Site-Specific Fluorogenic Protein Labelling Agent for Bioconjugation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
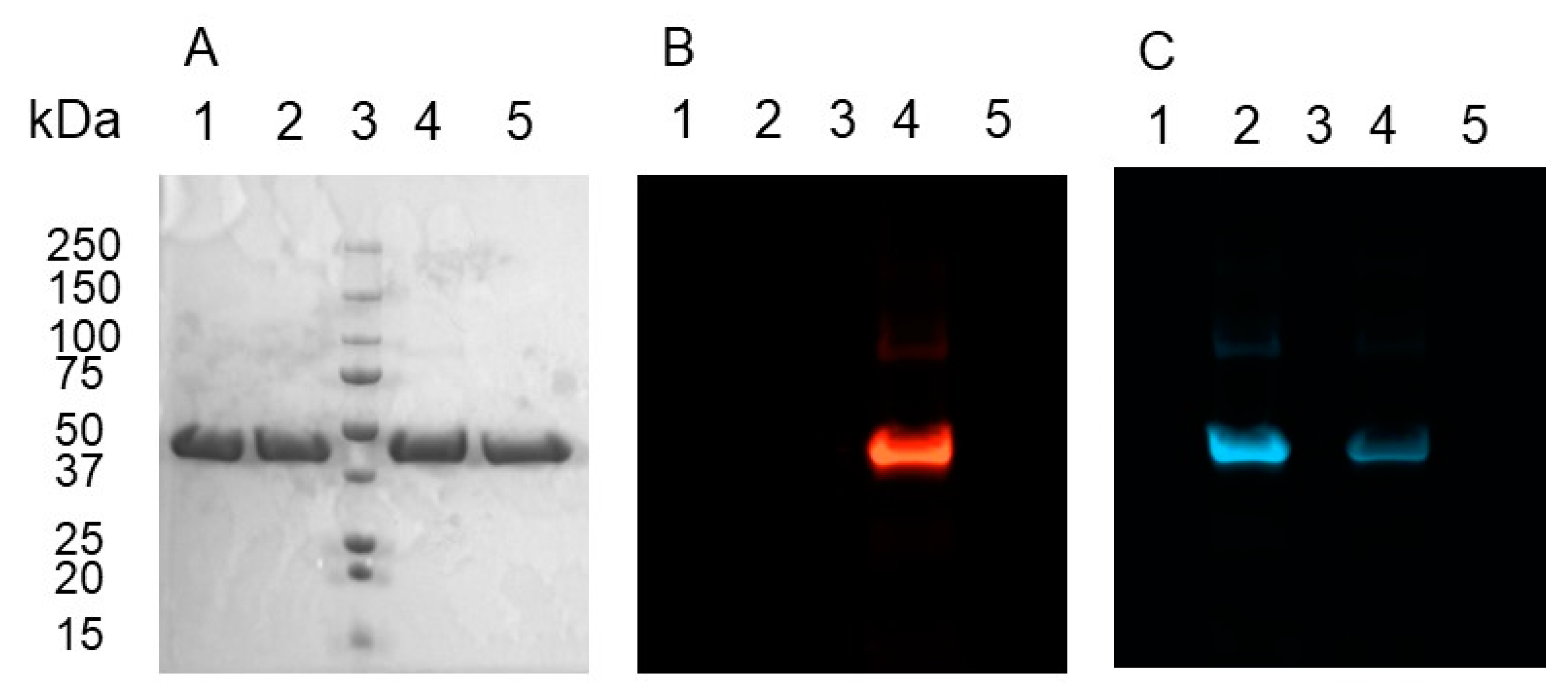
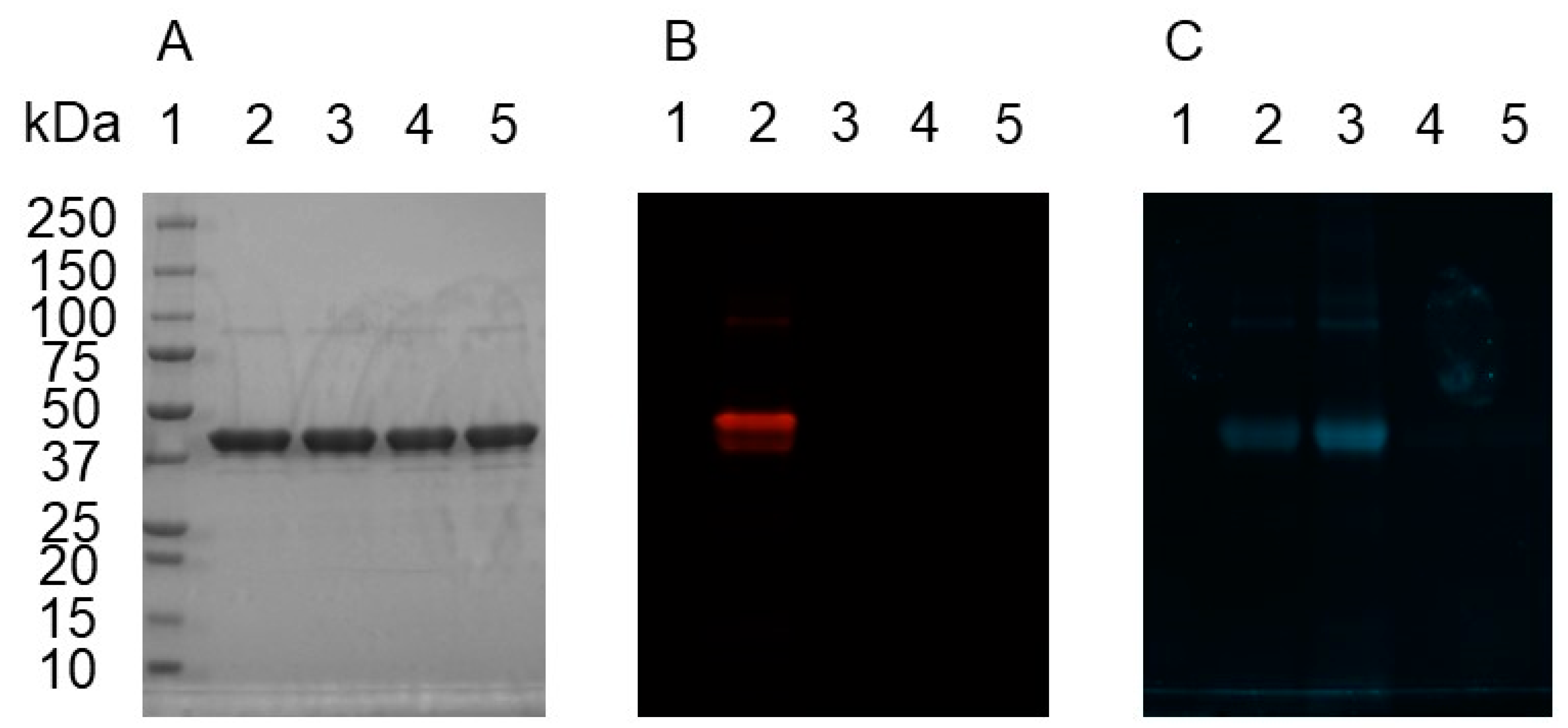
2.1. Expression and Purification of Test Protein
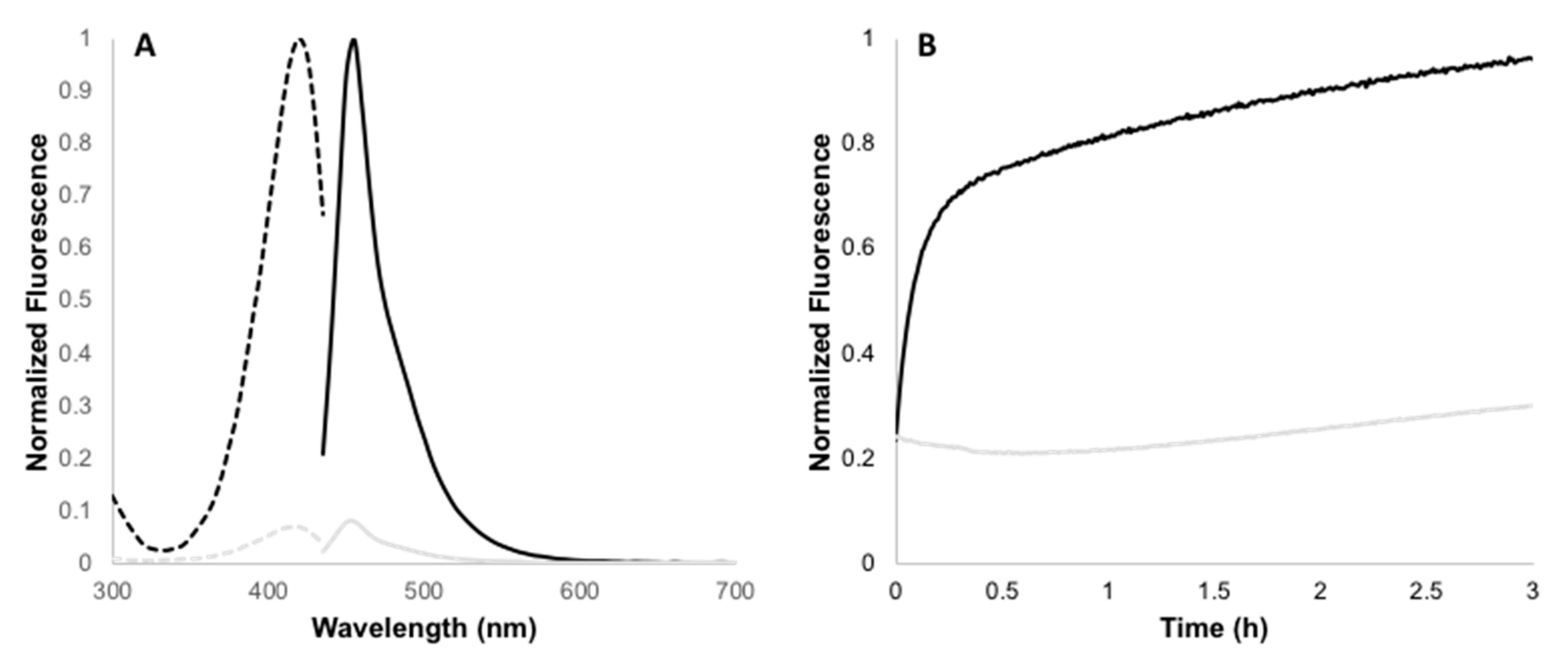
2.2. Determination of Fluorescence Enhancement Ratios
2.3. Kinetic Studies
2.4. Bioconjugation of Rhodamine B Azide to MBP-dC10*-15 or 6 Adduct
2.5. Bioconjugation of Rh-B-N3-15 Adduct to MBP-dC10*
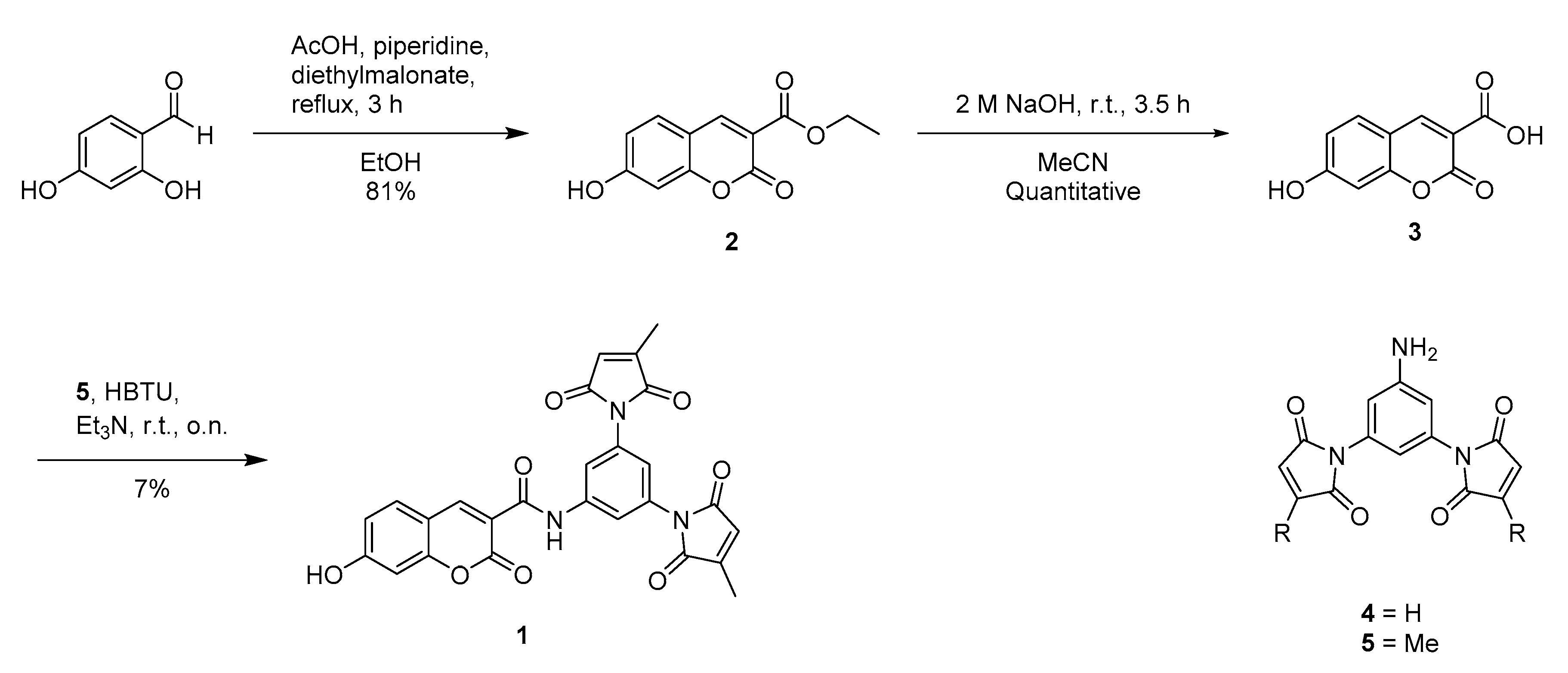
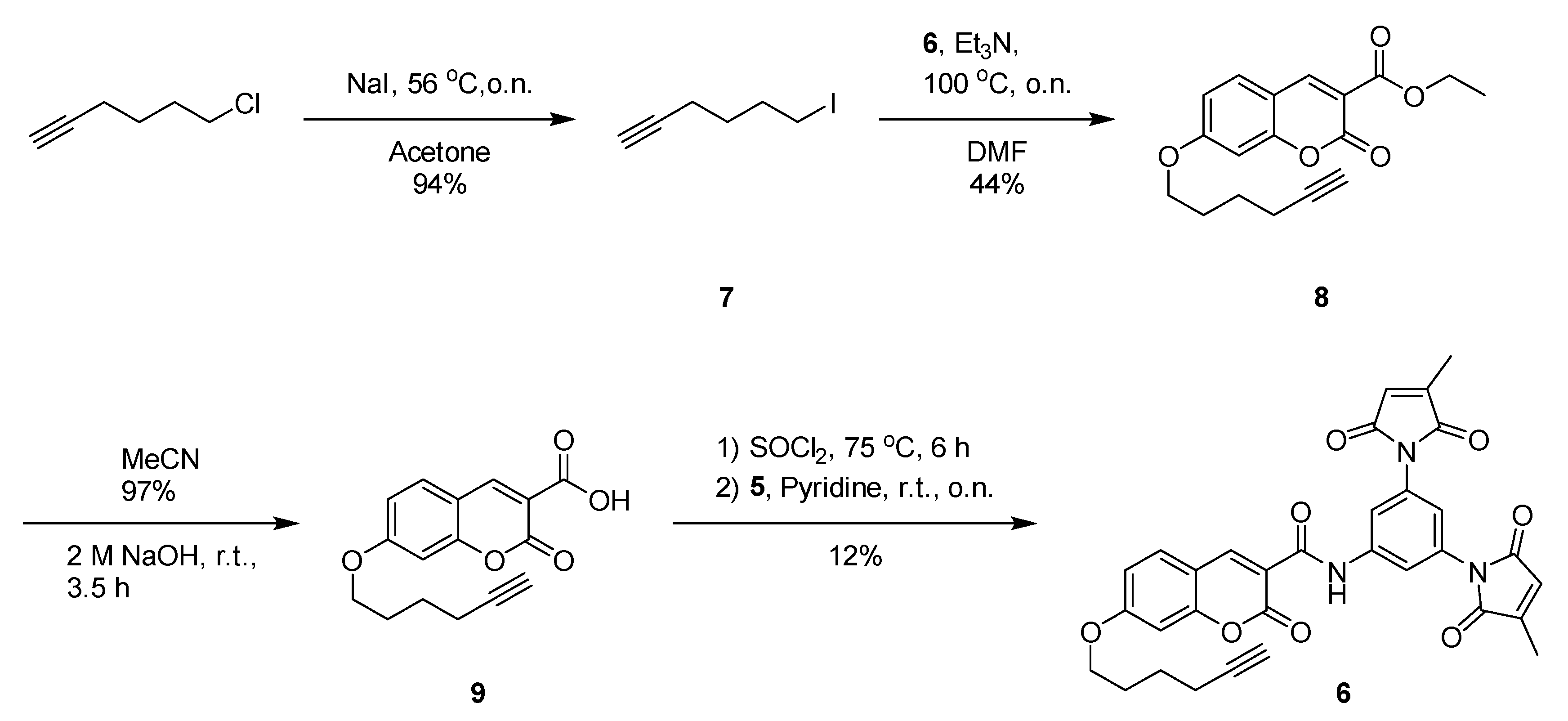
2.6. Synthesis
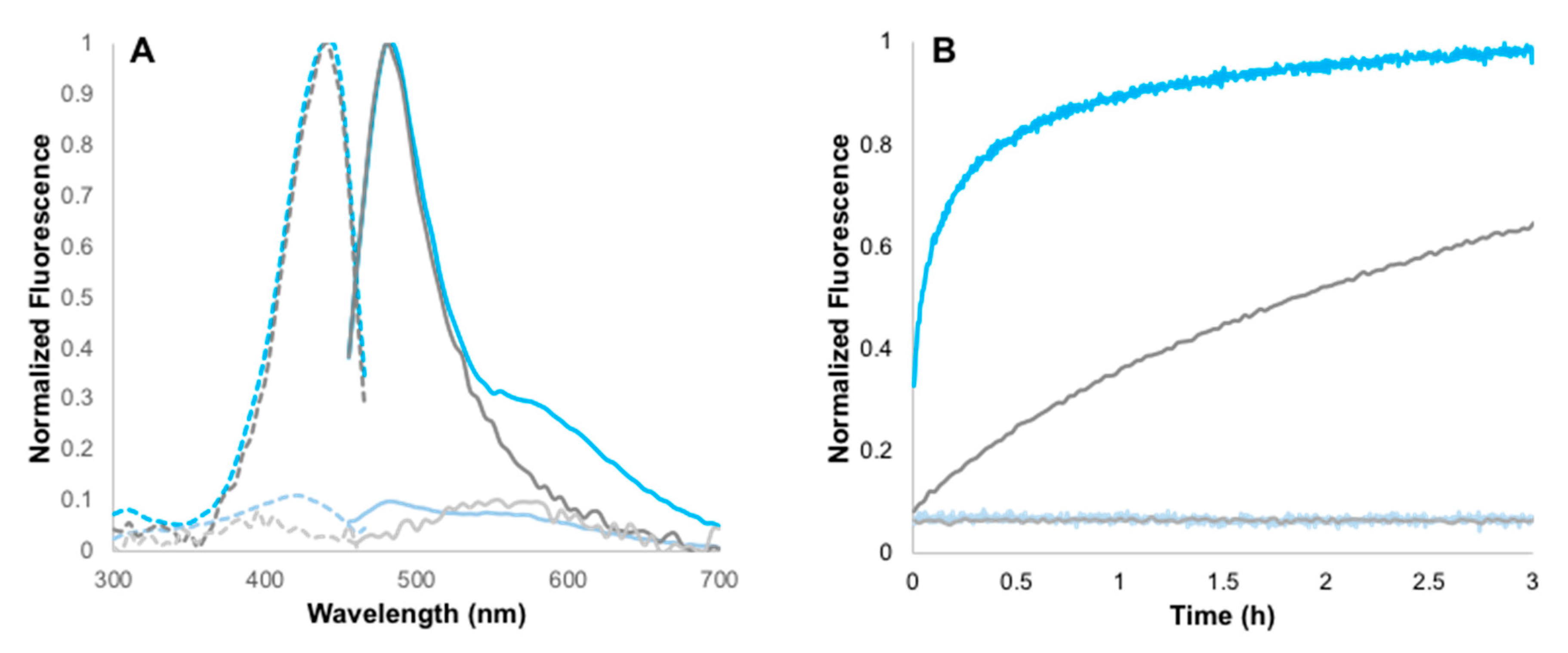
3. Results and Discussion
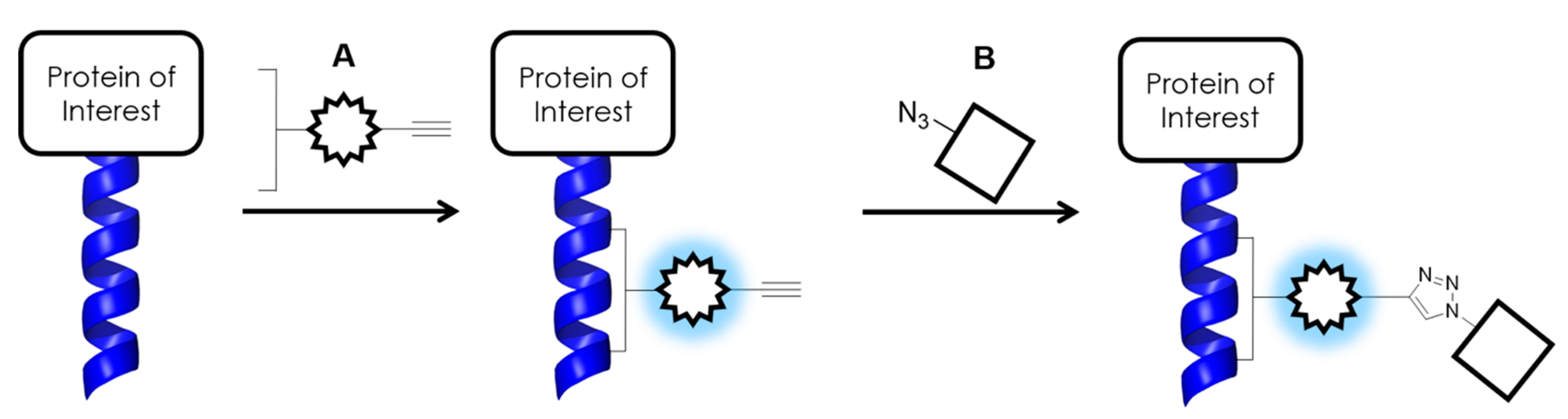
3.1. Initial Design
3.2. Improved Design
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pasut, G.; Veronese, F.M. PEG conjugates in clinical development or use as anticancer agents: An overview. Adv. Drug Deliv. Rev. 2009, 61, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Canalle, L.A.; Löwik, D.W.P.M.; Van Hest, J.C.M. Polypeptide-polymer bioconjugates. Chem. Soc. Rev. 2010, 39, 329–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Marklew, J. Emerging formats for next-generation antibody drug conjugates. Expert Opin. Drug Discov. 2015, 10, 463–481. [Google Scholar] [CrossRef]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody-drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef]
- Veronese, F.M. Peptide and protein PEGylation: a review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar] [CrossRef]
- Yamada, K.; Ito, Y. Recent Chemical Approaches for Site-Specific Conjugation of Native Antibodies: Technologies toward Next-Generation Antibody–Drug Conjugates. ChemBioChem 2019, 20, 2729–2737. [Google Scholar] [CrossRef]
- Kim, J.S.; Sirois, A.R.; Vazquez Cegla, A.J.; Jumai’An, E.; Murata, N.; Buck, M.E.; Moore, S.J. Protein-Polymer Conjugates Synthesized Using Water-Soluble Azlactone-Functionalized Polymers Enable Receptor-Specific Cellular Uptake toward Targeted Drug Delivery. Bioconjug. Chem. 2019, 30, 1220–1231. [Google Scholar] [CrossRef]
- Klok, H.A. Peptide/protein-synthetic polymer conjugates: Quo vadis. Macromolecules 2009, 42, 7990–8000. [Google Scholar] [CrossRef]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- Pelegri-Oday, E.M.; Lin, E.W.; Maynard, H.D. Therapeutic protein-polymer conjugates: Advancing beyond pegylation. J. Am. Chem. Soc. 2014, 136, 14323–14332. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody-drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Hu, J.; Liu, S. Emerging Applications of Fluorogenic and Non-fluorogenic Bifunctional Linkers. Chemistry. 2018, 24, 16484–16505. [Google Scholar] [CrossRef]
- Liu, G.; Shi, G.; Sheng, H.; Jiang, Y.; Liang, H.; Liu, S. Doubly Caged Linker for AND-Type Fluorogenic Construction of Protein/Antibody Bioconjugates and In Situ Quantification. Angew. Chem. Int. Ed. Engl. 2017, 56, 8686–8691. [Google Scholar] [CrossRef]
- Robin, M.P.; Wilson, P.; Mabire, A.B.; Kiviaho, J.K.; Raymond, J.E.; Haddleton, D.M.; O’Reilly, R.K. Conjugation-induced fluorescent labeling of proteins and polymers using dithiomaleimides. J. Am. Chem. Soc. 2013, 135, 2875–2878. [Google Scholar] [CrossRef]
- Dirks, A.T.J.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Monitoring Protein- Polymer Conjugation by a Fluorogenic Cu (I)-Catalyzed Azide- Alkyne 1, 3-Dipolar Cycloaddition. Bioconjug. Chem. 2009, 20, 1129–1138. [Google Scholar] [CrossRef]
- Chen, Y.; Tsao, K.; Acton, S.L.; Keillor, J.W. A Green BODIPY-Based, Super-Fluorogenic, Protein-Specific Labelling Agent. Angew. Chemie Int. Ed. 2018, 57, 12390–12394. [Google Scholar] [CrossRef]
- Chen, Y.; Clouthier, C.M.; Tsao, K.; Strmiskova, M.; Lachance, H.; Keillor, J.W. Coumarin-based fluorogenic probes for no-wash protein labeling. Angew. Chem. Int. Ed. Engl. 2014, 53, 13785–13788. [Google Scholar] [CrossRef]
- Chen, Y.; Tsao, K.; Keillor, J.W. Fluorogenic protein labelling: a review of photophysical quench mechanisms and principles of fluorogen design. Can. J. Chem. 2014, 398, 1–10. [Google Scholar] [CrossRef]
- Guy, J.; Caron, K.; Dufresne, S.; Michnick, S.W.; Skene, W.G.; Keillor, J.W. Convergent preparation and photophysical characterization of dimaleimide dansyl fluorogens: elucidation of the maleimide fluorescence quenching mechanism. J. Am. Chem. Soc. 2007, 129, 11969–11977. [Google Scholar] [CrossRef] [PubMed]
- Strmiskova, M.; Tsao, K.; Keillor, J.W. Rational design of a highly reactive dicysteine peptide tag for fluorogenic protein labelling. Org. Biomol. Chem. 2018, 16, 6332–6340. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Castonguay, R.; Campos-Reales Pineda, N.B.; Jacquier, V.; Caron, K.; Michnick, S.W.; Keillor, J.W. De novo helical peptides as target sequences for a specific, fluorogenic protein labelling strategy. Mol. Biosyst. 2010, 6, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Nasheri, N.; Joyce, M.; Rouleau, Y.; Yang, P.; Yao, S.; Tyrrell, D.L.; Pezacki, J.P. Modulation of fatty acid synthase enzyme activity and expression during hepatitis C virus replication. Chem. Biol. 2013, 20, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.Y.; Liu, K.; Ngai, M.H.; Lear, M.J.; Wenk, M.R.; Yao, S.Q. Activity-based proteome profiling of potential cellular targets of orlistat - An FDA-approved drug with anti-tumor activities. J. Am. Chem. Soc. 2010, 132, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Abdizadeh, T.; Kalani, M.R.; Abnous, K.; Tayarani-Najaran, Z.; Khashyarmanesh, B.Z.; Abdizadeh, R.; Ghodsi, R.; Hadizadeh, F. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur. J. Med. Chem. 2017, 132, 42–62. [Google Scholar] [CrossRef]
- Jiang, X.R.; Wang, P.; Smith, C.L.; Zhu, B.T. Synthesis of novel estrogen receptor antagonists using metal-catalyzed coupling reactions and characterization of their biological activity. J. Med. Chem. 2013, 56, 2779–2790. [Google Scholar] [CrossRef] [Green Version]
- Takakura, H.; Sasakura, K.; Ueno, T.; Urano, Y.; Terai, T.; Hanaoka, K.; Tsuboi, T.; Nagano, T. Development of luciferin analogues bearing an amino group and their application as BRET donors. Chem. Asian J. 2010, 5, 2053–2061. [Google Scholar] [CrossRef]
- Chen, Y.; Tsao, K.; De Francesco, É.; Keillor, J.W. Ring Substituent Effects on the Thiol Addition and Hydrolysis Reactions of N-Arylmaleimides. J. Org. Chem. 2015, 80, 12182–12192. [Google Scholar] [CrossRef]
- Jewett, J.C.; Bertozzi, C.R. Synthesis of a fluorogenic cyclooctyne activated by Cu-free click chemistry. Org. Lett. 2011, 13, 5937–5939. [Google Scholar] [CrossRef] [Green Version]
- Grimm, J.B.; English, B.P.; Chen, J.; Slaughter, J.P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J.J.; Normanno, D.; Singer, R.H.; et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 2015, 12, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Kwan, D.H.; Ernst, S.; Kötzler, M.P.; Withers, S.G. Chemoenzymatic Synthesis of a Type 2 Blood Group A Tetrasaccharide and Development of High-throughput Assays Enables a Platform for Screening Blood Group Antigen-cleaving Enzymes. Glycobiology 2015, 25, 806–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, F.; Petrella, A.; Chacón-Huete, F.; Covone, J.; Tsai, T.W.; Yu, C.C.; Forgione, P.; Kwan, D.H. A High-Throughput Glycosyltransferase Inhibition Assay for Identifying Molecules Targeting Fucosylation in Cancer Cell-Surface Modification. ACS Chem. Biol. 2019, 14, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Lin, W.; Zheng, K.; Zhu, S. FRET-based small-molecule fluorescent probes: Rational design and bioimaging applications. Acc. Chem. Res. 2013, 46, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-F.; Zhang, T.; Shen, S.-L.; Miao, J.-Y.; Zhao, B.-X. A ratiometric lysosomal pH probe based on the coumarin–rhodamine FRET system. RSC Adv. 2015, 5, 49115–49121. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsao, K.K.; Lee, A.C.; Racine, K.É.; Keillor, J.W. Site-Specific Fluorogenic Protein Labelling Agent for Bioconjugation. Biomolecules 2020, 10, 369. https://doi.org/10.3390/biom10030369
Tsao KK, Lee AC, Racine KÉ, Keillor JW. Site-Specific Fluorogenic Protein Labelling Agent for Bioconjugation. Biomolecules. 2020; 10(3):369. https://doi.org/10.3390/biom10030369
Chicago/Turabian StyleTsao, Kelvin K., Ann C. Lee, Karl É. Racine, and Jeffrey W. Keillor. 2020. "Site-Specific Fluorogenic Protein Labelling Agent for Bioconjugation" Biomolecules 10, no. 3: 369. https://doi.org/10.3390/biom10030369