossRef]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, N.K.; Leppla, S.H.; Moayeri, M. The cyclopentenone prostaglandin 15d-pgj2 inhibits the nlrp1 and nlrp3 inflammasomes. J. Immunol. 2015, 194, 2776–2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze-Topphoff, U.; Varrin-Doyer, M.; Pekarek, K.; Spencer, C.M.; Shetty, A.; Sagan, S.A.; Cree, B.A.; Sobel, R.A.; Wipke, B.T.; Steinman, L.; et al. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of nrf2. Proc. Natl. Acad. Sci. USA 2016, 113, 4777–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miglio, G.; Veglia, E.; Fantozzi, R. Fumaric acid esters prevent the nlrp3 inflammasome-mediated and atp-triggered pyroptosis of differentiated thp-1 cells. Int. Immunopharmacol. 2015, 28, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braley, T.J.; Huber, A.K.; Segal, B.M.; Kaplish, N.; Saban, R.; Washnock-Schmid, J.M.; Chervin, R.D. A randomized, subject and rater-blinded, placebo-controlled trial of dimethyl fumarate for obstructive sleep apnea. Sleep 2018, 41. [Google Scholar] [CrossRef] [Green Version]
- Shafer, D.A.; Chen, Z.-j.; Harris, T.; Tombes, M.B.; Shrader, E.; Strickler, K.; Ryan, A.A.; Dent, P.; Malkin, M.G. Phase i trial of dimethyl fumarate, temozolomide, and radiation therapy in glioblastoma multiforme. J. Clini. Oncol. 2017, 35, 2060. [Google Scholar] [CrossRef]
- Biogen. Clinical Trial Results: A Phase 2a, Randomized, Double-Blind, Placebo-Controlled, Multicenter Study to Evaluate the Efficacy, Safety, and Tolerability of bg00012 When given with Methotrexate to Subjects with Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Coventional Disease-Modifying Anti-Rheumatic Drug Therapy. Available online: https://www.clinicaltrialsregister.eu/ctr-search/rest/download/result/attachment/2008-004754-33/1/7891 (accessed on 7 January 2020).
- Kuhn, A.; Landmann, A.; Patsinakidis, N.; Ruland, V.; Nozinic, S.; Perusquia Ortiz, A.M.; Sauerland, C.; Luger, T.; Tsianakas, A.; Bonsmann, G. Fumaric acid ester treatment in cutaneous lupus erythematosus (cle): A prospective, open-label, phase ii pilot study. Lupus 2016, 25, 1357–1364. [Google Scholar] [CrossRef]
- Kavian, N.; Mehlal, S.; Jeljeli, M.; Saidu, N.E.B.; Nicco, C.; Cerles, O.; Chouzenoux, S.; Cauvet, A.; Camus, C.; Ait-Djoudi, M.; et al. The nrf2-antioxidant response element signaling pathway controls fibrosis and autoimmunity in scleroderma. Front. Immunol. 2018, 9, 1896. [Google Scholar] [CrossRef] [Green Version]
- Toyama, T.; Looney, A.P.; Baker, B.M.; Stawski, L.; Haines, P.; Simms, R.; Szymaniak, A.D.; Varelas, X.; Trojanowska, M. Therapeutic targeting of taz and yap by dimethyl fumarate in systemic sclerosis fibrosis. J. Invest. Dermatol. 2018, 138, 78–88. [Google Scholar] [CrossRef]
- Uddin, M.S.; Mamun, A.A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2019, 135624. [Google Scholar] [CrossRef]
- Houghton, C.A. Sulforaphane: Its “coming of age” as a clinically relevant nutraceutical in the prevention and treatment of chronic disease. Oxid. Med. Cell. Longev. 2019, 2019, 2716870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazarakis, N.; Snibson, K.; Licciardi, P.V.; Karagiannis, T.C. The potential use of l-sulforaphane for the treatment of chronic inflammatory diseases: A review of the clinical evidence. Clin. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | CTI | St | n | Ph | Design | Outcome | Ref |
|---|---|---|---|---|---|---|---|
| Obstructive Sleep Apnea | NCT02438137 | C | 65 | 2 | Rd-2B-P | Partial response | [145] |
| Adult Brain Glioblastoma | NCT02337426 | C | 12 | 1 | O | SafePhase 2 under consideration | [146] |
| Rheumatoid Arthritis | NCT00810836 | C | 153 | 2 | Rd-2B-P | Not effective | [147] |
| Cutaneous Lupus Erythematosus | NCT01352988 | C | 11 | 2 | O | Safe and effectiveRandomize trial required | [148] |
| Chronic Lymphocytic Leukemia | NCT02784834 | T | 2 | 1 | O | 1/2 patients lack of efficacy | |
| Systemic Sclerosis | NCT02981082 | R | 34 | 1 | Rd-4B-P | [149,150] | |
| Cutaneous T Cell Lymphoma | NCT02546440 | R | 25 | 2 | O | [75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hennig, P.; Fenini, G.; Di Filippo, M.; Beer, H.-D. Electrophiles against (Skin) Diseases: More Than Nrf2. Biomolecules 2020, 10, 271. https://doi.org/10.3390/biom10020271
Hennig P, Fenini G, Di Filippo M, Beer H-D. Electrophiles against (Skin) Diseases: More Than Nrf2. Biomolecules. 2020; 10(2):271. https://doi.org/10.3390/biom10020271
Chicago/Turabian StyleHennig, Paulina, Gabriele Fenini, Michela Di Filippo, and Hans-Dietmar Beer. 2020. "Electrophiles against (Skin) Diseases: More Than Nrf2" Biomolecules 10, no. 2: 271. https://doi.org/10.3390/biom10020271
APA StyleHennig, P., Fenini, G., Di Filippo, M., & Beer, H. -D. (2020). Electrophiles against (Skin) Diseases: More Than Nrf2. Biomolecules, 10(2), 271. https://doi.org/10.3390/biom10020271