Loss of Elongator- and KEOPS-Dependent tRNA Modifications Leads to Severe Growth Phenotypes and Protein Aggregation in Yeast

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
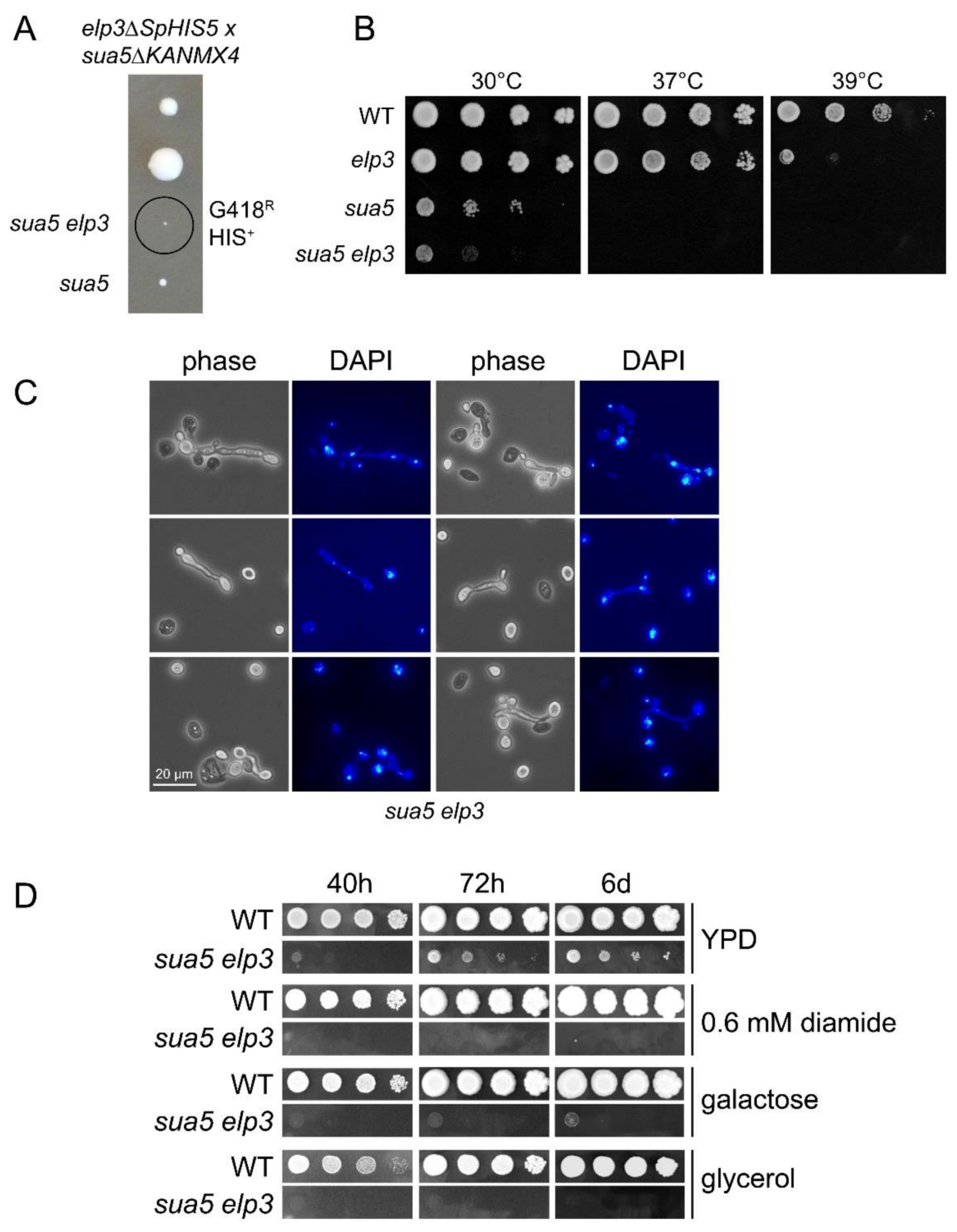
2.1. Strains, Plasmids and Growth Assays
2.2. Plasmid Construction
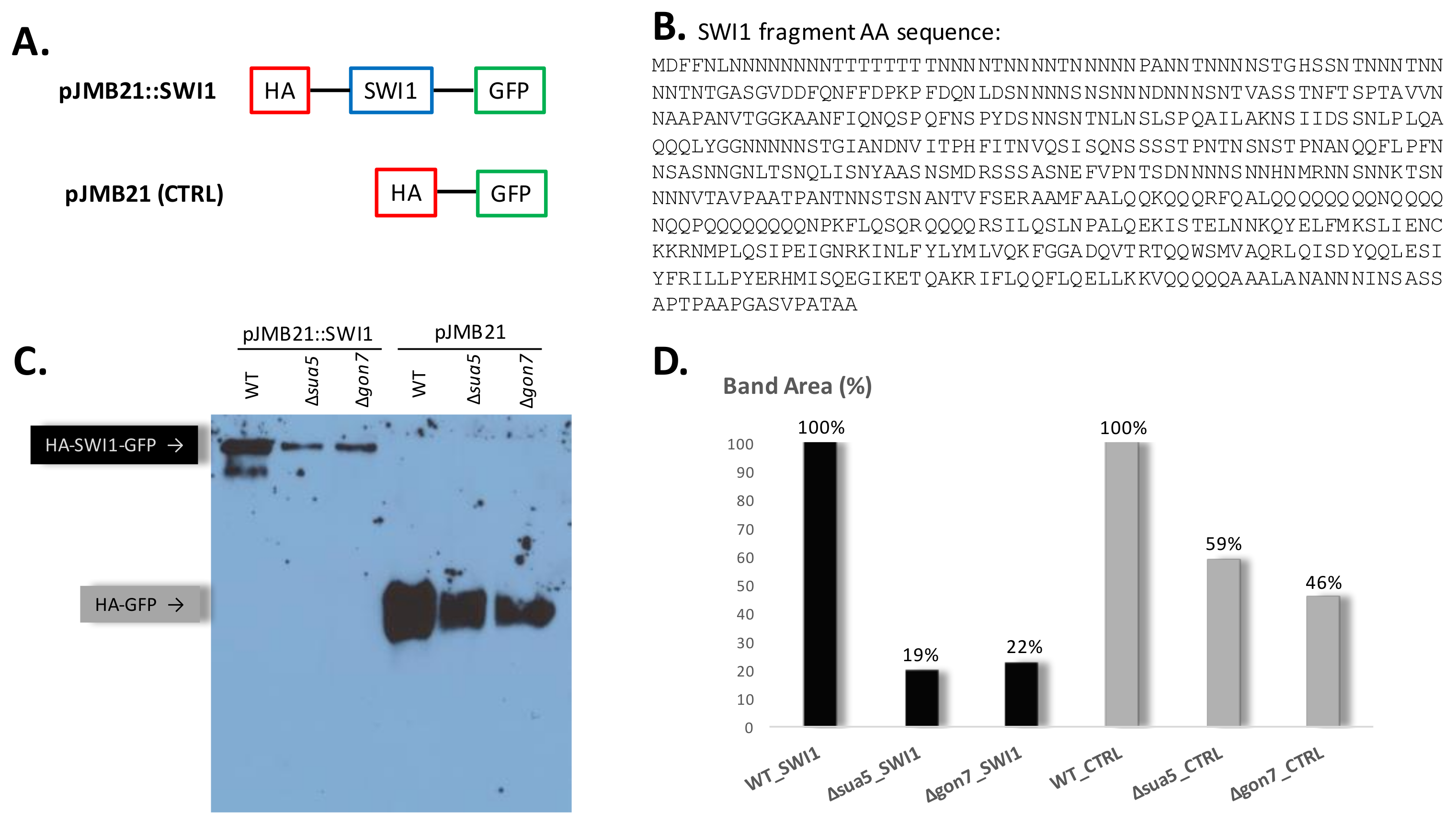
2.3. Detection of HA-SWI1-GFP Fusion in t6A Deficient Strains
2.4. Detection of Protein Aggregates by Different Methods
2.5. Proteomic Analyses
2.5.1. Isolation of Soluble and Insoluble Proteins
2.5.2. Protein Processing, Labeling with Isobaric Tags, and Peptide Fractionation
2.5.3. LC-MS Analysis of the Aggregated Yeast Proteome
2.5.4. Proteomics Data Analysis
2.5.5. Codon Usage Analysis
2.5.6. Statistical Analyses
2.6. Whole-Cell Analyses
3. Results
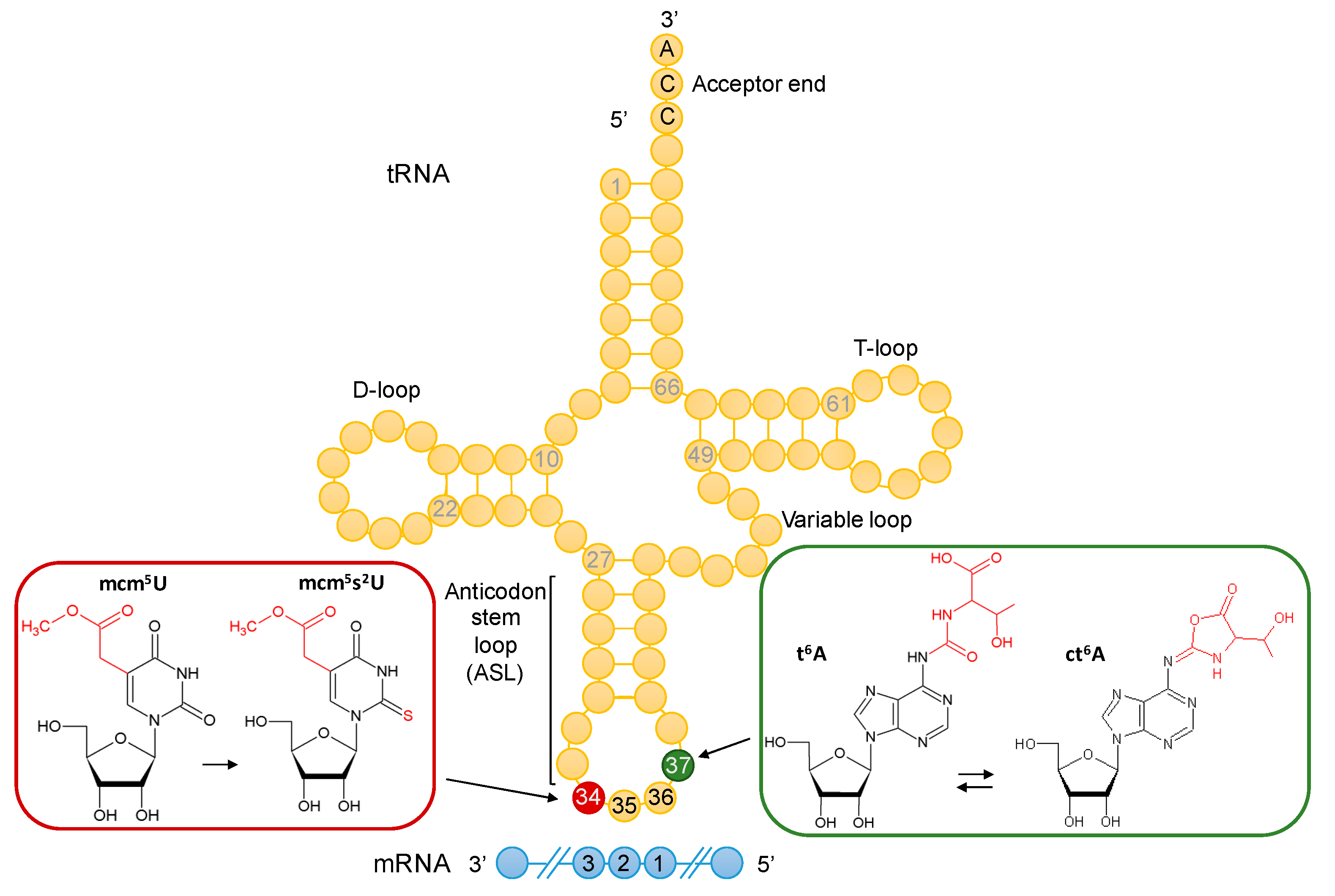
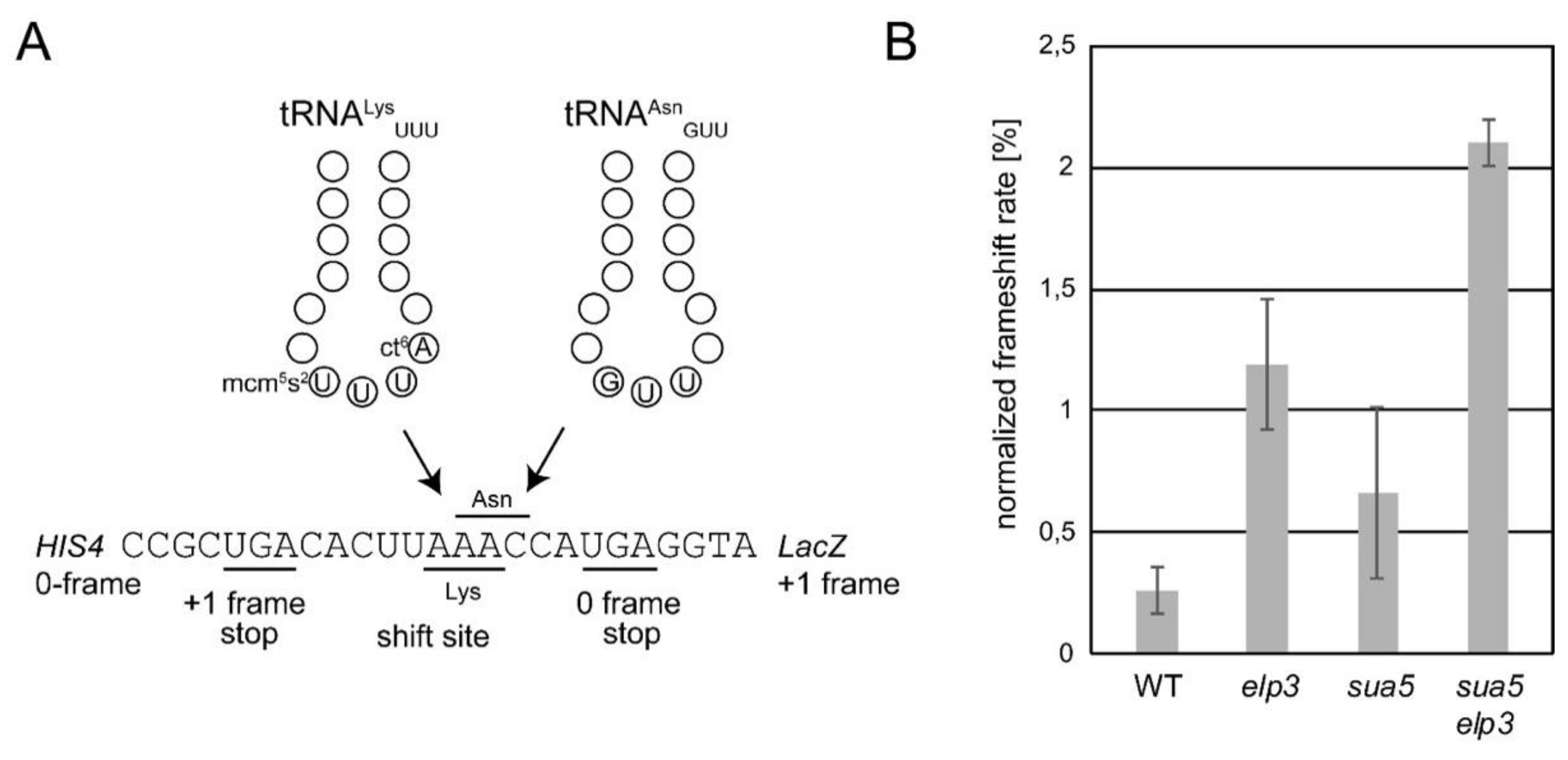
3.1. Absence of t6A and mcm5U Leads to Additive and Possibly Synergistic Translation Defects
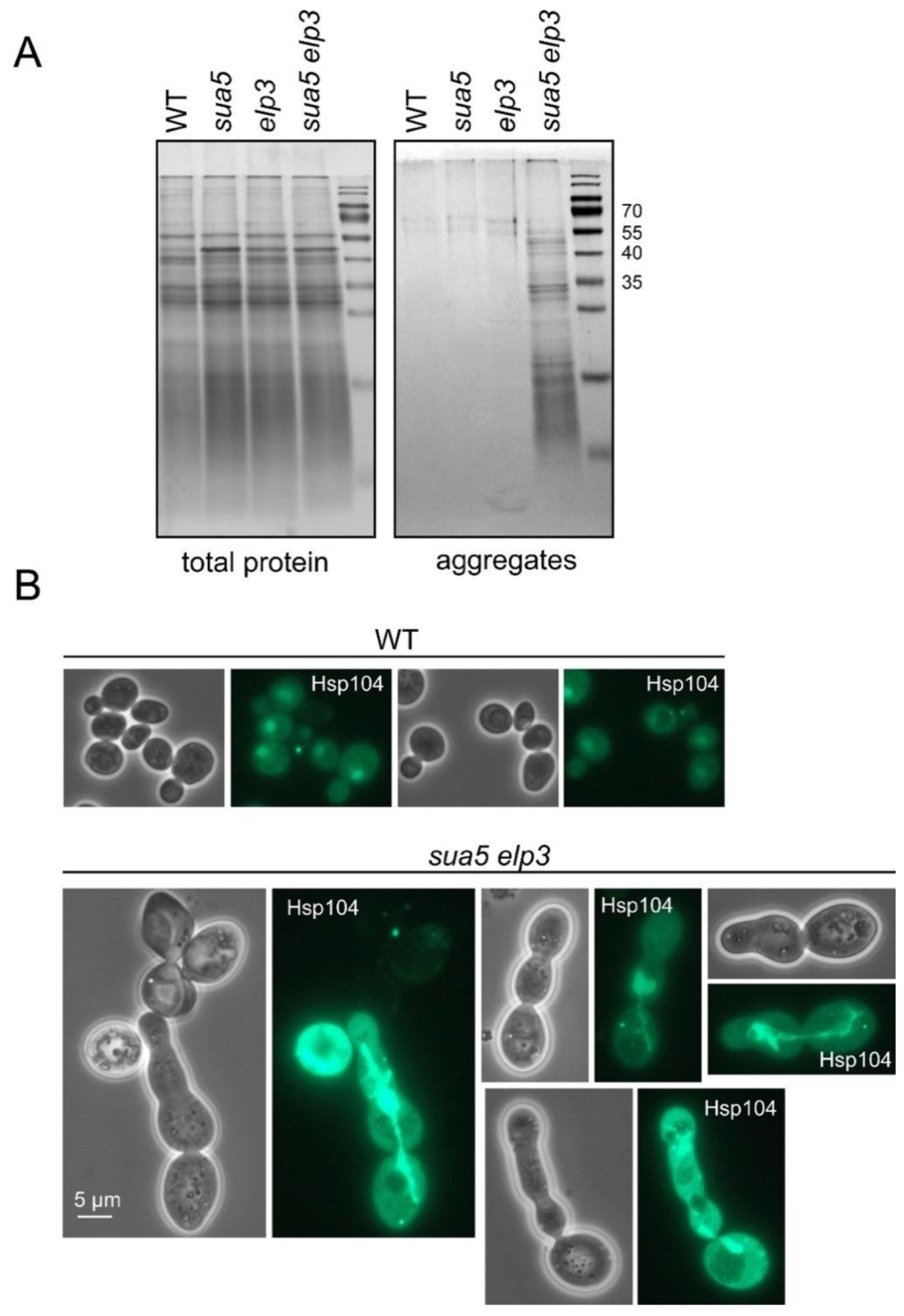
3.2. The Absence of both ASL Modifications Drastically Increases Formation of Protein Aggregates
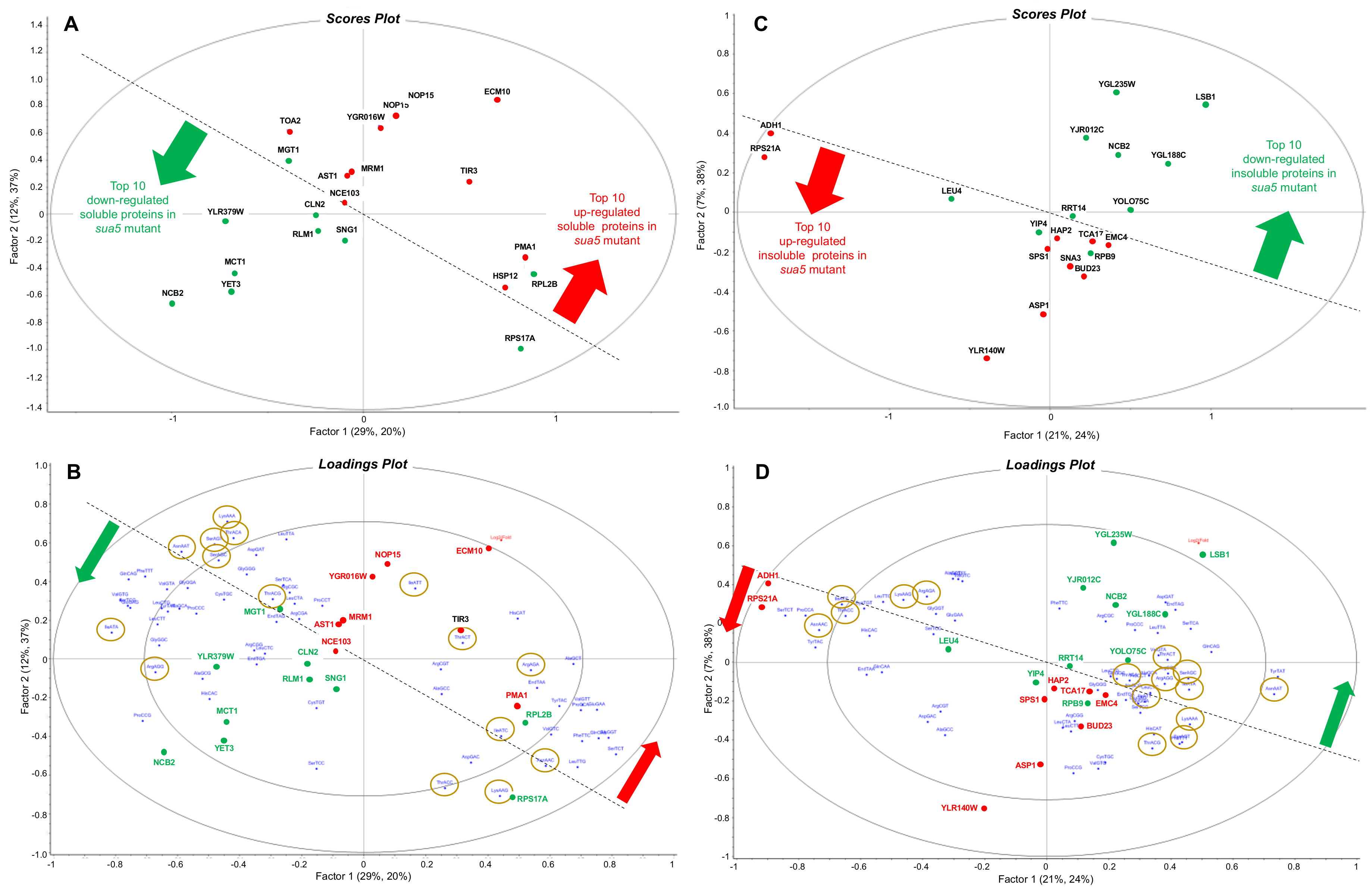
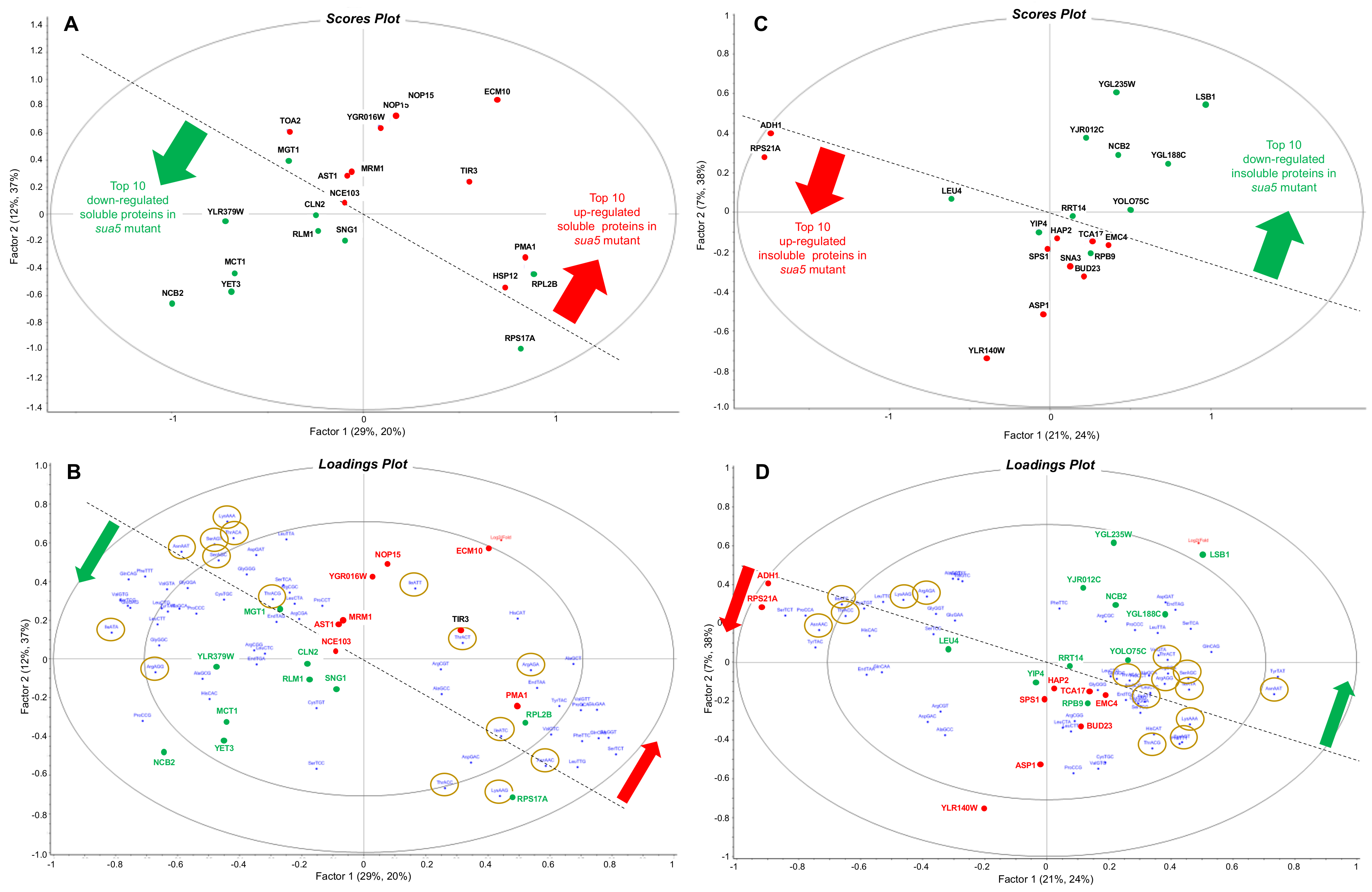
3.3. Loss of t6A Leads to Global Defects in Protein Folding and Mitochondrial Assembly
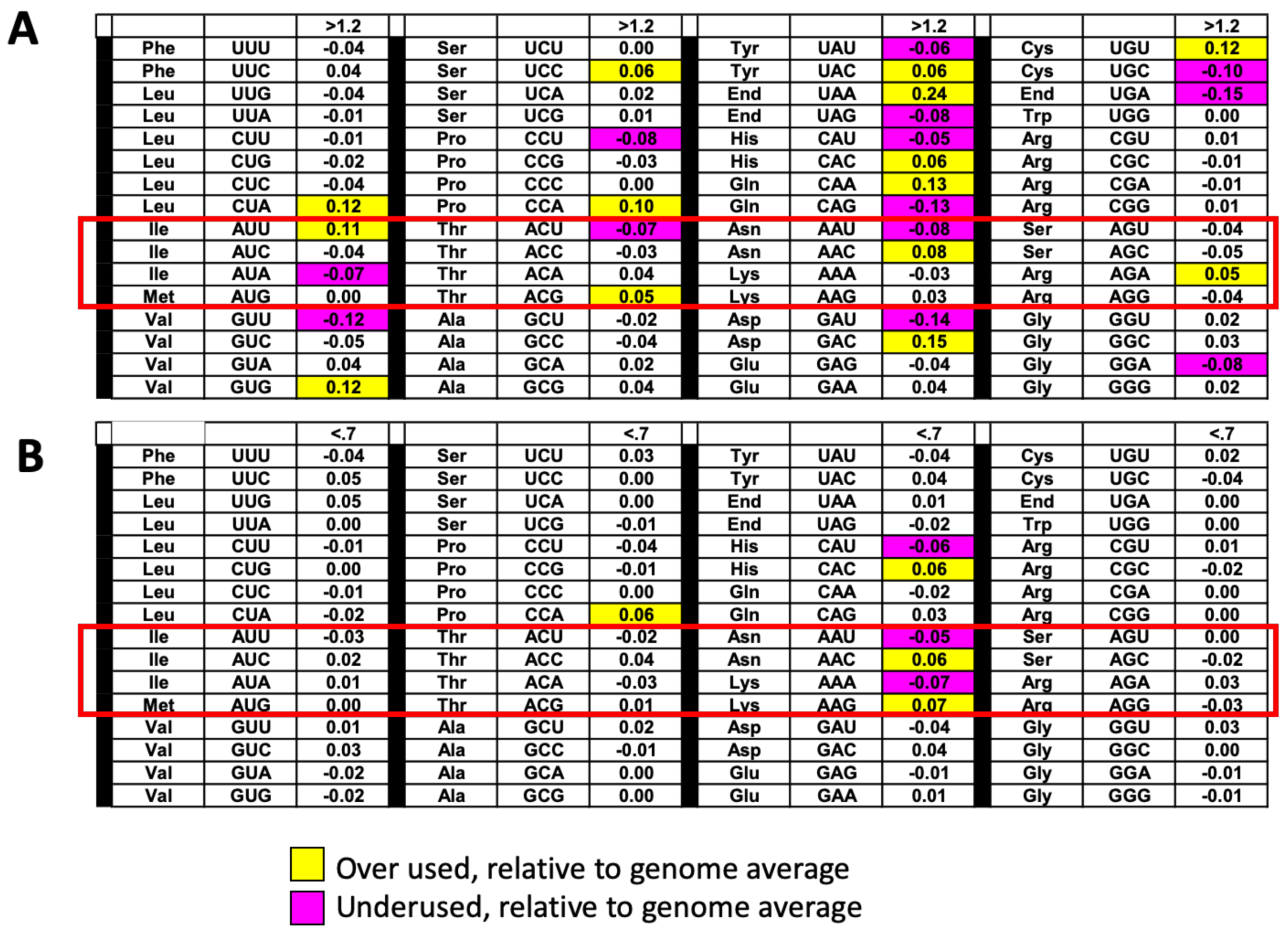
3.4. The Absence of t6A Does Not Specifically Affect the Translation of Prion Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Agris, P.F.; Eruysal, E.R.; Narendran, A.; Väre, V.Y.P.; Vangaveti, S.; Ranganathan, S.V. Celebrating wobble decoding: Half a century and still much is new. RNA Biol. 2018, 15, 537–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuorto, F.; Lyko, F. Genome recoding by tRNA modifications. Open Boil. 2016, 6, 160287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinshteyn, B.; Gilbert, W.V. Loss of a Conserved tRNA Anticodon Modification Perturbs Cellular Signaling. PLoS Genet. 2013, 9, e1003675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedialkova, D.D.; Leidel, S.A. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 2015, 161, 1606–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuorto, F.; Legrand, C.; Cirzi, C.; Federico, G.; Liebers, R.; Müller, M.; E Ehrenhofer-Murray, A.; Dittmar, G.; Gröne, H.; Lyko, F. Queuosine-modified tRNAs confer nutritional control of protein translation. EMBO J. 2018, 37, e99777. [Google Scholar] [CrossRef] [PubMed]
- Manickam, N.; Joshi, K.; Bhatt, M.J.; Farabaugh, P.J. Effects of tRNA modification on translational accuracy depend on intrinsic codon-anticodon strength. Nucleic Acids Res. 2016, 44, 1871–1881. [Google Scholar] [CrossRef] [Green Version]
- Kramer, E.B.; Farabaugh, P.J. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 2007, 13, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, S.; Cornu, D.; Hatin, I.; Grosjean, H.; Bertin, P.; Namy, O. Deciphering the reading of the genetic code by near-cognate tRNA. Proc. Natl. Acad. Sci. USA 2018, 115, 3018–3023. [Google Scholar] [CrossRef] [Green Version]
- Chou, H.-J.; Donnard, E.; Gustafsson, H.T.; Garber, M.; Rando, O.J. transcriptome-wide analysis of roles for tRNA modifications in translational regulation. Mol. Cell 2017, 68, 978–992. [Google Scholar] [CrossRef] [Green Version]
- Sokołowski, M.; Klassen, R.; Bruch, A.; Schaffrath, R.; Glatt, S. Cooperativity between different tRNA modifications and their modification pathways. Biochim. et Biophys. Acta (BBA) Bioenerg. 2018, 1861, 409–418. [Google Scholar] [CrossRef]
- Han, L.; Phizicky, E.M. A rationale for tRNA modification circuits in the anticodon loop. RNA 2018, 24, 1277–1284. [Google Scholar] [PubMed] [Green Version]
- Pereira, M.; Francisco, S.; Varanda, A.S.; Santos, M.; Santos, M.A.S.; Soares, A.R. Impact of tRNA modifications and tRNA-modifying enzymes on proteostasis and human disease. Int. J. Mol. Sci. 2018, 19, 3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendeix, F.A.P.; Murphy, F.V., IV; Cantara, W.A.; Leszczyńska, G.; Gustilo, E.M.; Sproat, B.; Malkiewicz, A.; Agris, P.F. Human tRNA(Lys3)(UUU) is pre-structured by natural modifications for cognate and wobble codon binding through keto-enol tautomerism. J. Mol. Biol. 2012, 416, 467–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weixlbaumer, A.; Murphy, F.V.; Dziergowska, A.; Malkiewicz, A.; Vendeix, F.A.P.; Agris, P.F.; Ramakrishnan, V. Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat. Struct. Mol. Boil. 2007, 14, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, F.V., IV; Ramakrishnan, V.; Malkiewicz, A.; Agris, P.F. The role of modifications in codon discrimination by tRNA(Lys)UUU. Nat. Struct. Mol. Biol. 2004, 11, 1186–1191. [Google Scholar] [CrossRef]
- Väre, V.Y.P.; Eruysal, E.R.; Narendran, A.; Sarachan, K.L.; Agris, P.F. Chemical and conformational diversity of modified nucleosides affects tRNA structure and function. Biomolecules 2017, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Sonawane, K.D.; Kamble, A.S.; Fandilolu, P.M. Preferences of AAA/AAG codon recognition by modified nucleosides, τm5s2U34 and t6A37 present in tRNALys. J. Biomol. Struct. Dyn. 2018, 36, 4182–4196. [Google Scholar] [CrossRef]
- Edvardson, S.; Prunetti, L.; Arraf, A.; Haas, D.; Bacusmo, J.M.; Hu, J.F.; Ta-Shma, A.; Dedon, P.C.; De Crécy-Lagard, V.; Elpeleg, O. tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur. J. Hum. Genet. 2017, 25, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Rao, J.; Mollet, G.; Schapiro, D.; Daugeron, M.-C.; Tan, W.; Gribouval, O.; Boyer, O.; Revy, P.; Jobst-Schwan, T.; et al. Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly. Nat. Genet. 2017, 49, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Schaffrath, R.; Leidel, S.A. Wobble uridine modifications-a reason to live, a reason to die?! RNA Boil. 2017, 14, 1209–1222. [Google Scholar] [CrossRef]
- Kojic, M.; Gaik, M.; Kiska, B.; Salerno-Kochan, A.; Hunt, S.; Tedoldi, A.; Mureev, S.; Jones, A.; Whittle, B.; Genovesi, L.A.; et al. Elongator mutation in mice induces neurodegeneration and ataxia-like behavior. Nat. Commun. 2018, 9, 3195. [Google Scholar] [CrossRef] [PubMed]
- Hawer, H.; Hammermeister, A.; Ravichandran, K.E.; Glatt, S.; Schaffrath, R.; Klassen, R. roles of elongator dependent tRNA modification pathways in neurodegeneration and cancer. Genes 2018, 10, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrochia, L.; Crozat, E.; Hecker, A.; Zhang, W.; Bareille, J.; Collinet, B.; Van Tilbeurgh, H.; Forterre, P.; Basta, T. In vitro biosynthesis of a universal t6A tRNA modification in Archaea and Eukarya. Nucleic Acids Res. 2013, 41, 1953–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Yacoubi, B.; Hatin, I.; Deutsch, C.; Kahveci, T.; Rousset, J.-P.; Iwata-Reuyl, D.; Murzin, A.G.; De Crécy-Lagard, V. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J. 2011, 30, 882–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Yacoubi, B.; Lyons, B.; Cruz, Y.; Reddy, R.; Nordin, B.; Agnelli, F.; Williamson, J.R.; Schimmel, P.; Swairjo, M.A.; De Crécy-Lagard, V. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res. 2009, 37, 2894–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björk, G.R.; Huang, B.; Persson, O.P.; Byström, A.S. A conserved modified wobble nucleoside (mcm5s2U) in lysyl-tRNA is required for viability in yeast. RNA 2007, 13, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Mehlgarten, C.; Jablonowski, D.; Wrackmeyer, U.; Tschitschmann, S.; Sondermann, D.; Jäger, G.; Gong, Z.; Byström, A.S.; Schaffrath, R.; Breunig, K.D. Elongator function in tRNA wobble uridine modification is conserved between yeast and plants. Mol. Microbiol. 2010, 76, 1082–1094. [Google Scholar] [CrossRef]
- Huang, B.; Johansson, M.J.; Byström, A.S. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 2005, 11, 424–436. [Google Scholar] [CrossRef] [Green Version]
- Klassen, R.; Ciftci, A.; Funk, J.; Bruch, A.; Butter, F.; Schaffrath, R. tRNA anticodon loop modifications ensure protein homeostasis and cell morphogenesis in yeast. Nucleic Acids Res. 2016, 44, 10946–10959. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, K.; Kimura, S.; Suzuki, T. A cyclic form of N6-threonylcarbamoyladenosine as a widely distributed tRNA hypermodification. Nat. Chem. Biol. 2013, 9, 105–111. [Google Scholar] [CrossRef]
- Boccaletto, P.; MacHnicka, M.A.; Purta, E.; Pitkowski, P.; Baginski, B.; Wirecki, T.K.; De Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- El Yacoubi, B.; Bailly, M.; De Crécy-Lagard, V. Biosynthesis and Function of Posttranscriptional Modifications of Transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Thiaville, P.C.; Iwata-Reuyl, D.; De Crécy-Lagard, V. Diversity of the biosynthesis pathway for threonylcarbamoyladenosine (t6A), a universal modification of tRNA. RNA Boil. 2014, 11, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolaj-Robin, O.; Séraphin, B. Structures and activities of the elongator complex and its cofactors. DNA Repair 2017, 41, 117–149. [Google Scholar]
- Lin, T.-Y.; Abbassi, N.E.H.; Zakrzewski, K.; Chramiec-Głąbik, A.; Jemioła-Rzemińska, M.; Różycki, J.; Glatt, S. The Elongator subunit Elp3 is a non-canonical tRNA acetyltransferase. Nat. Commun. 2019, 10, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazauric, M.-H.; Dirick, L.; Purushothaman, S.K.; Björk, G.R.; Lapeyre, B. Trm112p is a 15-kDa Zinc finger protein essential for the activity of two tRNA and one protein methyltransferases in Yeast*. J. Boil. Chem. 2010, 285, 18505–18515. [Google Scholar] [CrossRef] [Green Version]
- Thiaville, P.C.; Crécy-Lagard, V. The emerging role of complex modifications of tRNALysUUU in signaling pathways. Microb. Cell 2015, 2, 1–4. [Google Scholar] [CrossRef]
- Han, L.; Guy, M.P.; Kon, Y.; Phizicky, E.M. Lack of 2’-O-methylation in the tRNA anticodon loop of two phylogenetically distant yeast species activates the general amino acid control pathway. PLoS Genet. 2018, 14, e1007288. [Google Scholar] [CrossRef]
- Thiaville, P.C.; Legendre, R.; Rojas-Benítez, D.; Baudin-Baillieu, A.; Hatin, I.; Chalancon, G.; Glavic, A.; Namy, O.; De Crécy-Lagard, V. Global translational impacts of the loss of the tRNA modification t6A in yeast. Microb. Cell 2016, 3, 29–45. [Google Scholar] [CrossRef] [Green Version]
- Esberg, A.; Huang, B.; Johansson, M.J.; Byström, A.S. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell 2006, 24, 139–148. [Google Scholar] [CrossRef]
- Ranjan, N.; Rodnina, M.V. Thio-modification of tRNA at the wobble position as regulator of the kinetics of decoding and translocation on the ribosome. J. Am. Chem. Soc. 2017, 139, 5857–5864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezgui, V.A.N.; Tyagi, K.; Ranjan, N.; Konevega, A.L.; Mittelstaet, J.; Rodnina, M.V.; Peter, M.; Pedrioli, P.G.A. tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc. Natl. Acad. Sci. USA 2013, 110, 12289–12294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, K.; Bhatt, M.J.; Farabaugh, P.J. Codon-specific effects of tRNA anticodon loop modifications on translational misreading errors in the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 2018, 46, 10331–10339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, A.; Chan, C.T.; Dyavaiah, M.; Rooney, J.P.; Dedon, P.C.; Begley, T.J. Translational infidelity-induced protein stress results from a deficiency in Trm9-catalyzed tRNA modifications. RNA Boil. 2012, 9, 990–1001. [Google Scholar] [CrossRef] [Green Version]
- Rodnina, M.V. The ribosome in action: Tuning of translational efficiency and protein folding. Protein Sci. 2016, 25, 1390–1406. [Google Scholar] [CrossRef] [Green Version]
- Drummond, D.A.; Wilke, C.O. Mistranslation-Induced Protein Misfolding as a Dominant Constraint on Coding-Sequence Evolution. Cell 2008, 134, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Dewe, J.M.; Fuller, B.L.; Lentini, J.M.; Kellner, S.M.; Fu, D. TRMT1-catalyzed tRNA modifications are required for redox homeostasis to ensure proper cellular proliferation and oxidative stress survival. Mol. Cell. Boil. 2017, 37, e00214–e00217. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Benítez, D.; Eggers, C.; Glavic, A. Modulation of the proteostasis machinery to overcome stress caused by diminished levels of t6A-modified tRNAs in Drosophila. Biomolecules 2017, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Bruch, A.; Klassen, R.; Schaffrath, R. Independent suppression of ribosomal +1 frameshifts by different tRNA anticodon loop modifications. RNA Biol. 2017, 14, 1252–1259. [Google Scholar]
- Wickner, R.B.; Edskes, H.K.; Shewmaker, F.; Nakayashiki, T.; Engel, A.; McCann, L.; Kryndushkin, D. Yeast prions: Evolution of the prion concept. Prion 2007, 1, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Crow, E.T.; Li, L. Newly identified prions in budding yeast, and their possible functions. Semin. Cell Dev. Biol. 2011, 22, 452–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z. The complexity and implications of yeast prion domains AU. Prion 2011, 5, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncharoff, D.K.; Du, Z.; Li, L. A brief overview of the Swi1 prion—[SWI+]. FEMS Yeast Res. 2018, 18, 061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klassen, R.; Schaffrath, R. Collaboration of tRNA modifications and elongation factor eEF1A in decoding and nonsense suppression. Sci. Rep. 2018, 8, 12749. [Google Scholar] [CrossRef] [PubMed]
- Thiaville, P.C.; El Yacoubi, B.; Köhrer, C.; Thiaville, J.J.; Deutsch, C.; Iwata-Reuyl, D.; Bacusmo, J.M.; Armengaud, J.; Bessho, Y.; Wetzel, C.; et al. Essentiality of threonylcarbamoyladenosine (t6A), a universal tRNA modification, in bacteria. Mol. Microbiol. 2015, 98, 1199–1221. [Google Scholar] [CrossRef] [Green Version]
- Sherman, F. Getting started with yeast. In Guide to Yeast Genetics and Molecular and Cell Biology - Part B; Guthrie, C., Fink, G.R.B.T.-M., Eds.; Academic Press: Amsterdam, The Netherlands, 2002; Volume 350, pp. 3–41. [Google Scholar]
- Gueldener, U. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002, 30, 23. [Google Scholar] [CrossRef] [Green Version]
- Van Driessche, B.; Tafforeau, L.; Hentges, P.; Carr, A.M.; Vandenhaute, J. Additional vectors for PCR-based gene tagging in Saccharomyces cerevisiae and Schizosaccharomyces pombe using nourseothricin resistance. Yeast 2005, 22, 1061–1068. [Google Scholar] [CrossRef]
- Thiaville, P.C.; El Yacoubi, B.; Perrochia, L.; Hecker, A.; Prigent, M.; Thiaville, J.J.; Forterre, P.; Namy, O.; Basta, T.; De Crécy-Lagard, V. Cross kingdom functional conservation of the core universally conserved threonylcarbamoyladenosine tRNA synthesis enzymes. Eukaryot. Cell 2014, 13, 1222–1231. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B.; Neupert, W. Mitochondria-targeted green fluorescent proteins: Convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast 2000, 16, 1421–1427. [Google Scholar] [CrossRef]
- Zhang, T.; Lei, J.; Yang, H.; Xu, K.; Wang, R.; Zhang, Z. An improved method for whole protein extraction from yeast Saccharomyces cerevisiae. Yeast 2011, 28, 795–798. [Google Scholar] [CrossRef]
- Koplin, A.; Preissler, S.; Ilina, Y.; Koch, M.; Scior, A.; Erhardt, M.; Deuerling, E. A dual function for chaperones SSB–RAC and the NAC nascent polypeptide–associated complex on ribosomes. J. Cell Boil. 2010, 189, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.M. The Bicinchoninic Acid (BCA) Assay for Protein Quantitation—Basic Protein and Peptide Protocols; Humana Press: Totowa, NJ, USA, 1994; pp. 5–8. [Google Scholar]
- Kozlowski, L.P. Proteome-pI: Proteome isoelectric point database. Nucleic Acids Res. 2017, 45, D1112–D1116. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.A.; Raval, A.; Wilke, C.O. A single determinant dominates the rate of yeast protein evolution. Mol. Boil. Evol. 2005, 23, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Perez, M.; Villén, J. Determinants and regulation of protein turnover in yeast. Cell Syst. 2017, 5, 283–294.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belle, A.; Tanay, A.; Bitincka, L.; Shamir, R.; O’Shea, E.K. Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. USA 2006, 103, 13004–13009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, B.; Baryshnikova, A.; Brown, G.W. Unification of protein abundance datasets yields a quantitative Saccharomyces cerevisiae proteome. Cell Syst. 2018, 6, 192–205.e3. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [Green Version]
- Begley, U.; Dyavaiah, M.; Patil, A.; Rooney, J.P.; DiRenzo, D.; Young, C.M.; Conklin, U.S.; Zitomer, R.S.; Begley, T.J. Trm9-Catalyzed tRNA Modifications Link Translation to the DNA Damage Response. Mol. Cell 2007, 28, 860–870. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Tükenmez, H.; Xu, H.; Esberg, A.; Byström, A.S. The role of wobble uridine modifications in +1 translational frameshifting in eukaryotes. Nucleic Acids Res. 2015, 43, 9489–9499. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Larsson, L.; Caballero, A.; Hao, X.; Oling, D.; Grantham, J.; Nyström, T. The polarisome is required for segregation and retrograde transport of protein aggregates. Cell 2010, 140, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Slaughter, B.D.; Unruh, J.R.; Eldakak, A.; Rubinstein, B.; Li, R. Motility and segregation of Hsp104-associated protein aggregates in budding yeast. Cell 2011, 147, 1186–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkach, J.M.; Glover, J.R. Nucleocytoplasmic trafficking of the molecular chaperone hsp104 in unstressed and heat-shocked cells. Traffic 2008, 9, 39–56. [Google Scholar] [CrossRef]
- Weids, A.J.; Ibstedt, S.; Tamás, M.J.; Grant, C.M. Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 2016, 6, 24554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.T.Y.; Deng, W.; Li, F.; DeMott, M.S.; Babu, I.R.; Begley, T.J.; Dedon, P.C. Highly predictive reprogramming of tRNA modifications is linked to selective expression of codon-biased genes. Chem. Res. Toxicol. 2015, 28, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.T.; Pang, Y.L.J.; Deng, W.; Babu, I.R.; Dyavaiah, M.; Begley, T.J.; Dedon, P.C. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012, 3, 937. [Google Scholar] [CrossRef] [Green Version]
- Chionh, Y.H.; McBee, M.; Babu, I.R.; Hia, F.; Lin, W.; Zhao, W.; Cao, J.; Dziergowska, A.; Malkiewicz, A.; Begley, T.J.; et al. tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat. Commun. 2016, 7, 13302. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.S.; Sinha, A.; Aniweh, Y.; Nah, Q.; Babu, I.R.; Gu, C.; Chionh, Y.H.; Dedon, P.C.; Preiser, P.R. tRNA epitranscriptomics and biased codon are linked to proteome expression in Plasmodium falciparum. Mol. Syst. Boil. 2018, 14, e8009. [Google Scholar] [CrossRef]
- Rothenberg, D.A.; Taliaferro, J.M.; Huber, S.M.; Begley, T.J.; Dedon, P.C.; White, F.M. A Proteomics Approach to Profiling the Temporal Translational Response to Stress and Growth. iScience 2018, 9, 367–381. [Google Scholar] [CrossRef] [Green Version]
- Doyle, F.; Leonardi, A.; Endres, L.; Tenenbaum, S.A.; Dedon, P.C.; Begley, T.J.; Doyle, F. Gene- and genome-based analysis of significant codon patterns in yeast, rat and mice genomes with the CUT Codon UTilization tool. Methods 2016, 107, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Grunewald, P.; Thuring, K.L.; Eichler, C.; Helm, M.; Schaffrath, R. Loss of anticodon wobble uridine modifications affects tRNA(Lys) function and protein levels in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0119261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Walvekar, A.S.; Liang, S.; Rashida, Z.; Shah, P.; Laxman, S. A tRNA modification balances carbon and nitrogen metabolism by regulating phosphate homeostasis. Elife 2019, 8, e44795. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Miyauchi, K.; Harada, T.; Okita, R.; Takeshita, E.; Komaki, H.; Fujioka, K.; Yagasaki, H.; Goto, Y.-I.; Yanaka, K.; et al. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun. 2018, 9, 1875. [Google Scholar] [CrossRef] [PubMed]
- Chernova, T.A.; Romanyuk, A.V.; Karpova, T.S.; Shanks, J.R.; Ali, M.; Moffatt, N.; Howie, R.L.; O’Dell, A.; McNally, J.G.; Liebman, S.W.; et al. Prion induction by the short-lived, stress-induced protein Lsb2 is regulated by ubiquitination and association with the actin cytoskeleton. Mol. Cell 2011, 43, 242–252. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | References/Sources |
|---|---|---|
| BY4741 | MATa, his3Δ, leu2Δ, met15Δ, ura3Δ | Euroscarf, Frankfurt |
| BY4742 | MATα, his3Δ, leu2Δ, lys2Δ, ura3Δ | Euroscarf, Frankfurt |
| Y02742 | BY4741 elp3::KANMX4 | Euroscarf, Frankfurt |
| RK311 | BY4741 HSP104-GFP::natMX6 | This study |
| RK477 | BY4741 elp3::SpHIS5 | This study |
| VDC9100 | BY4742 sua5::KANMX4 | [59] |
| Y07017 | BY4741 gon7::KANMX4 | Euroscarf, Frankfurt [39] |
| RK340 | BY4742 sua5::KANMX4 elp3::SpHIS5 | This study |
| RK357 | BY4742 sua5::KANMX4 elp3::SpHIS5 HSP104-GFP::natMX6 | This study |
| RK359 | BY4742 elp3::SpHIS5 HSP104-GFP::natMX6 | This study |
| RK360 | BY4742 sua5::KANMX5 HSP104-GFP::natMX6 | This study |
| LPO0180 | BY4741 pJMB21 | This study |
| LPO0181 | BY4741 pJMB21::SWI1 | This study |
| LPO0085 | BY4742 sua5::KANMX5 pJMB21 | This study |
| LPO0087 | BY4742 sua5::KANMX5 pJMB21::SWI1 | This study |
| LPO0089 | BY4741 gon7::KANMX5 pJMB21 | This study |
| LPO0091 | BY4741 gon7::KANMX5 pJMB21::SWI1 | This study |
| Go Category | # Genes | P-Value |
|---|---|---|
| Protein binding involved in protein folding | 7 | 6.53 × 10−8 |
| Misfolded protein binding | 7 | 4.24 × 10−7 |
| Heat shock protein binding | 7 | 3.37 × 10−6 |
| ATPase activity, coupled | 12 | 6.40 × 10−4 |
| Purine ribonucleotide triphosphate binding | 25 | 1.23 × 10−3 |
| Unfolded protein binding | 8 | 2.08 × 10−3 |
| Protein | ORF | Description | Fold-Change |
|---|---|---|---|
| Mitochondrial heat shock protein, SSC3 | YEL030W | Refolding imported precursors | 3.0 |
| rRNA methyltransferase 2, mitochondrial | YGL136C | Peptidyl transferase domain | 2.3 |
| Exosome complex component RRP40 | YOL142W | Exoribonuclease | 1.9 |
| Mitochondrial heat-shock protein SSC1 | YJR045C | Binds to precursor preprotein | 1.8 |
| Interacting with cytoskeleton protein 1 ICY1 | YMR195W | Required for the viability of cells lacking mtDNA | 1.7 |
| Plasma membrane ATPase 2 | YPLO36W | Nutrient active transport by H+ symport | 1.7 |
| rRNA-processing protein CGR1 | YGL029W | Involved in nucleolar integrity, required for processing 60S pre-RNA | 1.7 |
| Mitochondrial import receptor subunit TOM5 | YPR133W-A | Component of receptor complex responsible for recognizing, translocating cytosolically synthesized mitochondrial preproteins | 1.7 |
| Endoplasmic reticulum chaperone BiP (aka KAR2) | YJL034W | Role in facilitating assembly of multimeric protein complexes in ER—required for secretory polypeptide translocation | 1.7 |
| Cytochrome b-c1 complex subunit 10 QCR10 | YHR001W-A | Part of the mitochondrial respiratory chain that generates electrochemical potential coupled to ATP synthesis | 1.7 |
| V-Type proton ATPase subunit B | YBR127C | Non-catalytic subunit of V-ATPase: electrogenic proton pump generating proton motive force of 180 mV | 1.7 |
| Sulfiredoxin | YKL086W | Contributes to oxidative stress resistance by reducing cysteine-sulfinic acid formed by oxidants in the peroxiredoxin TSA1 | 1.6 |
| Vacuolar morphogenesis protein 10 | YOR068C | Required for vacuolar fusion; involved in the early steps of the fusion pathway | 1.6 |
| Threonine-tRNA ligase, mitochondrial | YKL194C | - | 1.6 |
| Mitochondrial peroxiredoxin PRX1 | YBL064C | Involved in mitochondrial protection from oxidative stress | 1.6 |
| Inheritance of peroxisomes protein 1 | YMR204C | Inhibition of peroxisomes | 1.6 |
| Elongation factor 3A | YLR249W | Release of deacylated tRNA from ribosomal E-site during synthesis | 1.6 |
| Heat shock protein SSA2 | YLLO24C | Transport polypeptides both across the mitochondrial membranes and into the ER | 1.6 |
| Glutathione peroxidase-like peroxiredoxin 2 GPX2 | YBR224W | Protects cells from phospholipid hydroperoxides and nonphospholipid peroxides during oxidative stress | 1.6 |
| Glutathione peroxidase-like peroxiredoxin HYR1 | YIRO37W | Oxidative stress response pathway | 1.4 |
| ATP synthase subunit f, mitochondrial | YDR377W | Mitochondrial membrane ATP synthase | 1.4 |
| Heat shock protein SSA1 | YALOO5C | Role in the transport of polypeptides both across the mitochondrial membranes and into the endoplasmic reticulum | 1.4 |
| GO Term ID | Term Description | Observed Gene Count | Background Gene Count | False Discovery Rate |
|---|---|---|---|---|
| GO:0046034 | ATP metabolic process | 31 | 94 | 2.55 × 10−12 |
| GO:0009167 | purine ribonucleoside monophosphate metabolic process | 32 | 118 | 1.74 × 10−11 |
| GO:0009161 | ribonucleoside monophosphate metabolic process | 33 | 136 | 6.23 × 10−11 |
| GO:0022900 | electron transport chain | 25 | 74 | 1.09 × 10−10 |
| GO:0009150 | purine ribonucleotide metabolic process | 32 | 147 | 1.29 × 10−09 |
| GO:0006119 | oxidative phosphorylation | 18 | 39 | 2.27 × 10−09 |
| GO:0009259 | ribonucleotide metabolic process | 33 | 162 | 2.27 × 10−09 |
| GO:0019693 | ribose phosphate metabolic process | 35 | 182 | 2.27 × 10−09 |
| GO:0042775 | mitochondrial ATP synthesis coupled electron transport | 17 | 37 | 7.14 × 10−09 |
| GO:0022904 | respiratory electron transport chain | 18 | 45 | 1 1.07 × 10−08 |
| GO:0009117 | nucleotide metabolic process | 39 | 250 | 2.69 × 10−08 |
| GO:1902600 | proton transmembrane transport | 25 | 108 | 4.38 × 10−08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollo-Oliveira, L.; Klassen, R.; Davis, N.; Ciftci, A.; Bacusmo, J.M.; Martinelli, M.; DeMott, M.S.; Begley, T.J.; Dedon, P.C.; Schaffrath, R.; et al. Loss of Elongator- and KEOPS-Dependent tRNA Modifications Leads to Severe Growth Phenotypes and Protein Aggregation in Yeast. Biomolecules 2020, 10, 322. https://doi.org/10.3390/biom10020322
Pollo-Oliveira L, Klassen R, Davis N, Ciftci A, Bacusmo JM, Martinelli M, DeMott MS, Begley TJ, Dedon PC, Schaffrath R, et al. Loss of Elongator- and KEOPS-Dependent tRNA Modifications Leads to Severe Growth Phenotypes and Protein Aggregation in Yeast. Biomolecules. 2020; 10(2):322. https://doi.org/10.3390/biom10020322
Chicago/Turabian StylePollo-Oliveira, Leticia, Roland Klassen, Nick Davis, Akif Ciftci, Jo Marie Bacusmo, Maria Martinelli, Michael S. DeMott, Thomas J. Begley, Peter C. Dedon, Raffael Schaffrath, and et al. 2020. "Loss of Elongator- and KEOPS-Dependent tRNA Modifications Leads to Severe Growth Phenotypes and Protein Aggregation in Yeast" Biomolecules 10, no. 2: 322. https://doi.org/10.3390/biom10020322
APA StylePollo-Oliveira, L., Klassen, R., Davis, N., Ciftci, A., Bacusmo, J. M., Martinelli, M., DeMott, M. S., Begley, T. J., Dedon, P. C., Schaffrath, R., & de Crécy-Lagard, V. (2020). Loss of Elongator- and KEOPS-Dependent tRNA Modifications Leads to Severe Growth Phenotypes and Protein Aggregation in Yeast. Biomolecules, 10(2), 322. https://doi.org/10.3390/biom10020322