Role of an Aromatic–Aromatic Interaction in the Assembly and Trafficking of the Zebrafish Panx1a Membrane Channel


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and Mutagenesis
2.2. Cell Culture and Transfection
2.3. Western Blot
2.4. Immunofluorescence, Confocal Microscopy, and Co-localization
2.5. His60 Ni Gravity Column Pull Down
2.6. Cell Surface Biotinylation Assay
2.7. Pharmacology
2.8. Dye Uptake Assay
2.9. Fluorescence Recovery After Photobleaching (FRAP)
2.10. Förster Resonance Energy Transfer (FRET)
2.11. Quantitative Real-Time PCR
2.12. Statistical Analysis
3. Results
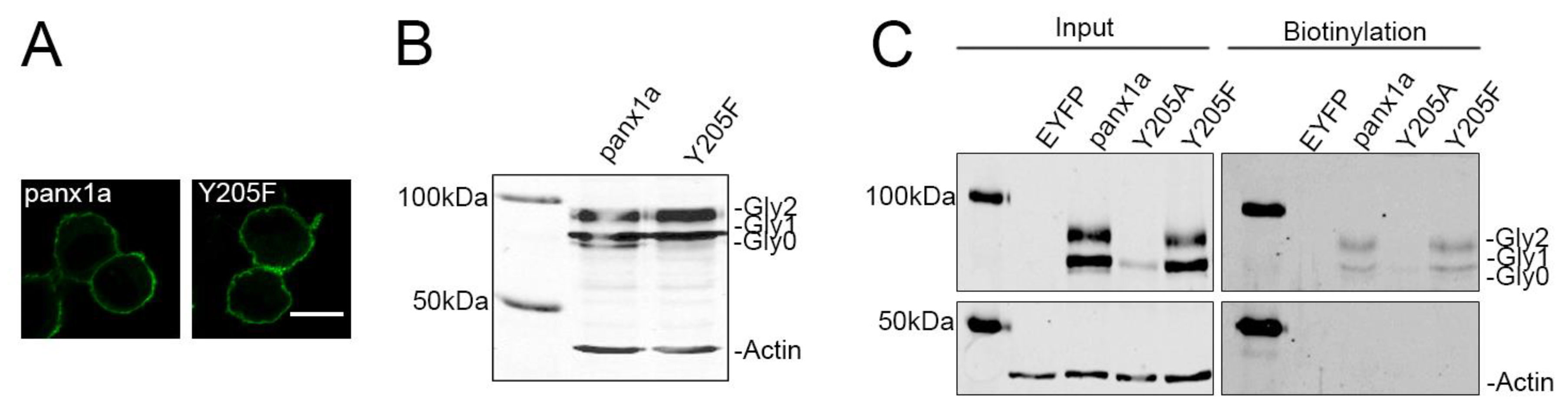
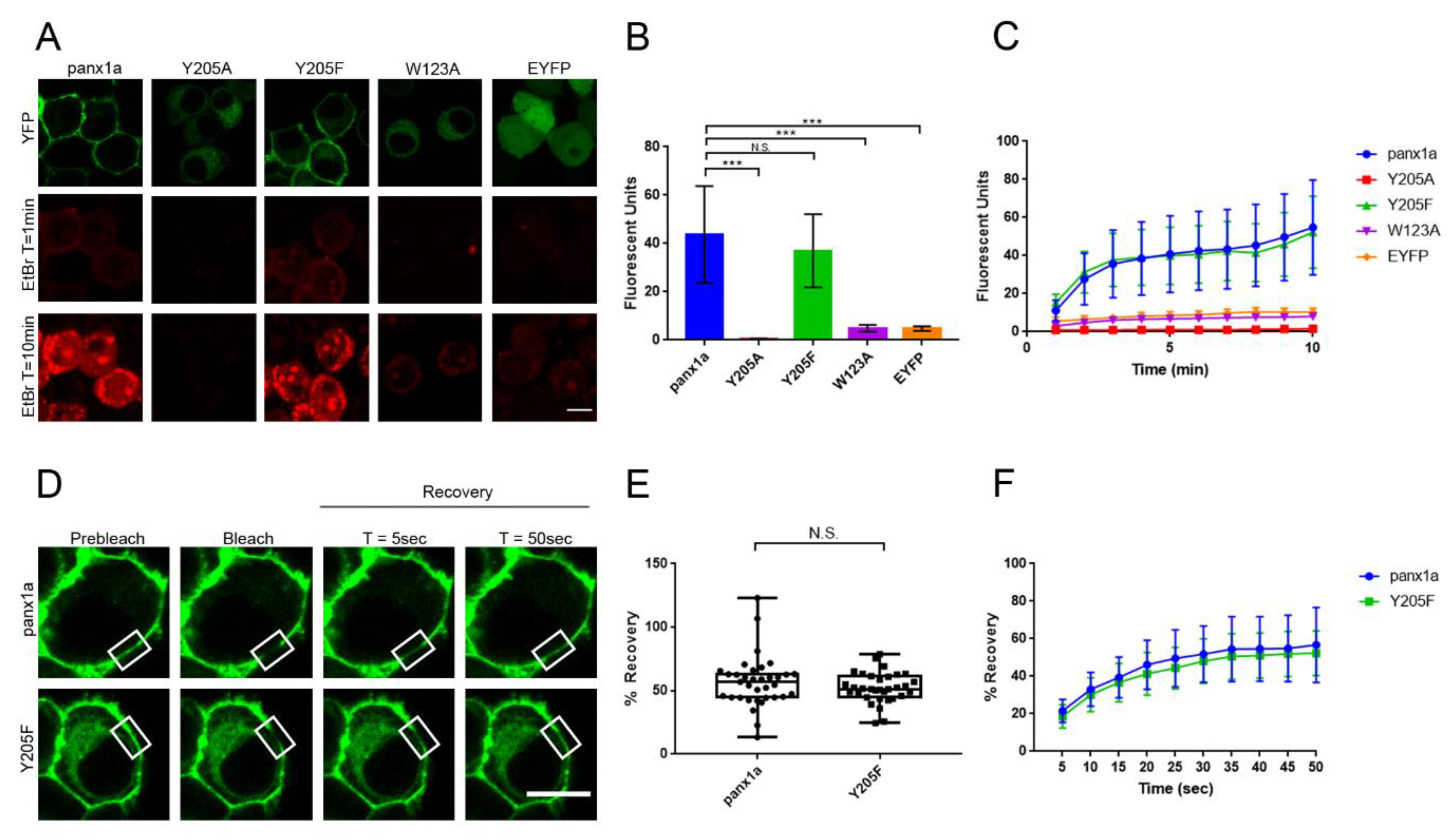
3.1. Mutation of Aromatic Amino Acids Alters Trafficking of Panx1a to the Cell Membrane
3.2. Y205 Phosphorylation Is Not Required for Membrane Expression
3.3. The Y205F Mutation Restores Panx1a Channel Function and Cell Surface Transport
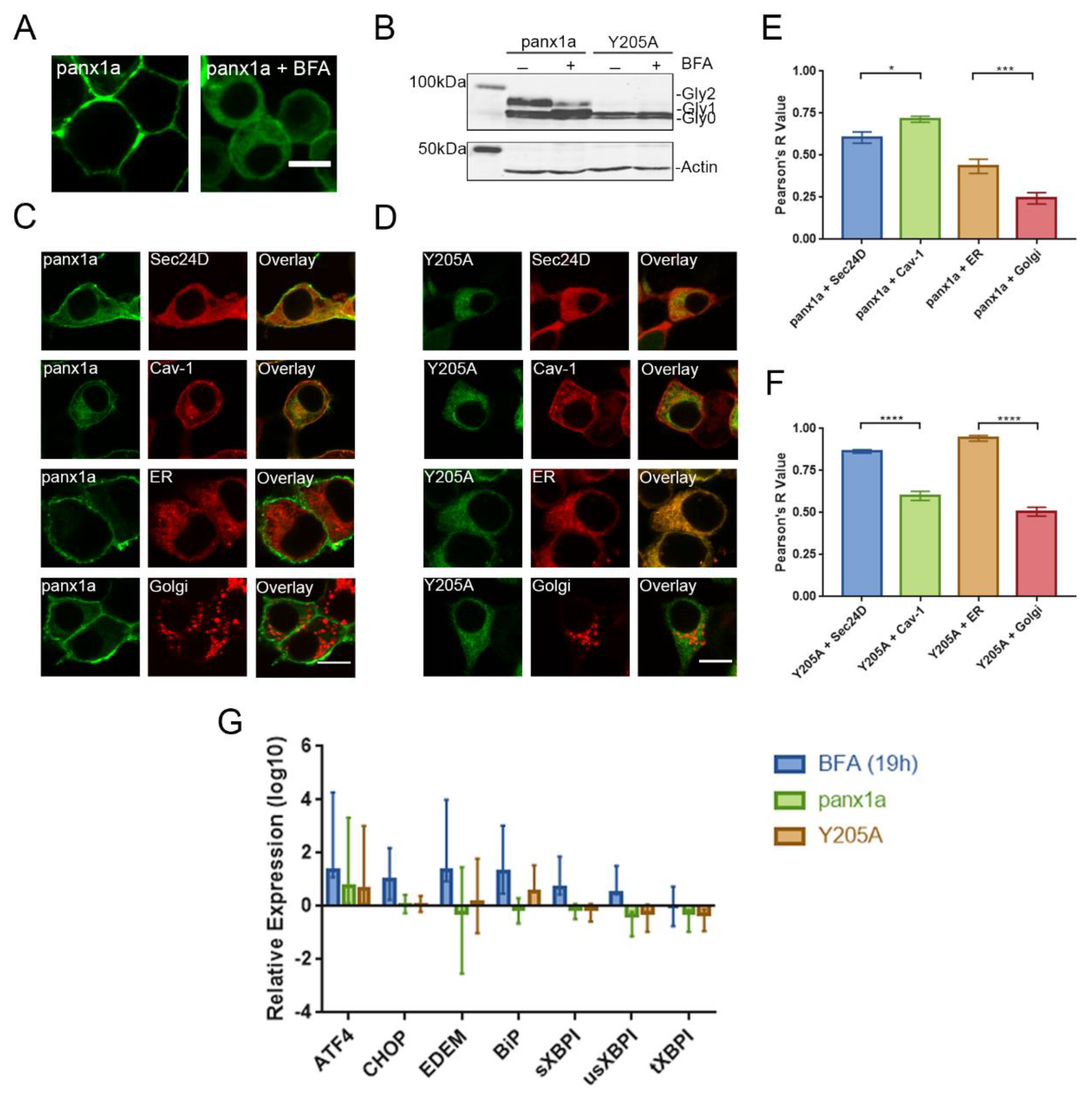
3.4. Y205A Is Retained in Intracellular Compartments but Does Not Induce ER Stress
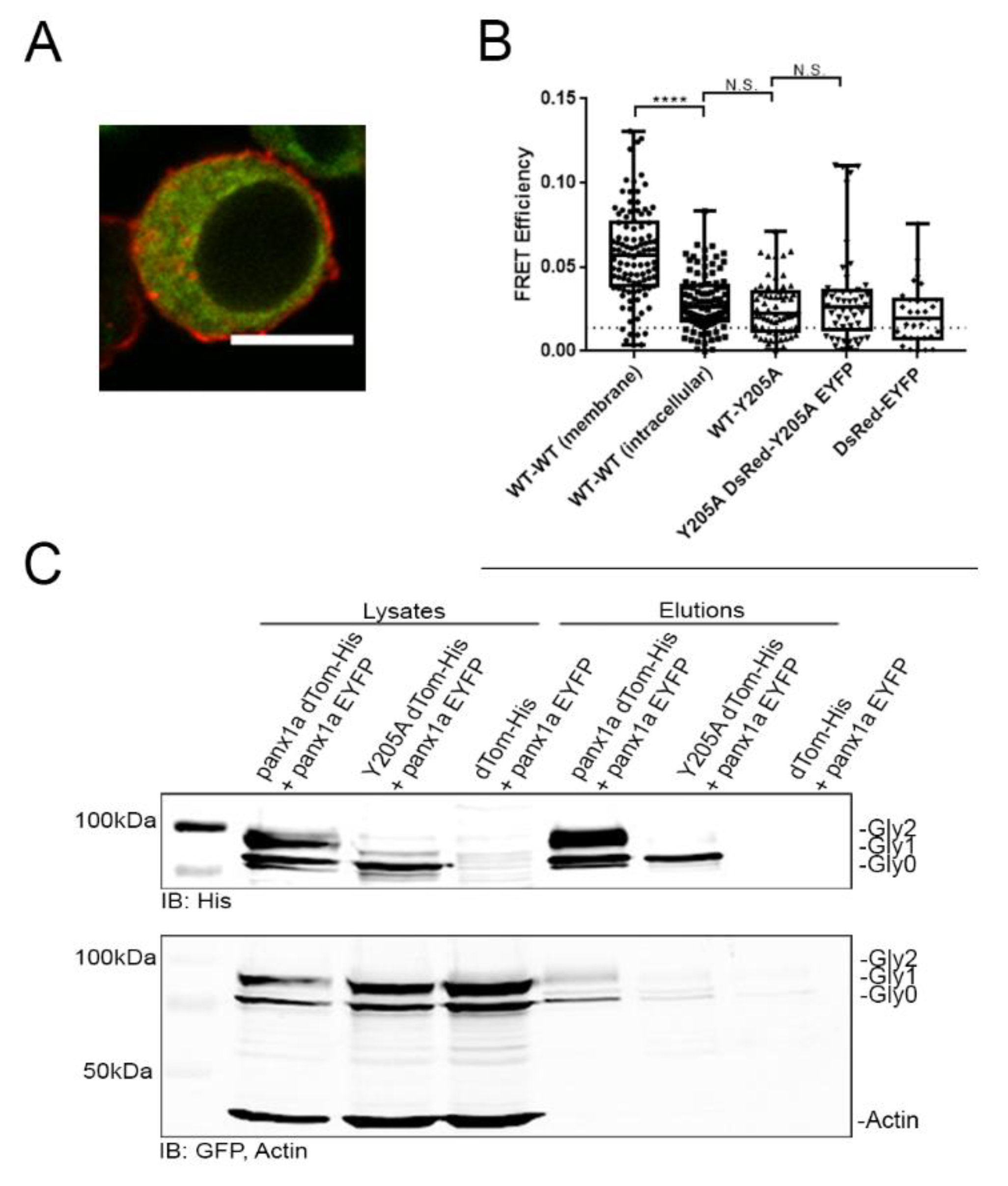
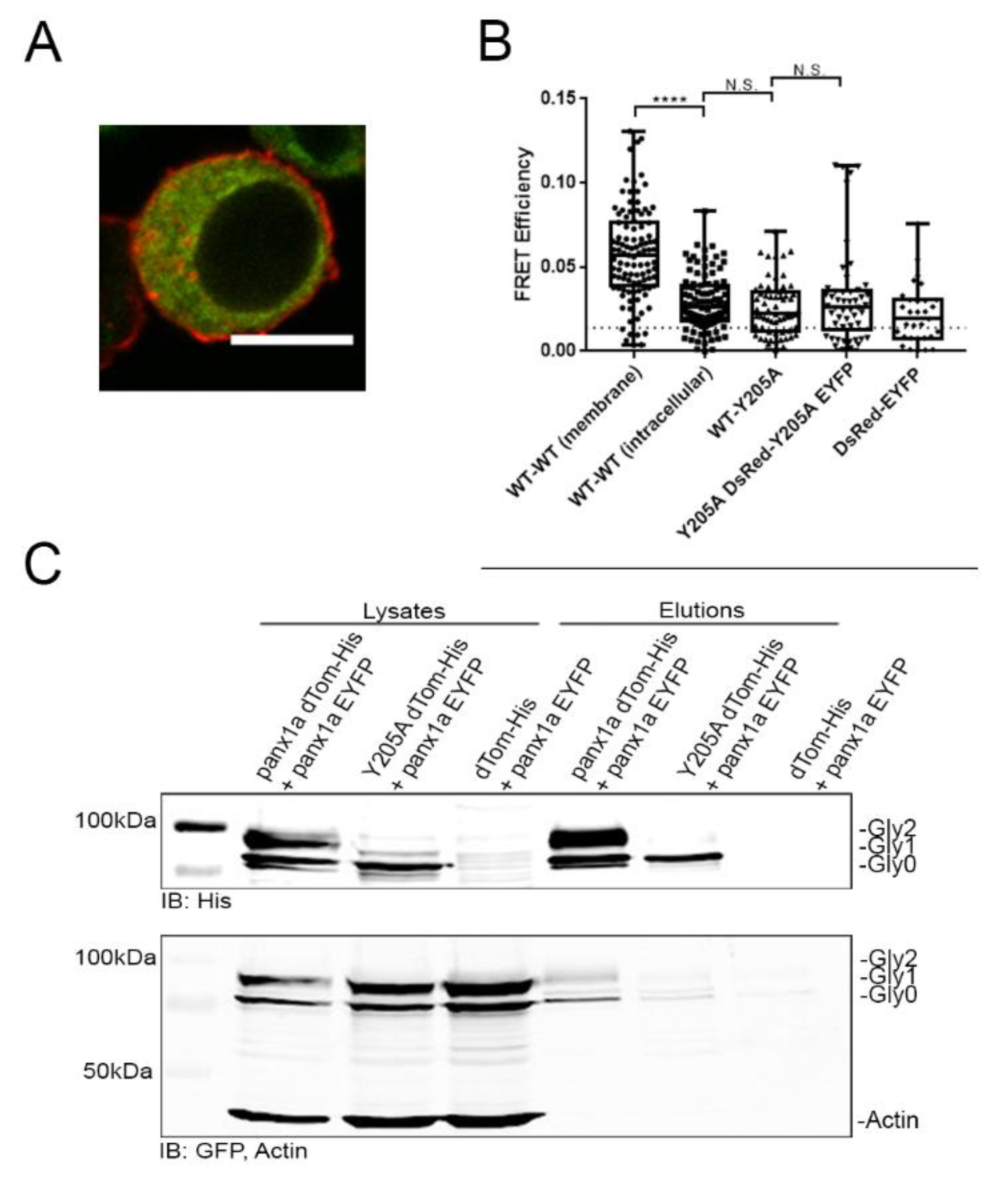
3.5. Trafficking of Y205A to the Cell Surface Cannot Be Rescued by WT Panx1a
4. Discussion
4.1. Panx1a Requires Aromatic Amino Acid Residues for Folding and Stabilization
4.2. Lack of the Aromatic Amino Acid Residues Disrupts Trafficking and Limits Post-Translational Processing of Panx1a
4.3. The Y205A Trafficking Deficiency Was Not Caused by ER Stress
4.4. The Y205A Mutation Disrupted the Oligomerization State of the Panx1a Channel Assembly
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Zoidl, G.; Weickert, S.; Wahle, P.; Dermietzel, R. Site-specific and developmental expression of pannexin1 in the mouse nervous system. Eur. J. Neurosci. 2005, 21, 3277–3290. [Google Scholar] [CrossRef]
- Vogt, A.; Hormuzdi, S.G.; Monyer, H. Pannexin1 and Pannexin2 expression in the developing and mature rat brain. Mol. Brain Res. 2005, 141, 113–120. [Google Scholar] [CrossRef]
- Dahl, G.; Muller, K.J. Innexin and pannexin channels and their signaling. FEBS Lett. 2014, 588, 1396–1402. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, C.; Gassmann, O.; Pranskevich, J.N.; Boassa, D.; Smock, A.; Wang, J.; Dahl, G.; Steinem, C.; Sosinsky, G.E. Pannexin1 and Pannexin2 channels show quaternary similarities to connexons and different oligomerization numbers from each other. J. Biol. Chem. 2010, 285, 24420–24431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004, 572, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, G. ATP release through pannexon channels. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dahl, G. Pannexin1: A multifunction and multiconductance and/or permeability membrane channel. Am. J. Physiol. Cell Physiol. 2018, 315, C290–C299. [Google Scholar] [CrossRef] [PubMed]
- Boassa, D.; Qiu, F.; Dahl, G.; Sosinsky, G. Trafficking dynamics of glycosylated pannexin 1 proteins. Cell Commun. Adhes. 2008, 15, 119–132. [Google Scholar] [CrossRef]
- Penuela, S.; Bhalla, R.; Gong, X.Q.; Cowan, K.N.; Celetti, S.J.; Cowan, B.J.; Bai, D.; Shao, Q.; Laird, D.W. Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J. Cell Sci. 2007, 120, 3772–3783. [Google Scholar] [CrossRef] [Green Version]
- Penuela, S.; Bhalla, R.; Nag, K.; Laird, D.W. Glycosylation regulates pannexin intermixing and cellular localization. Mol. Biol. Cell 2009, 20, 4313–4323. [Google Scholar] [CrossRef] [PubMed]
- Sosinsky, G.E.; Boassa, D.; Dermietzel, R.; Duffy, H.S.; Laird, D.W.; MacVicar, B.; Naus, C.C.; Penuela, S.; Scemes, E.; Spray, D.C.; et al. Pannexin channels are not gap junction hemichannels. Channels 2011, 5, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Scemes, E.; Suadicani, S.O.; Dahl, G.; Spray, D.C. Connexin and pannexin mediated cell-cell communication. Neuron Glia Biol. 2007, 3, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtenbach, S.; Prochnow, N.; Kurtenbach, S.; Klooster, J.; Zoidl, C.; Dermietzel, R.; Kamermans, M.; Zoidl, G. Pannexin1 channel proteins in the zebrafish retina have shared and unique properties. PLoS ONE 2013, 8, e77722. [Google Scholar] [CrossRef] [Green Version]
- Prochnow, N.; Hoffmann, S.; Vroman, R.; Klooster, J.; Bunse, S.; Kamermans, M.; Dermietzel, R.; Zoidl, G. Pannexin1 in the outer retina of the zebrafish, Danio rerio. Neuroscience 2009, 162, 1039–1054. [Google Scholar] [CrossRef]
- Bond, S.R.; Wang, N.; Leybaert, L.; Naus, C.C. Pannexin 1 ohnologs in the teleost lineage. J. Membr. Biol. 2012, 245, 483–493. [Google Scholar] [CrossRef]
- Dahl, G.; Qiu, F.; Wang, J. The bizarre pharmacology of the ATP release channel pannexin1. Neuropharmacology 2013, 75, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Whyte-Fagundes, P.; Zoidl, G. Mechanisms of pannexin1 channel gating and regulation. Biochim. Biophys. Acta Biomembr. 2018, 1860, 65–71. [Google Scholar] [CrossRef]
- Gebhardt, M.; Hoffgaard, F.; Hamacher, K.; Kast, S.M.; Moroni, A.; Thiel, G. Membrane anchoring and interaction between transmembrane domains are crucial for K+ channel function. J. Biol. Chem. 2011, 286, 11299–11306. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.M.; Hecht, K.; Deber, C.M. Aromatic and cation-pi interactions enhance helix-helix association in a membrane environment. Biochemistry 2007, 46, 9208–9214. [Google Scholar] [CrossRef]
- Siu, R.C.; Smirnova, E.; Brown, C.A.; Zoidl, C.; Spray, D.C.; Donaldson, L.W.; Zoidl, G. Structural and Functional Consequences of Connexin 36 (Cx36) Interaction with Calmodulin. Front. Mol. Neurosci. 2016, 9, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte-Fagundes, P.; Kurtenbach, S.; Zoidl, C.; Shestopalov, V.I.; Carlen, P.L.; Zoidl, G. A Potential Compensatory Role of Panx3 in the VNO of a Panx1 Knock Out Mouse Model. Front. Mol. Neurosci. 2018, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.M.; Galliardt, H.; Schneider, J.; Barisas, B.G.; Seidel, T. Quantification of Forster resonance energy transfer by monitoring sensitized emission in living plant cells. Front. Plant Sci. 2013, 4, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A. Structure of an innexin gap junction channel and cryo-EM sample preparation. Microscopy 2017, 66, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Oshima, A.; Tani, K.; Fujiyoshi, Y. Atomic structure of the innexin-6 gap junction channel determined by cryo-EM. Nat. Commun. 2016, 7, 13681. [Google Scholar] [CrossRef]
- Omasits, U.; Ahrens, C.H.; Muller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [Green Version]
- Killian, J.A.; von Heijne, G. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 2000, 25, 429–434. [Google Scholar] [CrossRef]
- Cowan, S.W.; Schirmer, T.; Rummel, G.; Steiert, M.; Ghosh, R.; Pauptit, R.A.; Jansonius, J.N.; Rosenbusch, J.P. Crystal structures explain functional properties of two E. coli porins. Nature 1992, 358, 727–733. [Google Scholar] [CrossRef]
- Ulmschneider, M.B.; Sansom, M.S. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta 2001, 1512, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lampert, A.; O’Reilly, A.O.; Dib-Hajj, S.D.; Tyrrell, L.; Wallace, B.A.; Waxman, S.G. A pore-blocking hydrophobic motif at the cytoplasmic aperture of the closed-state Nav1.7 channel is disrupted by the erythromelalgia-associated F1449V mutation. J. Biol. Chem. 2008, 283, 24118–24127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Dahl, G. SCAM analysis of Panx1 suggests a peculiar pore structure. J. Gen. Physiol. 2010, 136, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochnow, N.; Hoffmann, S.; Dermietzel, R.; Zoidl, G. Replacement of a single cysteine in the fourth transmembrane region of zebrafish pannexin 1 alters hemichannel gating behavior. Exp. Brain Res. 2009, 199, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Bhalla-Gehi, R.; Penuela, S.; Churko, J.M.; Shao, Q.; Laird, D.W. Pannexin1 and pannexin3 delivery, cell surface dynamics, and cytoskeletal interactions. J. Biol. Chem. 2010, 285, 9147–9160. [Google Scholar] [CrossRef] [Green Version]
- DeLalio, L.J.; Keller, A.S.; Chen, J.; Boyce, A.K.J.; Artamonov, M.V.; Askew-Page, H.R.; Keller, T.C.S.; Johnstone, S.R.; Weaver, R.B.; Good, M.E.; et al. Interaction Between Pannexin 1 and Caveolin-1 in Smooth Muscle Can Regulate Blood Pressure. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2065–2078. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Urano, F. The binary switch that controls the life and death decisions of ER stressed beta cells. Curr. Opin. Cell Biol. 2011, 23, 207–215. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer ID | Primers * |
|---|---|---|
| panx1a | Y205A_Fwd | ACTGATCCGCgccCTTCTTTGCC |
| Y205A_Rev | AATCCTTTAGAGTAGCGC | |
| R204Y:Y205R_Fwd | ATTACTGATCtaccgcCTTCTTTGCCGCAC | |
| R204Y:Y205R _Rev | CCTTTAGAGTAGCGCTTG | |
| I203Y:R204I:Y205R _Fwd | ccgcCTTCTTTGCCGCACCATC | |
| I203Y:R204I:Y205R _Rev | atgtaCAGTAATCCTTTAGAGTAGCG | |
| W123A_Fwd | AGCGTTGTTTgcgCGGTTTACAG | |
| W123A_Rev | GGCATGTAAACTGACACTG | |
| Y205F_Fwd | ACTGATCCGCttcCTTCTTTGCC | |
| Y205F_Rev | AATCCTTTAGAGTAGCGCTTG |
| Primer ID | Primers |
|---|---|
| ATF4_Fwd | GGGTTCTGTCTTCCACTCCA |
| ATF4_Rev | AAGCAGCAGAGTCAGGCTTTC |
| CHOP_Fwd | CCACCACACCTGAAAGCAGAA |
| CHOP_Rev | AGGTGAAAGGCAGGGACTCA |
| EDEM_Fwd | CTACCTGCGAAGAGGCCG |
| EDEM_Rev | GTTCATGAGCTGCCCACTGA |
| BiP_Fwd | TTCAGCCAATTATCAGCAAACTCT |
| BiP_Rev | TTTTCTGATGTATCCTCTTCACCAGT |
| sXBP1_Fwd | CTGAGTCCGAATCAGGTGCAG |
| sXBP1_Rev | GTCCATGGGAAGATGTTCTGG |
| usXBP1_Fwd | CAGCACTCAGACTATGTGCA |
| usXBP1_Rev | GTCCATGGGAAGATGTTCTGG |
| tXBP1_Fwd | TGGCCGGGTCTGCTGAGTCCG |
| tXBP1_Rev | GTCCATGGGAAGATGTTCTGG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timonina, K.; Kotova, A.; Zoidl, G. Role of an Aromatic–Aromatic Interaction in the Assembly and Trafficking of the Zebrafish Panx1a Membrane Channel. Biomolecules 2020, 10, 272. https://doi.org/10.3390/biom10020272
Timonina K, Kotova A, Zoidl G. Role of an Aromatic–Aromatic Interaction in the Assembly and Trafficking of the Zebrafish Panx1a Membrane Channel. Biomolecules. 2020; 10(2):272. https://doi.org/10.3390/biom10020272
Chicago/Turabian StyleTimonina, Ksenia, Anna Kotova, and Georg Zoidl. 2020. "Role of an Aromatic–Aromatic Interaction in the Assembly and Trafficking of the Zebrafish Panx1a Membrane Channel" Biomolecules 10, no. 2: 272. https://doi.org/10.3390/biom10020272
APA StyleTimonina, K., Kotova, A., & Zoidl, G. (2020). Role of an Aromatic–Aromatic Interaction in the Assembly and Trafficking of the Zebrafish Panx1a Membrane Channel. Biomolecules, 10(2), 272. https://doi.org/10.3390/biom10020272