The Challenge of Disease-Modifying Therapies in Parkinson’s Disease: Role of CSF Biomarkers



Abstract
:1. Introduction
2. Molecular Targets for Disease-Modifying Strategies in PD
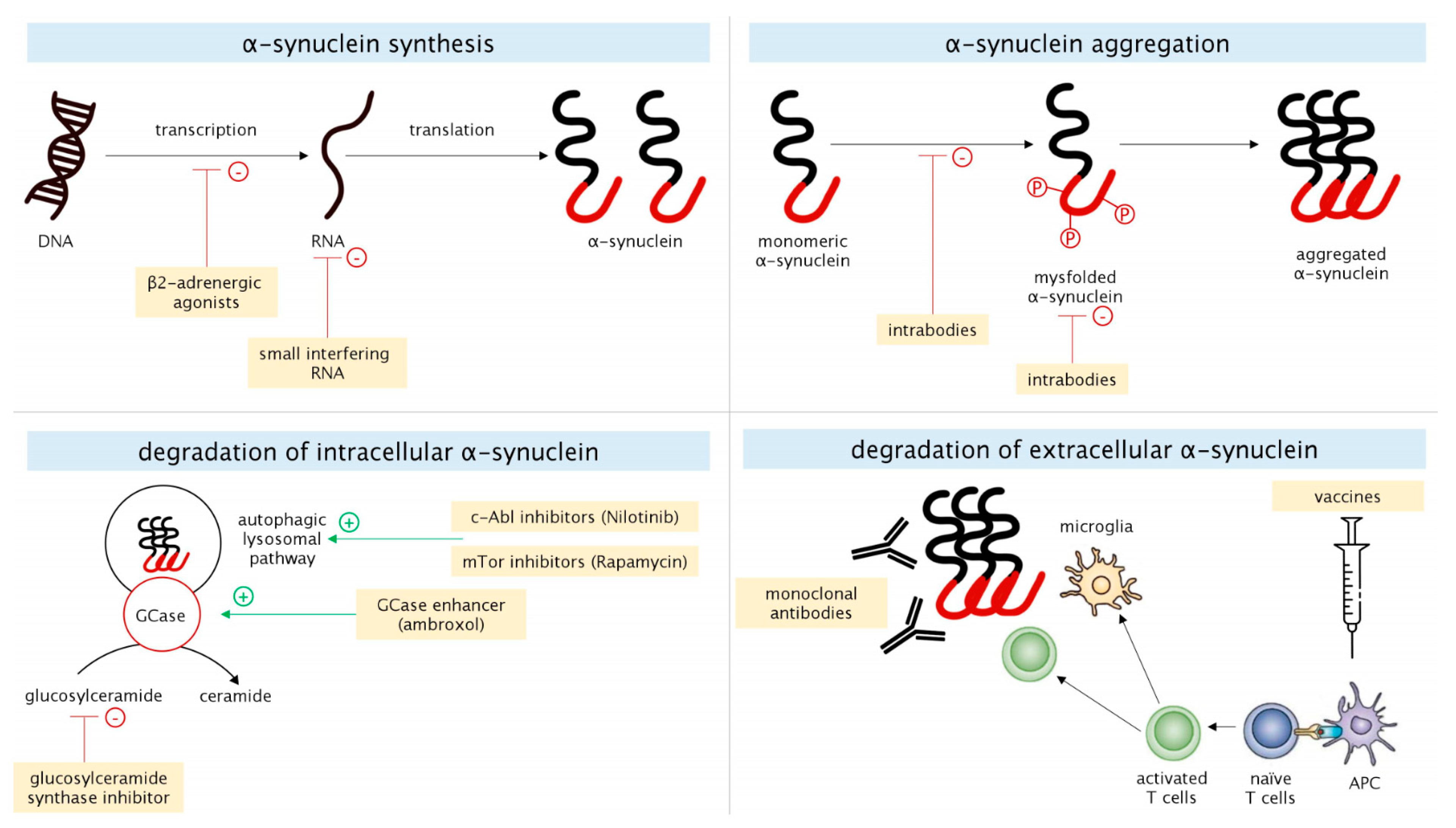
2.1. α-Synuclein
2.1.1. α-Synuclein Synthesis
2.1.2. α-Synuclein Aggregation
2.1.3. Degradation of Intracellular α-Synuclein
2.1.4. Degradation of Extracellular α-Synuclein
2.2. GBA
2.2.1. GCase Activity
2.2.2. GBA-Related Glycosphingolipids Metabolism
2.3. LRRK2
2.4. Other Molecular Targets
3. CSF Biomarkers: An Overview
4. Disease-Modifying Therapies in PD: Challenges, Open Issues and Potential Role of CSF Biomarkers
4.1. The Issue of Pathophysiological and Clinical Heterogeneity
Role of CSF Biomarkers
4.2. The Issue of Pre-Motor Diagnosis
Role of CSF Biomarkers
4.3. The Issue of Reliable Outcomes for Treatment Monitoring
Role of CSF Biomarkers
4.4. State of Art of Ongoing Clinical Trials Adopting CSF Biomarkers as Outcome Measures
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Parkinson study group. Datatop: A multicenter controlled clinical trial in early parkinson’s disease. Arch. Neurol. 1989, 46, 1052–1060. [Google Scholar] [CrossRef]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Marek, K.; Seibyl, J.; Lang, A.; Olanow, C.W.; Tanner, C.; et al. Levodopa and the progression of parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [PubMed]
- Rascol, O.; Fitzer-Attas, C.J.; Hauser, R.; Jankovic, J.; Lang, A.; Langston, J.W.; Melamed, E.; Poewe, W.; Stocchi, F.; Tolosa, E.; et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): Prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol. 2011, 10, 415–423. [Google Scholar] [CrossRef]
- Verschuur, C.V.M.; Suwijn, S.R.; Boel, J.A.; Post, B.; Bloem, B.R.; Van Hilten, J.J.; Van Laar, T.; Tissingh, G.; Munts, A.G.; Deuschl, G.; et al. Randomized delayed-start trial of levodopa in Parkinson’s disease. N. Engl. J. Med. 2019, 11, 145–152. [Google Scholar] [CrossRef]
- Sardi, S.P.; Cedarbaum, J.M.; Brundin, P. Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov. Disord. 2018, 33, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-synuclein oligomers-neurotoxic molecules in Parkinson’s disease and other lewy body disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- McCormack, A.L.; Mak, S.K.; Henderson, J.M.; Bumcrot, D.; Farrer, M.; Di Monte, N.A. α-Synuclein Suppression by Targeted Small Interfering RNA in the Primate Substantia Nigra. PLoS ONE 2010, 5, e12122. [Google Scholar] [CrossRef]
- Zharikov, A.D.; Cannon, J.R.; Tapias, V.; Bai, Q.; Horowitz, M.P.; Shah, V.; El Ayadi, A.; Hastings, T.G.; Greenamyre, J.T.; Burton, E.A. ShRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Invest. 2015, 125, 2721–2735. [Google Scholar] [CrossRef] [Green Version]
- Cole, T.; Paumier, K.; Zhao, H.; Weihofen, A.; Kordasiewicz, H.; Swayze, E. Snca targeted antisense oligonucleotides mediate progression of pathological deposition in alpha synuclein rodent transmission models of Parkinson’s disease (P6.239). Neurology 2016, 86, P6.239. [Google Scholar]
- Mittal, S.; Bjornevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, M.A.; Messer, A.; Kordower, J.H. Can intrabodies serve as neuroprotective therapies for parkinson’s disease? Beginning thoughts. J. Parkinsons Dis. 2013, 3, 581–591. [Google Scholar] [CrossRef]
- Zhang, G.; Xia, Y.; Wan, F.; Ma, K.; Guo, X.; Kou, L.; Yin, S.; Han, C.; Liu, L.; Huang, J.; et al. New Perspectives on Roles of Alpha-Synuclein in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 370. [Google Scholar] [CrossRef] [Green Version]
- Gorenberg, E.L.; Chandra, S.S. The Role of Co-chaperones in Synaptic Proteostasis and Neurodegenerative Disease. Front. Mol. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.V.; Gorenberg, E.L.; Nagy, M.; Thrasher, D.; Fenton, W.A.; Volpicelli-Daley, L.; Horwich, A.L.; Chandra, S.S. Hsp110 mitigates α-synuclein pathology in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 24310–24316. [Google Scholar]
- Karuppagounder, S.S.; Brahmachari, S.; Lee, Y.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep. 2014, 4, 4874. [Google Scholar] [CrossRef] [Green Version]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Park. Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Arellano, J.; Howard, H.H.; et al. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharmacol Res. Perspect 2019, 7, e00470. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Björklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Tyson, T.; George, S.; Hildebrandt, E.N.; Steiner, J.A.; Madaj, Z.; Schulz, E.; MacHiela, E.; McDonald, W.G.; Galvis, M.L.E.; et al. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci. Transl. Med. 2016, 8, 368ra174. [Google Scholar] [CrossRef]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti–α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti–Synuclein Monoclonal Antibody, in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; et al. Randomized phase I clinical trial of anti–α-synuclein antibody BIIB054. Mov. Disord. 2019, 34, 1154–1163. [Google Scholar] [CrossRef]
- Brys, M.; Ellenbogen, A.; Fanning, L.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; Makh, S.; et al. Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose Study of Anti-Alpha-Synuclein Antibody BIIB054 in Patients with Parkinson’s Disease (S26.001). Neurology 2018, 34, 1154–1163. [Google Scholar]
- Zella, S.M.A.; Metzdorf, J.; Ciftci, E.; Ostendorf, F.; Muhlack, S.; Gold, R.; Tönges, L. Emerging Immunotherapies for Parkinson Disease. Neurol. Ther. 2019, 8, 29–44. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, O.; Svennerholm, L. Characterization and quantitative determination of gangliosides and neutral glycosphingolipids in human liver. J. Lipid Res. 1982, 23, 327–334. [Google Scholar]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Parnetti, L.; Bellomo, G. The issue of waste disposal in Parkinson’s disease pathogenesis. Mov. Disord. 2019, 34, 985. [Google Scholar] [CrossRef]
- Sardi, S.P.; Cedarbaum, J.M.; Brundin, P. Targeting lysosomal glucocerebrosidase defects in the treatment of parkinson’s disease: From genetics to therapeutics. Neurodegener. Dis. 2017, 33, 684–696. [Google Scholar]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.C.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.J.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [Green Version]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef]
- Aflaki, E.; Borger, D.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Zheng, J.; Jeon, S.; Jiang, W.; Burbulla, L.F.; Ysselstein, D.; Oevel, K.; Krainc, D.; Silverman, R.B. Correction to: Conversion of quinazoline modulators from inhibitors to activators of β-glucocerebrosidase. J. Med. Chem. 2019, 62, 1218–1230. [Google Scholar] [CrossRef]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J. Genetic Analysis of Pathways to Parkinson Disease. Neuron 2010, 68, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-J.; Kim, N.-K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef] [Green Version]
- Schapansky, J.; Nardozzi, J.D.; Lavoie, M.J. The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson’s disease. Neuroscience 2015, 302, 74–88. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.; Lim, K.S.; Kim, M.-K.; Kim, H.S.; Hong, J.; Jhee, S.; Kim, J.; Yoo, S.; Chung, Y.-T.; Lee, J.M.; et al. A first-in-human study to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of KM-819 (FAS-associated factor 1 inhibitor), a drug for Parkinson’s disease, in healthy volunteers. Drug Des. Dev. Ther. 2019, 13, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.H.; Suh, J.H.; Kim, E.H.; Kim, J.T.; Yoo, S.-E.; Kang, N. The discovery of inhibitors of Fas-mediated cell death pathway using the combined computational method. Bioorg. Med. Chem. Lett. 2013, 23, 5155–5164. [Google Scholar] [CrossRef] [Green Version]
- Webb, M.; Sideris, D.P.; Biddle, M. Modulation of mitochondrial dysfunction for treatment of disease. Bioorg. Med. Chem. Lett. 2019, 29, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Jucaite, A.; Svenningsson, P.; Rinne, J.O.; Cselényi, Z.; Varnäs, K.; Johnström, P.; Amini, N.; Kirjavainen, A.K.; Helin, S.; Minkwitz, M.; et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: A PET study in Parkinson’s disease. Brain 2015, 138, 2687–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.E.; Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov. Disord. 2018, 33, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Chiasserini, D.; Bellomo, G.; Giannandrea, D.; de Carlo, C.; Qureshi, M.M.; Ardah, M.T.; Varghese, S.; Bonanni, L.; Borroni, B.; et al. Cerebrospinal fluid Tau/α-synuclein ratio in Parkinson’s disease and degenerative dementias. Mov. Disord. 2011, 26, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnetti, L.; Farotti, L.; Eusebi, P.; Chiasserini, D.; De Carlo, C.; Giannandrea, D.; Salvadori, N.; Lisetti, V.; Tambasco, N.; Rossi, A.; et al. Differential role of CSF alpha-synuclein species, tau, and Aβ42 in Parkinson’s disease. Front. Aging Neurosci. 2014, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length α-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef]
- Majbour, N.K.; Vaikath, N.N.; Eusebi, P.; Chiasserini, D.; Ardah, M.; Varghese, S.; Haque, M.E.; Tokuda, T.; Auinger, P.; Calabresi, P.; et al. Longitudinal changes in CSF alpha-synuclein species reflect Parkinson’s disease progression. Mov. Disord. 2016, 31, 1535–1542. [Google Scholar] [CrossRef]
- Hall, S.; Surova, Y.; Öhrfelt, A.; Blennow, K.; Zetterberg, H.; Hansson, O. Longitudinal Measurements of Cerebrospinal Fluid Biomarkers in Parkinson’s Disease. Mov. Disord. 2016, 31, 898–905. [Google Scholar] [CrossRef]
- Parnetti, L.; Cicognola, C.; Eusebi, P.; Chiasserini, D. Value of cerebrospinal fluid α-synuclein species as biomarker in Parkinson’s diagnosis and prognosis. Biomark. Med. 2016, 10, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Eusebi, P.; Giannandrea, D.; Biscetti, L.; Abraha, I.; Chiasserini, D.; Orso, M.; Calabresi, P.; Parnetti, L. Diagnostic utility of cerebrospinal fluid α-synuclein in Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2017, 32, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Compta, Y.; Parkkinen, L.; O’Sullivan, S.S.; Vandrovcova, J.; Holton, J.L.; Collins, C.; Lashley, T.; Kallis, C.; Williams, D.R.; De Silva, R.; et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 2011, 134, 1493–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, G.; Lange, J.; Blennow, K.; Zetterberg, H.; Andreasson, U.; Førland, M.G.; Tysnes, O.B.; Larsen, J.P.; Pedersen, K.F. CSF Aβ 42 predicts early-onset dementia in Parkinson disease. Neurology 2014, 82, 1784–1790. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Cicognola, C.; Brinkmalm, G.; Wahlgren, J.; Portelius, E.; Gobom, J.; Cullen, N.C.; Hansson, O.; Parnetti, L.; Constantinescu, R.; Wildsmith, K.; et al. Novel tau fragments in cerebrospinal fluid: Relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, L.; Höglund, K.; Parnetti, L.; Pujol-Calderón, F.; Becker, B.; Eusebi, P.; Sarchielli, P.; Calabresi, P.; Di Filippo, M.; Zetterberg, H.; et al. A new enzyme-linked immunosorbent assay for neurofilament light in cerebrospinal fluid: Analytical validation and clinical evaluation. Alzheimer’s Res. Ther. 2018, 10, 8. [Google Scholar] [CrossRef]
- Bridel, C.; Van Wieringen, W.N.; Zetterberg, H.; Tijms, B.M.; Teunissen, C.E.; Alvarez-Cermeño, J.C.; Andreasson, U.; Axelsson, M.; Bäckström, D.C.; Bartos, A.; et al. Diagnostic Value of Cerebrospinal Fluid Neurofilament Light Protein in Neurology: A Systematic Review and Meta-analysis. JAMA Neurol. 2019. [Google Scholar] [CrossRef]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef]
- Hong, Z.; Shi, M.; Chung, K.A.; Quinn, J.; Peskind, E.R.; Galasko, U.; Jankovic, J.; Zabetian, C.; Leverenz, J.; Baird, G.; et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 2010, 133, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Schröder, J.B.; Pawlowski, M.; Meyer zu Hörste, G.; Gross, C.C.; Wiendl, H.; Meuth, S.G.; Ruck, T.; Warnecke, T. Immune Cell Activation in the Cerebrospinal Fluid of Patients With Parkinson’s Disease. Front. Neurol. 2018, 9, 1081. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, G.; Singh, K.; Tiwari, M.N.; Singh, M.P. Proteomics in Parkinson’s disease: Current trends, translational snags and future possibilities. Expert Rev. Proteom. 2010, 7, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Calne, D.B.; Mizuno, Y. The neuromythology of Parkinson’s Disease. Parkinsonism Relat. Disord. 2004, 10, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Brundin, P.; Lang, A.E. Precision medicine for disease modification in Parkinson disease. Nat. Rev. Neurol. 2017, 13, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wider, C.; Dickson, D.W.; Wszolek, Z. Leucine-rich repeat kinase 2 gene-associated disease: Redefining genotype-phenotype correlation. Neurodegener. Dis. 2010, 7, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Oertel, W.H.; Schulz, J.B. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J. Neurochem. 2016, 139, 325–337. [Google Scholar] [CrossRef]
- Kang, J.-H.; Mollenhauer, B.; Coffey, C.S.; Toledo, J.; Weintraub, D.; Galasko, U.R.; Irwin, D.J.; Van Deerlin, V.; Chen-Plotkin, A.S.; Caspell-Garcia, C.; et al. CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: The Parkinson’s Progression Markers Initiative study. Acta Neuropathol. 2016, 131, 935–949. [Google Scholar] [CrossRef]
- Goldman, J.G.; Andrews, H.; Amara, A.; Naito, A.; Alcalay, R.N.; Shaw, L.M.; Taylor, P.; Xie, T.; Tuite, P.; Henchcliffe, C.; et al. Cerebrospinal fluid, plasma, and saliva in the BioFIND study: Relationships among biomarkers and Parkinson’s disease Features. Mov. Disord. 2018, 33, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Compta, Y.; Martí, M.J.; Ibarretxe-Bilbao, N.; Junqué, C.; Valldeoriola, F.; Muñoz, E.; Ezquerra, M.; Ríos, J.; Tolosa, E. Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson’s disease. Mov. Disord. 2009, 24, 2203–2210. [Google Scholar] [CrossRef]
- Siderowf, A.; Xie, S.X.; Hurtig, H.; Weintraub, D.; Duda, J.; Chen-Plotkin, A.; Shaw, L.M.; Van Deerlin, V.; Trojanowski, J.Q.; Clark, C. CSF amyloid β 1–42 predicts cognitive decline in Parkinson disease (e–Pub ahead of print). Neurology 2010, 75, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Fereshtehnejad, S.-M.; Zeighami, Y.; Dagher, A.; Postuma, R.B. Clinical criteria for subtyping Parkinson’s disease: Biomarkers and longitudinal progression. Brain 2017, 140, 1959–1976. [Google Scholar] [CrossRef]
- Mahlknecht, P.; Seppi, K.; Poewe, W. The Concept of Prodromal Parkinson’s Disease. J. Park. Dis. 2015, 5, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compta, Y.; Valente, T.; Saura, J.; Segura, B.; Iranzo, Á.; Serradell, M.; Junqué, C.; Tolosa, E.; Valldeoriola, F.; Muñoz, E.; et al. Correlates of cerebrospinal fluid levels of oligomeric- and total-α-synuclein in premotor, motor and dementia stages of Parkinson’s disease. J. Neurol. 2015, 262, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Ishii, R.; Mollenhauer, B.; Soto, C.; Mendez, N.; Trenkwalder, C. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of α-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 2017, 74, 163. [Google Scholar] [CrossRef]
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818. [Google Scholar] [CrossRef]
- Tokuda, T.; Salem, S.A.; Allsop, D.; Mizuno, T.; Nakagawa, M.; Qureshi, M.M.; Locascio, J.J.; Schlossmacher, M.G.; El-Agnaf, O. Decreased α-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 349, 162–166. [Google Scholar] [CrossRef]
- Kang, J.-H.; Irwin, D.J.; Chen-Plotkin, A.S.; Siderowf, A.; Caspell, C.; Coffey, C.S.; Waligorska, T.; Taylor, P.; Pan, S.; Frasier, M.; et al. Association of cerebrospinal fluid β-amyloid 1–42, T-tau, P-tau181, and α-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol. 2013, 70, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Aasly, J.; Johansen, K.K.; Brønstad, G.; Warø, B.J.; Majbour, N.K.; Varghese, S.; Alzahmi, F.; Paleologou, K.E.; Amer, D.A.M.; Al-Hayani, A.; et al. Elevated levels of cerebrospinal fluid α-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front. Aging Neurosci. 2014, 6, 248. [Google Scholar] [CrossRef]
- Stewart, T.; Sossi, V.; Aasly, J.; Wszolek, Z.; Uitti, R.J.; Hasegawa, K.; Yokoyama, T.; Zabetian, C.; Leverenz, J.; Stoessl, A.J.; et al. Phosphorylated α-synuclein in Parkinson’s disease: Correlation depends on disease severity. Acta Neuropathol. Commun. 2015, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.; Surova, Y.; Öhrfelt, A.; Zetterberg, H.; Lindqvist, D.; Hansson, O. CSF biomarkers and clinical progression of Parkinson disease. Neurology 2015, 84, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Lleó, A.; Cavedo, E.; Parnetti, L.; Vanderstichele, H.; Herukka, S.K.; Andreasen, N.; Ghidoni, R.; Lewczuk, P.; Jeromin, A.; Winblad, B.; et al. Cerebrospinal fluid biomarkers in trials for Alzheimer and Parkinson diseases. Nat. Rev. Neurol. 2015, 11, 41–55. [Google Scholar] [CrossRef]
- Farotti, L.; Paciotti, S.; Tasegian, A.; Eusebi, P.; Parnetti, L. Discovery, validation and optimization of cerebrospinal fluid biomarkers for use in Parkinson’s disease. Expert Rev. Mol. Diagn. 2017, 17, 771–780. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CSF Biomarkers | Targeting Mechanism | Drug | CinicalTrial.gov Identifier | Phase | Recruited Subjects | Period |
|---|---|---|---|---|---|---|
| α-syn (t-α-syn; o-α-syn) | c-Abl inhibition | Nilotinib | NCT022814774 | I | 12 PDD patients | Nov. 2014–May 2015 |
| Nilotinib | NCT02954978 | II | 75 PD patients | Jan. 2017–ongoing | ||
| Anti-α-syn antibody | MEDI1341 | NCT03272165 | I | 48 healthy volunteers | Oct. 2017–ongoing | |
| Anti-α-syn vaccine | AFFITOPE® PD01A | NCT01568099 | I | 32 PD patients | Feb. 2012–May 2014 | |
| NCT02216188 | I | 28 PD patients | Aug. 2014–Aug. 2015 | |||
| GCase activation | Ambroxol | NCT02914366 | II | 75 PDD patients | Nov. 2015–ongoing | |
| FAF1 inhibition | KM-819 | NCT03022799 | I | 88 healthy volunteers | Oct. 2016–Oct. 2017 | |
| Tau proteins (t-tau; p-tau) | c-Abl inhibition | Nilotinib | NCT022814774 | I | 12 PDD patients | Nov. 2014–May 2015 |
| GCase activation | Ambroxol | NCT02914366 | II | 75 PDD patients | Nov. 2015–ongoing | |
| FAF1 inhibition | KM-819 | NCT03022799 | I | 88 healthy volunteers | Oct. 2016–Oct. 2017 | |
| Aβ-42 | c-Abl inhibition | Nilotinib | NCT022814774 | I | 12 PDD patients | Nov. 2014–May 2015 |
| GCase activation | Ambroxol | NCT02914366 | II | 75 PDD patients | Nov. 2015–ongoing | |
| GCase activity | GCase activation | Ambroxol | NCT02941822 | II | 20 PD patients (10 GBA+, 10 GBA-) | Dec. 2016–Apr. 2018 |
| Neuronal and glial death biomarkers (NSE; S100B) | c-Abl inhibition | Nilotinib | NCT022814774 | I | 12 PDD patients | Nov. 2014–May 2015 |
| Dopamine metabolites (HVA; DOPAC) | c-Abl inhibition | Nilotinib | NCT022814774 | I | 12 PDD patients | Nov. 2014–May 2015 |
| c-Abl inhibition | Nilotinib | NCT02954978 | II | 75 PD patients | Jan. 2017–ongoing |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolini Paoletti, F.; Gaetani, L.; Parnetti, L. The Challenge of Disease-Modifying Therapies in Parkinson’s Disease: Role of CSF Biomarkers. Biomolecules 2020, 10, 335. https://doi.org/10.3390/biom10020335
Paolini Paoletti F, Gaetani L, Parnetti L. The Challenge of Disease-Modifying Therapies in Parkinson’s Disease: Role of CSF Biomarkers. Biomolecules. 2020; 10(2):335. https://doi.org/10.3390/biom10020335
Chicago/Turabian StylePaolini Paoletti, Federico, Lorenzo Gaetani, and Lucilla Parnetti. 2020. "The Challenge of Disease-Modifying Therapies in Parkinson’s Disease: Role of CSF Biomarkers" Biomolecules 10, no. 2: 335. https://doi.org/10.3390/biom10020335
APA StylePaolini Paoletti, F., Gaetani, L., & Parnetti, L. (2020). The Challenge of Disease-Modifying Therapies in Parkinson’s Disease: Role of CSF Biomarkers. Biomolecules, 10(2), 335. https://doi.org/10.3390/biom10020335